
Synthesis, Characterization, DFT, and In Silico Investigation of Two Newly Synthesized β-Diketone Derivatives as Potent COX-2 Inhibitors

 ,
,  ,
,  ,
,  and
and
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Chemistry
2.2. General
General Procedure for the Synthesis of 2-(2-(Aryl)hydrazono)-5,5-dimethylcyclohexane-1,3-diones (1 and 2)
- 2-(2-(4-fluorophenyl)hydrazono)-5,5-dimethylcyclohexane-1,3-dione (1)
- 5,5-dimethyl-2-(2-(2-(trifluoromethyl)phenyl)hydrazono)cyclohexane-1,3-dione (2)
2.3. The DFT Optimizations
2.4. The Hirshfeld Surface and Energy Framework Analysis
2.5. Molecular Docking
2.5.1. The Preparations of the Ligands
2.5.2. The Preparations of the Targets
2.5.3. Protocol Used for Docking Using Glide
2.6. The ADMET Study
2.7. Molecular Dynamics Simulation
3. Results
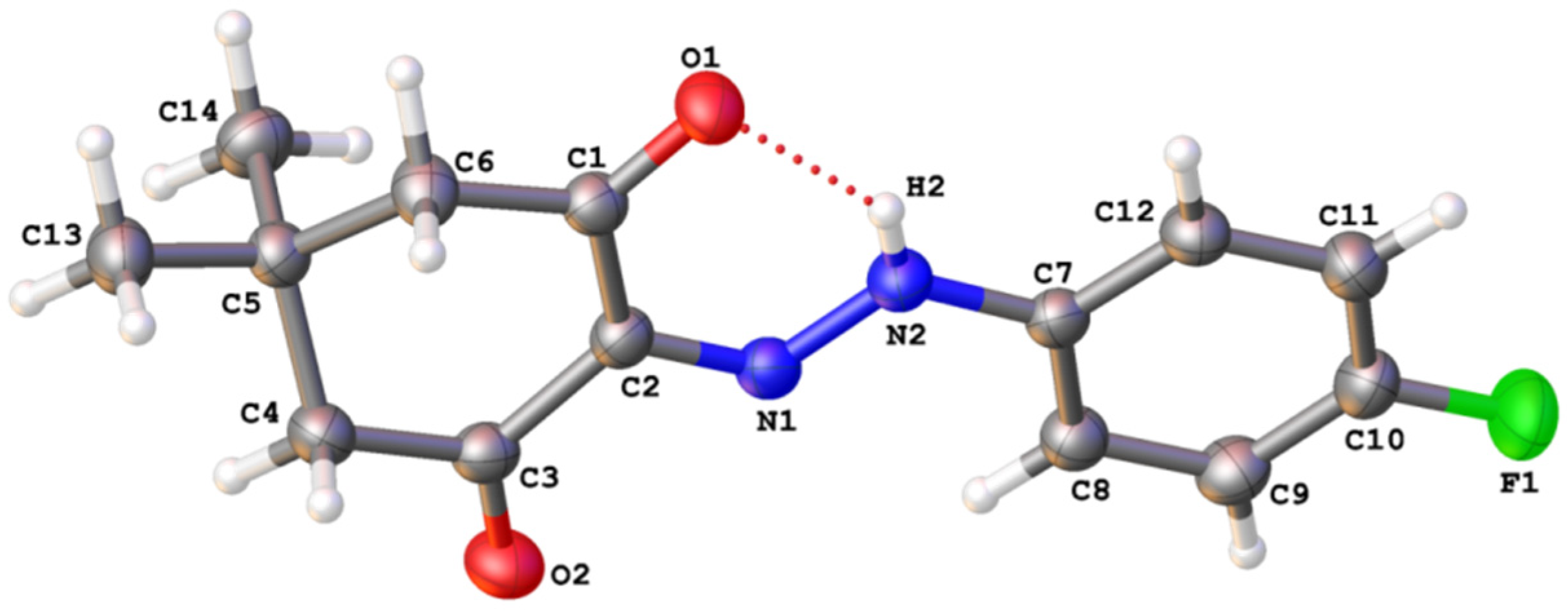
3.1. The Crystal Structures of the Studied Compounds
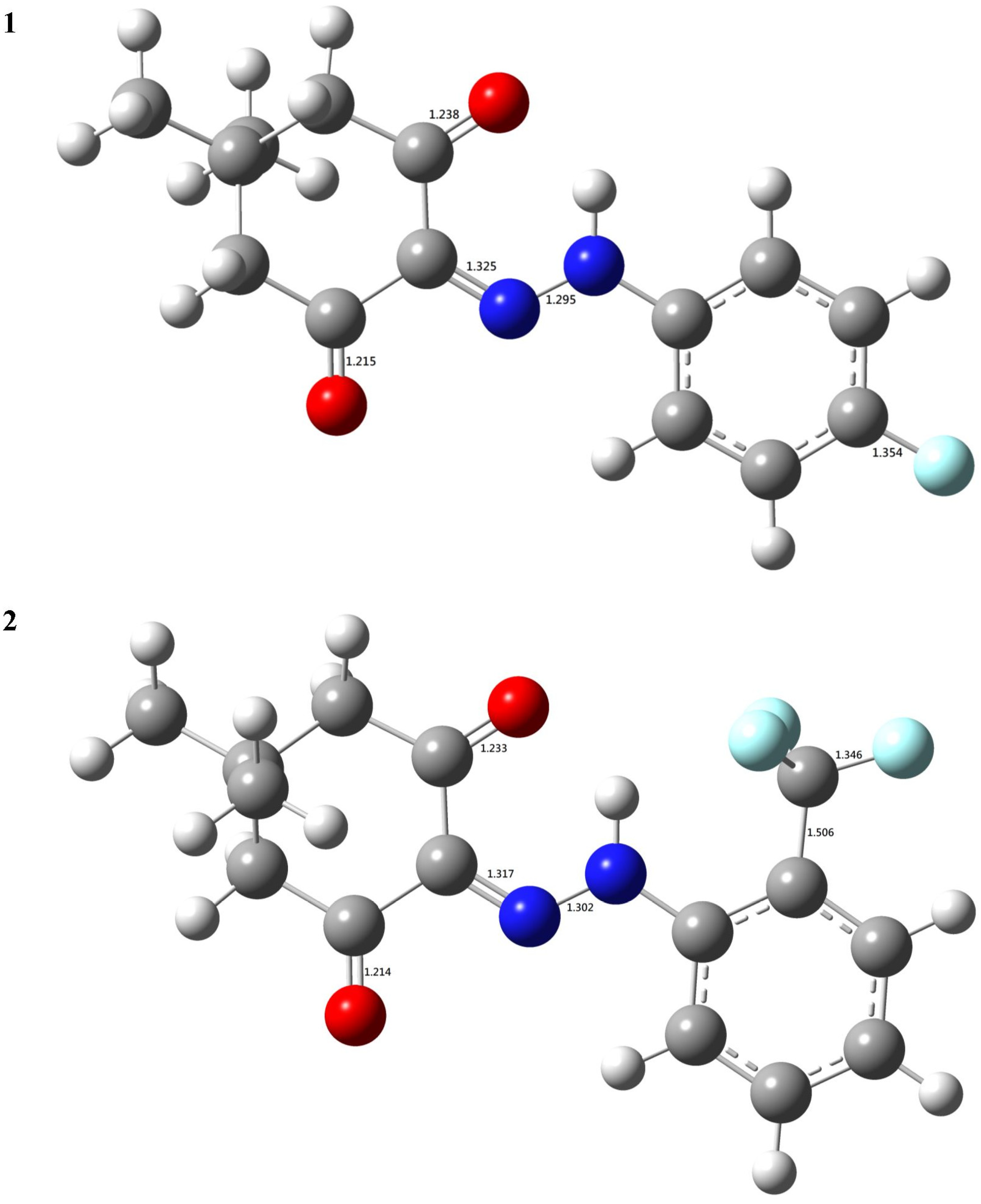
3.2. The DFT-B3LYP Study
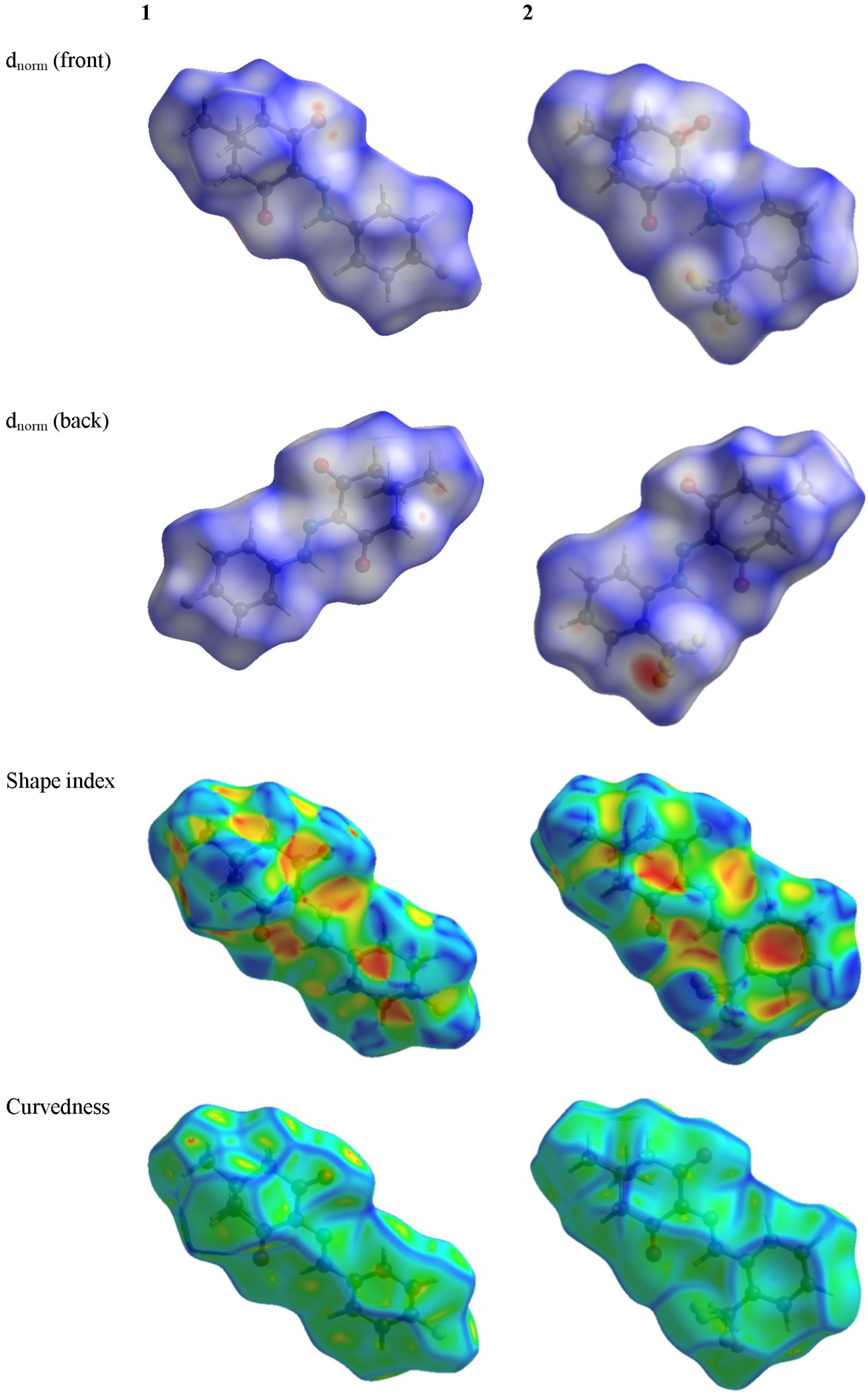
3.3. Hirshfeld Surface Analysis
3.4. Energy Framework Analysis
3.5. The Molecular Docking Study
3.6. Molecular Dynamics Analysis
3.7. The ADMET Study
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vaidya, S.R.; Shelke, V.A.; Jadhav, S.M.; Shankarwar, S.G.; Chondhekar, T.K. Synthesis and Characterization of β-Diketone Ligands and Their Antimicrobial Activity. Arch. Appl. Sci. Res. 2012, 4, 1839–1843. [Google Scholar]
- Annamalai, S.; Balasubramaniyam, A.P.; Sabeta, K.; Rajnikant, V. Synthesis, spectral and RAHB studies on some arylhydrazones of β-Diketones: Crystal and molecular structures of 2-(2-(3-pyridyl)hydrazono)-5,5-dimethylcyclohexane-1,3-dione and 2-(2-(2-methoxyphenyl)hydrazono)-5,5-dimethylcyclohexane-1,3-dione. Struct. Chem. 2011, 22, 23–33. [Google Scholar]
- Jadhav, S.M.; Shelke, V.A.; Munde, A.S.; Shankarwar, S.G.; Patharkar, V.R.; Chondhekar, T.K. Synthesis, characterization, potentiometry, and antimicrobial studies of transition metal complexes of a tridentate ligand. J. Coord. Chem. 2010, 23, 4153–4164. [Google Scholar] [CrossRef]
- Karvembu, R.; Jayabalakrishnan, C.; Natarajan, K. Thiobis(β-diketonato)-bridged binuclear ruthenium(III) complexes containing triphenylphosphine or triphenylarsine. Synthetic, spectral, catalytic and antimicrobial studies. Transit. Met. Chem. 2002, 27, 574–579. [Google Scholar] [CrossRef]
- Ryszard, G.; Erkki, K.; Henryk, J.; Reijo, K.; Maija, N.; Borys, O. Predominance of 2-arylhydrazones of 1,3-diphenylpropane-1,2,3-trione over its proton-transfer products. J. Phys. Org. Chem. 2001, 14, 797–803. [Google Scholar]
- Lasri, J.; Gajewski, G.; Guedes da Silva, M.; Fátima, C.; Kuznetsov, M.L.; Fernandes, R.; Pombeiro Armando, J.L. Solvent-dependent reactivities of acyclic nitrones with β-diketones: Catalyst-free syntheses of endiones and enones. Tetrahedron 2012, 68, 7019–7027. [Google Scholar] [CrossRef]
- Kumar, C.U.; Sethukumar, A.; Prakasam, B. Arul. Synthesis and spectral studies of some 4H-pyran derivatives: Crystal and molecular structure of isobutyl 6-amino-5-cyano-2-methyl-4-phenyl-4H-pyran-3-carboxylate. J. Mol. Struct. 2013, 1036, 257–266. [Google Scholar] [CrossRef]
- Lingaiah, B.P.V.; Reddy, G.; Venkat; Yakaiah, T.; Narsaiah, B.; Reddy, S.N.; Yadla, R.; Rao, P. Shanthan. Efficient and Convenient Method for the Synthesis of Poly Functionalised 4H-Pyrans. Synth. Commun. 2004, 34, 4431–4437. [Google Scholar] [CrossRef]
- Gou, S.-B.; Wang, S.-X.; Li, J.-T. D,L-Proline-Catalyzed One-Pot Synthesis of Pyrans and Pyrano [2,3-c]pyrazole Derivatives by a Grinding Method under Solvent-Free Conditions. Synth. Commun. 2007, 37, 2111–2120. [Google Scholar]
- John, V.D.; Krishanankutty, K. Antitumour activity of synthetic curcuminoid analogues (1,7-diaryl-1,6-heptadiene-3,5-diones) and their copper complexes. Appl. Organomet. Chem. 2006, 20, 477–482. [Google Scholar] [CrossRef]
- Pihlaja, K.; Taskinen, A.; Gawinecki, R.; Janota, H. Behaviour of 1,3-diphenyl-2-arylhydrazono-1,3-propanediones under electron ionisation. Rapid Commun. Mass Spectrom. 2003, 17, 104–106. [Google Scholar] [CrossRef] [PubMed]
- Maurya, R.C.; Rajput, S. Oxovanadium(IV) complexes of bioinorganic and medicinal relevance: Synthesis, characterization, and 3D molecular modeling and analysis of some oxovanadium(IV) complexes involving O,O-donor environment. J. Mol. Struct. 2004, 687, 35–44. [Google Scholar] [CrossRef]
- Hinckley, C.C. Paramagnetic shifts in solutions of cholesterol and the dipyridine adduct of trisdipivalomethanatoeuropium(III). A shift reagent. J. Am. Chem. Soc. 1969, 91, 5160–5162. [Google Scholar] [CrossRef] [PubMed]
- Marciniak, B.; Buono-Core, G.E. Photochemical properties of 1,3-diketonate transition metal chelates. J. Photochem. Photobiol. A 1990, 52, 1–25. [Google Scholar] [CrossRef]
- Legan, M. Cyclooxygenase-2, p53 and glucose transporter-1 as predictors of malignancy in the development of gallbladder carcinomas. Bosn, J. Basic Med. Sci. 2010, 10, 192–196. [Google Scholar] [CrossRef] [PubMed]
- Menter, D.G.; Schilsky, R.L.; DuBois, R.N. Cyclooxygenase-2 and cancer treatment: Understanding the risk should be worth the reward. Clin. Cancer Res. 2010, 16, 1384–1390. [Google Scholar] [CrossRef]
- Wang, D.; Patel, V.V.; Ricciotti, E.; Zhou, R.; Levin, M.D.; Gao, E.; Yu, Z.; Ferrari, V.A.; Lu, M.M.; Xu, J.; et al. Cardiomyocyte cyclooxygenase-2 influences cardiac rhythm and function. Proc. Natl. Acad. Sci. USA. 2009, 106, 7548–7852. [Google Scholar] [CrossRef]
- Hawash, M.; Jaradat, N.; Abualhasan, M.; Şüküroğlu, M.K.; Mohammed, T.; Qaoud, M.T.; Kahraman, D.C.; Heba Daraghmeh, H.; Maslamani, L.; Mais Sawafta, M.; et al. Design, synthesis, molecular docking studies and biological evaluation of thiazole carboxamide derivatives as COX inhibitors. BMC Chem. 2023, 17, 11. [Google Scholar] [CrossRef]
- Zarghi, A.; Arfaei, S. Selective COX-2 Inhibitors: A Review of Their Structure-Activity Relationships. Iran J. Pharm. Res. 2011, 10, 655–683. [Google Scholar]
- Beck, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B Condens. Matter. 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed]
- McLean, A.D.; Chandler, G.S. Contracted Gaussian basis sets for molecular calculations. I. Second row atoms, Z = 11–18. J. Chem. Phys. 1980, 72, 5639–5648. [Google Scholar] [CrossRef]
- Mohammed Hawash, M.; Qaoud, M.T.; Jaradat, N.; Abdallah, S.; Issa, S.; Adnan, N.; Hoshya, M.; Sobuh, S.; Hawash, Z. Anticancer Activity of Thiophene Carboxamide Derivatives as CA-4 Biomimetics: Synthesis, Biological Potency, 3D Spheroid Model, and Molecular Dynamics Simulation. Biomimetics 2022, 7, 247. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H. Gaussian 16, Revision C.01/C.02; Gaussian, Inc.: Wallingford, CT, USA, 2016.
- Spackman, M.A.; Byrom, P.G. A novel definition of a molecule in a crystal. Chem. Phys. Lett. 1997, 267, 215–220. [Google Scholar] [CrossRef]
- McKinnon, J.J.; Spackman, M.A.; Mitchell, A.S. Novel tools for visualizing and exploring intermolecular interactions in molecular crystals. Acta Crystallogr. B Struct. Sci. Cryst. Eng. Mater. 2004, 60, 627–668. [Google Scholar] [CrossRef] [PubMed]
- Spackman, M.A.; Jayatilaka, D. Hirshfeld surface analysis. CrystEngComm 2009, 11, 19–28. [Google Scholar] [CrossRef]
- Mackenzie, C.F.; Spackman, P.R.; Jayatilaka, D.M.A.; Spackman, M.A. CrystalExplorer model energies and energy frameworks: Extension to metal coordination compounds, organic salts, solvates and open-shell systems. IUCrJ 2017, 4, 575–587. [Google Scholar] [CrossRef]
- Turner, M.J.; McKinnon, J.J.; Wolff, S.K.; Grimwood, D.J.; Spackman, P.R.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer17; University of Western Australia: Crawley, Australia.
- Salih, T. A Comparative Study for the Accuracy of Three Molecular Docking Programs Using HIV-1 Protease Inhibitors as a Model. Iraqi. J. Pharm. Sci. 2022, 31, 160–168. [Google Scholar] [CrossRef]
- Castro-Alvarez, A.; Costa, A.M.; Vilarrasa, J. The Performance of Several Docking Programs at Reproducing Protein-Macrolide-Like Crystal Structures. Molecules 2017, 22, 136. [Google Scholar] [CrossRef]
- Ivanova, L.; Karelson, M. The Impact of Software Used and the Type of Target Protein on Molecular Docking Accuracy. Molecules 2022, 27, 9041. [Google Scholar] [CrossRef]
- Wang, Z.; Sun, H.; Yao, X.; Li, D.; Xu, L.; Li, Y.; Tian, S.; Hou, T. Comprehensive evaluation of ten docking programs on a diverse set of protein–ligand complexes: The prediction accuracy of sampling power and scoring power. Phys. Chem. Chem. Phys. 2016, 18, 12964–12975. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.-M.; Chen, C.-C. GEMDOCK: A generic evolutionary method for molecular docking. Proteins Struct. Funct. Genet. 2004, 55, 288–304. [Google Scholar] [CrossRef] [PubMed]
- Hsu, K.-C.; Chen, Y.-F.; Lin, S.-R.; Yang, J.-M. iGEMDOCK: A graphical environment of enhancing GEMDOCK using pharmacological interactions and post-screening analysis. BMC Bioinform. 2011, 12, S33. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. Autodock4 and AutoDockTools4: Automated docking with selective receptor flexiblity. J. Comput. Chem. 2009, 16, 2785–2791. [Google Scholar] [CrossRef]
- Eberhardt, J.; Santos-Martins, D.; Tillack, A.F.; Forli, S. AutoDock Vina 1.2.0: New Docking Methods, Expanded Force Field, and Python Bindings. J. Chem. Inf. Model. 2021, 61, 3891–3898. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Valdés-Tresanco, M.S.; Valdés-Tresanco, M.E.; Valiente, P.A.; Moreno, E. AMDock: A versatile graphical tool for assisting molecular docking with Autodock Vina and Autodock4. Biol. Direct 2020, 15, 12. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Geoffrey, R.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Meng, E.C.; Couch, G.S.; Croll, T.I.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Sci. 2021, 30, 70–82. [Google Scholar] [CrossRef]
- Goddard, T.D.; Huang, C.C.; Meng, E.C.; Pettersen, E.F.; Couch, G.S.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Meeting modern challenges in visualization and analysis. Protein Sci. 2018, 27, 14–25. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed]
- Alghamdi, A.; Abouzied, A.S.; Alamri, A.; Anwar, S.; Ansari, M.; Khadra, I.; Zaki, Y.H.; Gomha, S.M. Synthesis, Molecular Docking, and Dynamic Simulation Targeting Main Protease (Mpro) of New, Thiazole Clubbed Pyridine Scaffolds as Potential COVID-19 Inhibitors. Curr. Issues Mol. Biol. 2023, 45, 1422–1442. [Google Scholar] [CrossRef] [PubMed]
- Mali, S.N.; Pandey, A. Synthesis of new hydrazones using a biodegradable catalyst, their biological evaluations and molecular modeling studies (Part-II). J. Comput. Biophys. Chem. 2022, 21, 857–882. [Google Scholar] [CrossRef]
- Ghosh, S.; Mali, S.N.; Bhowmick, D.N.; Pratap, A.P. Neem oil as natural pesticide: Pseudo ternary diagram and computational study. J. Indian Chem. Soc. 2021, 98, 100088. [Google Scholar] [CrossRef]
- Mali, S.N.; Sawant, S.; Chaudhari, H.K.; Mandewale, M.C. In silico appraisal, synthesis, antibacterial screening and DNA cleavage for 1, 2, 5-thiadiazole derivative. Curr. Comput. Aided. Drug Des. 2019, 15, 445–455. [Google Scholar] [CrossRef]
- Mali, S.N.; Pandey, A. Multiple QSAR and molecular modelling for identification of potent human adenovirus inhibitors. J. Indian Chem. Soc. 2021, 98, 100082. [Google Scholar] [CrossRef]
- Mali, S.N.; Pandey, A.; Bhandare, R.R.; Shaik, A.B. Identification of hydantoin based Decaprenylphosphoryl-β-d-Ribose Oxidase (DprE1) inhibitors as antimycobacterial agents using computational tools. Sci. Rep. 2022, 12, 16368. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissTargetPrediction: Updated data and new features for efficient prediction of protein targets of small molecules. Nucleic Acids Res. 2019, 47, W357–W364. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Lipinski, C.A. Lead- and drug-like compounds: The rule-of-five revolution. Drug Discov. Today Technol. 2004, 1, 337–341. [Google Scholar] [CrossRef]
- Cheng, T.; Zhao, Y.; Li, X.; Lin, F.; Xu, Y.; Zhang, X.; Li, Y.; Wang, R.; Lai, L. Computation of Octanol−Water Partition Coefficients by Guiding an Additive Model with Knowledge. J. Chem. Inf. Model. 2007, 47, 2140–2148. [Google Scholar] [CrossRef] [PubMed]
- Ertl, P.; Rohde, B.; Selzer, P. Fast Calculation of Molecular Polar Surface Area as a Sum of Fragment-Based Contributions and Its Application to the Prediction of Drug Transport Properties. J. Med. Chem. 2000, 43, 3714–3717. [Google Scholar] [CrossRef] [PubMed]
- Ali, J.; Camilleri, P.; Brown, M.B.; Hutt, A.J.; Kirton, S.B. Revisiting the General Solubility Equation: In Silico Prediction of Aqueous Solubility Incorporating the Effect of Topographical Polar Surface Area. J. Chem. Inf. Model. 2012, 52, 420–428. [Google Scholar] [CrossRef] [PubMed]
- Delaney, J.S. ESOL: Estimating Aqueous Solubility Directly from Molecular Structure. J. Chem. Inf. Comput. 2004, 44, 1000–1005. [Google Scholar] [CrossRef]
- Ritchie, T.J.; Ertl, P.; Lewis, R. The graphical representation of ADME-related molecule properties for medicinal chemists. Drug Discov. Today 2011, 16, 65–72. [Google Scholar] [CrossRef]
- Lovering, F.; Bikker, J.; Humblet, C. Escape from Flatland: Increasing Saturation as an Approach to Improving Clinical Success. J. Med. Chem. 2009, 52, 6752–6756. [Google Scholar] [CrossRef]
- Daina, A.; Zoete, V.A. A BOILED-Egg To Predict Gastrointestinal Absorption and Brain Penetration of Small Molecules. ChemMedChem 2016, 11, 1117–1121. [Google Scholar] [CrossRef]
- Di, L. The role of drug metabolizing enzymes in clearance. Expert Opin. Drug Metab. Toxicol. 2014, 10, 379–393. [Google Scholar] [CrossRef]
- Potts, R.O.; Guy, R.H. Predicting Skin Permeability. Pharm. Res. 1992, 9, 663–669. [Google Scholar] [CrossRef]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
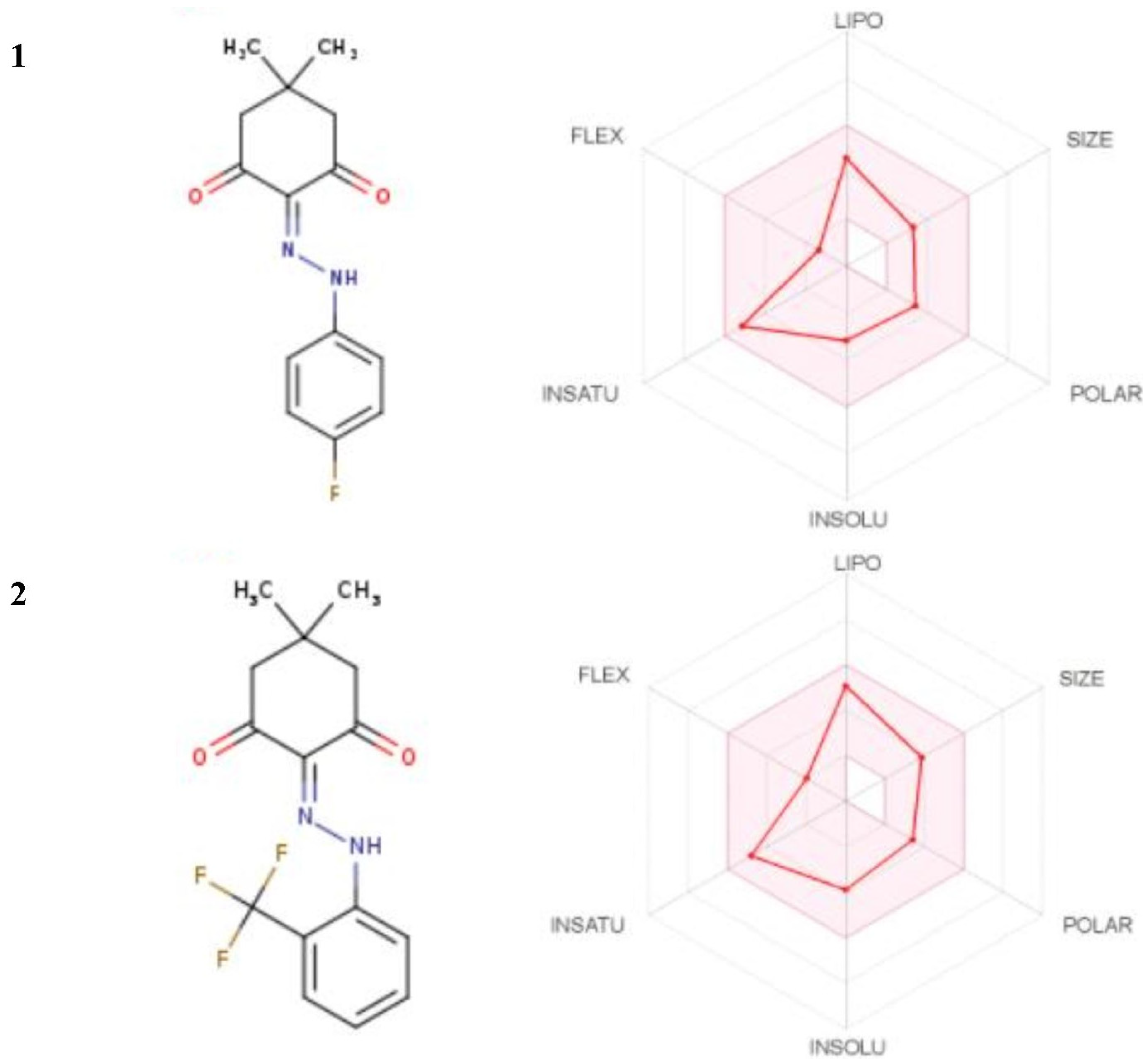{kind=link}
{kind=link}
| N | Symop | R | Electron Density | E_ele | E_pol | E_dis | E_rep | E_tot |
|---|---|---|---|---|---|---|---|---|
| 2 | x, y, z | 10.45 | B3LYP/6-31G(d,p) | −0.2 | −0.1 | −10.1 | 3.4 | −7.1 |
| 1 | −x, −y, −z | 13.23 | B3LYP/6-31G(d,p) | −6 | −0.4 | −7 | 0 | −12.8 |
| 2 | x, y, z | 5.99 | B3LYP/6-31G(d,p) | −12.4 | −3.8 | −37.8 | 23 | −34.6 |
| 1 | −x, −y, −z | 5.48 | B3LYP/6-31G(d,p) | −19.8 | −6.3 | −51.1 | 34.8 | −48.6 |
| 1 | −x, −y, −z | 6.89 | B3LYP/6-31G(d,p) | −6.5 | −5.5 | −14.7 | 9.8 | −17.6 |
| 1 | −x, −y, −z | 6.55 | B3LYP/6-31G(d,p) | −22.8 | −5.5 | −24.3 | 33.2 | −28.9 |
| 1 | −x, −y, −z | 5.63 | B3LYP/6-31G(d,p) | −5.7 | −1.2 | −49.7 | 22.7 | −36.2 |
| 2 | x, y, z | 12.82 | B3LYP/6-31G(d,p) | −5.5 | −0.4 | −11 | 0 | −15.7 |
| 1 | −x, −y, −z | 12.43 | B3LYP/6-31G(d,p) | −2.5 | −0.8 | −15.4 | 0 | −16.6 |
| 1 | −x, −y, −z | 15.97 | B3LYP/6-31G(d,p) | −1 | −0.1 | −1.2 | 0 | −2.1 |
| 1 | −x, −y, −z | 15.11 | B3LYP/6-31G(d,p) | −0.3 | 0 | −4.2 | 0 | −4 |
| N | Symop | R | Electron Density | E_ele | E_pol | E_dis | E_rep | E_tot |
|---|---|---|---|---|---|---|---|---|
| 0 | x + 1/2, −y + 1/2, z + 1/2 | 12.67 | B3LYP/6-31G(d,p) | 0 | −5.8 | 0 | 0 | −4.3 |
| 0 | −x + 1/2, y + 1/2, −z + 1/2 | 7.88 | B3LYP/6-31G(d,p) | −2.4 | −6.8 | −27.9 | 8 | −26.8 |
| 0 | x, y, z | 6.11 | B3LYP/6-31G(d,p) | 22.8 | −18.8 | −44.2 | 16.7 | −18 |
| 0 | −x, −y, −z | 8.12 | B3LYP/6-31G(d,p) | 15.3 | −24.3 | −49 | 27.4 | −27.5 |
| 0 | X + 1/2, −y + 1/2, z + 1/2 | 11.78 | B3LYP/6-31G(d,p) | −14.2 | −5.1 | −13.5 | 4.4 | −27.9 |
| 0 | −x, −y, −z | 7.94 | B3LYP/6-31G(d,p) | 35.1 | −17.5 | −23.3 | 1.4 | 4.8 |
| 0 | −x + 1/2, y + 1/2, −z + 1/2 | 8.84 | B3LYP/6-31G(d,p) | −15.4 | −4.1 | −16.4 | 5.5 | −30.2 |
| 0 | −x, −y, −z | 9.03 | B3LYP/6-31G(d,p) | 3.9 | −17.7 | −17.2 | 7.2 | −19.5 |
| 0 | −x, −y, −z | 8.87 | B3LYP/6-31G(d,p) | −4.2 | −5.6 | −21.5 | 30.2 | −8.7 |
| k_ele | k_pol | k_disp | k_rep | |
|---|---|---|---|---|
| 1 | 1.057 | 0.651 | 0.901 | 0.811 |
| 2 | 1.057 | 0.740 | 0.871 | 0.618 |
| 1 | 2 | IBF | |
|---|---|---|---|
| Energy | −86.7525 | −85.0493 | −81.2119 |
| Van Der Waals | −70.4011 | −75.6551 | −81.2119 |
| Hydrogen Bond | −16.3514 | −9.39419 | 0 |
| Electrostatic | 0 | 0 | 0 |
| 1 | 2 | IBF | ||||
|---|---|---|---|---|---|---|
| Autodock 4 | Autodock Vina | Autodock 4 | Autodock Vina | Autodock 4 | Autodock Vina | |
| Affinity (kcal/mol) | −8.57 | −8.4 | −8.4 | −8.9 | −7.7 | −6.7 |
| Estimated Ki | 522.58 | 696.25 | 696.25 | 299.41 | 2.27 | 12.27 |
| Ki units | nM | nM | nM | nM | uM | uM |
| Ligand Efficiency | −0.45 | −0.44 | −0.38 | −0.40 | −0.51 | −0.45 |
| 1 | 2 | |
|---|---|---|
| Physicochemical Properties | ||
| Molecular weight in g/mol (≤500) | 262.28 | 312.29 |
| Saturation: fraction of carbons in the sp3 hybridization (not less than 0.25) | 0.36 | 0.40 |
| Lipophilicity: XLOGP3 (desirable between −0.7 and +5.0) | 2.56 | 3.34 |
| No. rotatable bonds (not more than 9 rotatable bonds) | 2 | 3 |
| No. H-bond acceptors (H-bond acceptor ≤ 10) | 4 | 6 |
| No. H-bond donors (H-bond donors ≤ 5) | 1 | 1 |
| Topological polar surface area TPSA (between 20 and 130 Å2) | 58.53 | 58.53 |
| Solubility | ||
| log S (Ali) | −3.44 | −4.25 |
| log S (ESOL) | −3.18 | −3.88 |
| Pharmacokinetic properties | ||
| GI absorption | High | High |
| P-glycoprotein substrate | No | No |
| Skin permeation (logKP in cm/s) | −6.08 | −5.83 |
| BBB permeation | Yes | Yes |
| Cytochromes P450 1A2, 2C19, 2C9, 2D6. 3A4 inhibitor | Only for 1A2 | Only for 2C19 |
| Bioavailability score | 0.55 | 0.55 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kurbanova, M.M.; Maharramov, A.M.; Sadigova, A.Z.; Gurbanova, F.Z.; Mali, S.N.; Al-Salahi, R.; El Bakri, Y.; Lai, C.-H. Synthesis, Characterization, DFT, and In Silico Investigation of Two Newly Synthesized β-Diketone Derivatives as Potent COX-2 Inhibitors. Bioengineering 2023, 10, 1361. https://doi.org/10.3390/bioengineering10121361
Kurbanova MM, Maharramov AM, Sadigova AZ, Gurbanova FZ, Mali SN, Al-Salahi R, El Bakri Y, Lai C-H. Synthesis, Characterization, DFT, and In Silico Investigation of Two Newly Synthesized β-Diketone Derivatives as Potent COX-2 Inhibitors. Bioengineering. 2023; 10(12):1361. https://doi.org/10.3390/bioengineering10121361
Chicago/Turabian StyleKurbanova, Malahat Musrat, Abel Mammadali Maharramov, Arzu Zabit Sadigova, Fidan Zaur Gurbanova, Suraj Narayan Mali, Rashad Al-Salahi, Youness El Bakri, and Chin-Hung Lai. 2023. "Synthesis, Characterization, DFT, and In Silico Investigation of Two Newly Synthesized β-Diketone Derivatives as Potent COX-2 Inhibitors" Bioengineering 10, no. 12: 1361. https://doi.org/10.3390/bioengineering10121361