A Systematic AQbD Approach for Optimization of the Most Influential Experimental Parameters on Analysis of Fish Spoilage-Related Volatile Amines



Abstract
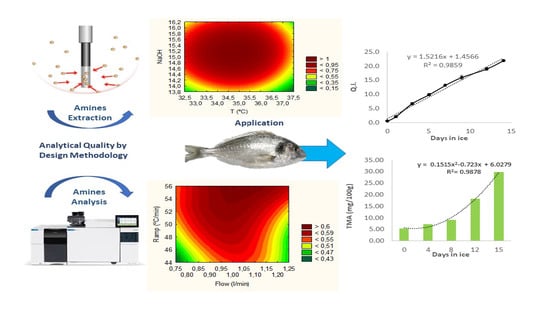
:
1. Introduction
2. Materials and Methods
2.1. Reagents and Materials
2.2. Degradation Trial
2.3. Sample Preparation for Amine Analysis
2.4. HS-SPME Procedure
2.5. GC–MS Conditions
2.6. Method Validation
2.7. Shelf Life Estimation
2.8. Statistical Analysis
3. Results and Discussion
3.1. Establishing the Quality by Design Bases for Analysis
3.1.1. Defining the Analytical Target Profile and Method Scouting
3.1.2. Method Critical Quality Attribute Definition Methods
3.1.3. Critical Method Parameters and Quality Risk Assessment
3.2. Method Knowledge of Space
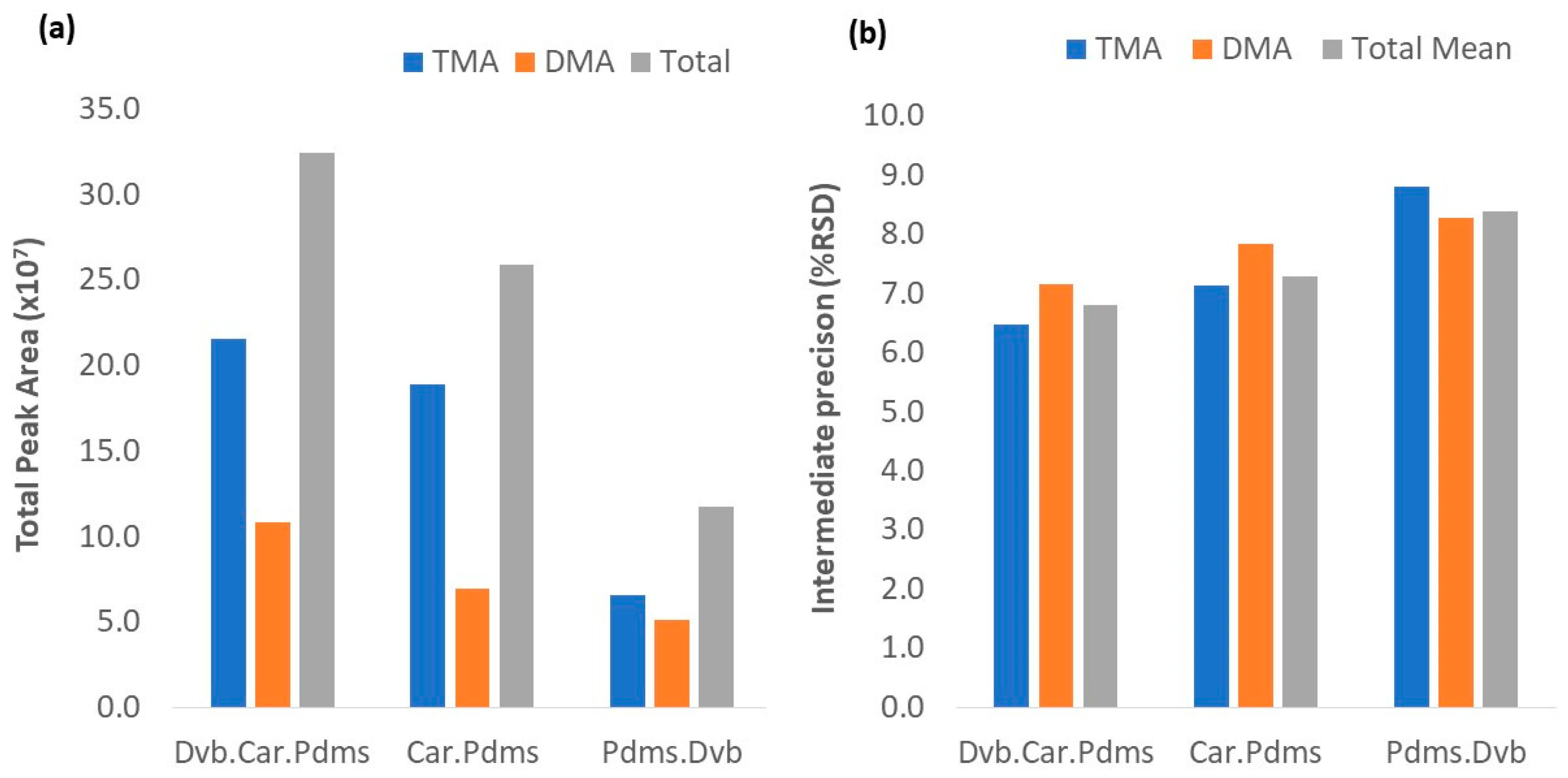
3.2.1. HS-SPME Fibers
- Molecular weight and analyte size related to analyte speed; the smaller the analytes, the faster it moves, and the retention might be harder. Highly cross-linked fibers could better retain fast-moving analytes.
- Analyte polarity is defined by the type of coating and its ability to retain analytes according to polarity. However, each fiber has a certain capacity that might be better suited for ones than others.
- Analyte concentration level and range-thicker fibers might require higher concentrations of an analyte to be more efficient.
- The complexity of the sample-related to the analyte displacement by the other sample components with higher affinity or concentration.
3.2.2. GC-MS Conditions
3.3. Determination of Method Operable Design Region
3.3.1. HS-SPME Extraction
3.3.2. Chromatographic Conditions
3.4. Robustness and Method Control
3.4.1. Amines Extraction
3.4.2. Chromatographic Analysis
3.4.3. Method Control
3.5. Analytical Method Validation
3.6. Method Application and Shelf Life Estimation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Oehlenschläger, J. Seafood Quality Assessment. In Seafood Processing: Technology, Quality and Safety; Wiley: Hoboken, NJ, USA, 2014; pp. 359–386. ISBN 9781118346174. [Google Scholar]
- Prabhakar, P.; Vatsa, S.; Srivastav, P.; Pathak, S. A comprehensive review on freshness of fish and assessment: Analytical methods and recent innovations. Food Res. Int. 2020, 133, 109157. [Google Scholar] [CrossRef]
- Freitas, J.; Vaz-Pires, P.; Câmara, J.S. From aquaculture production to consumption: Freshness, safety, traceability and authentication, the four pillars of quality. Aquaculture 2020, 518, 734857. [Google Scholar] [CrossRef]
- Howgate, P. A Critical review of total volatile bases and trimethylamine as indices of freshness of fish. Part 1. Determination. Electron. J. Environ. Agric. Food Chem. 2010, 9, 29–57. [Google Scholar]
- Yousefi, H.; Su, H.M.; Imani, S.M.; Alkhaldi, K.; Filipe, C.D.; Didar, T.F. Intelligent Food Packaging: A Review of Smart Sensing Technologies for Monitoring Food Quality. ACS Sensors 2019, 4, 808–821. [Google Scholar] [CrossRef] [PubMed]
- Baixas-Nogueras, S.; Bover-Cid, S.; Vidal-Carou, M.G.; Veciana-Nogués, M.T.; Mariné-Font, A. Trimethylamine and total volatile basic nitrogen determination by flow injection/gas diffusion in Mediterranean hake (Merluccius merluccius). J. Agric. Food Chem. 2001, 49, 1681–1686. [Google Scholar] [CrossRef]
- Béné, A.; Hayman, A.; Reynard, E.; Luisier, J.L.; Villettaz, J.C. New method for the rapid determination of volatile substances: The SPME-direct method. Part II. Determination of the freshness of fish. Sensors Actuators B Chem. 2001, 72, 204–207. [Google Scholar] [CrossRef]
- Howgate, P. A Critical review of total volatile bases and trimethylamine as indices of freshness of fish. Part 2. Formation of the bases, and application in quality assurance. Electron. J. Environ. Agric. Food Chem. 2010, 9, 58–88. [Google Scholar]
- Bello, F.D.; Aigotti, R.; Zorzi, M.; Giaccone, V.; Medana, C. Multi-analyte ms based investigation in relation to the illicit treatment of fish products with hydrogen peroxide. Toxics 2020, 8, 2. [Google Scholar] [CrossRef] [Green Version]
- Esposito, G.; Sciuto, S.; Acutis, P.L. Quantification of TMA in fishery products by direct sample analysis with high resolution mass spectrometry. Food Control 2018, 94, 162–166. [Google Scholar] [CrossRef]
- Nevigato, T.; Masci, M.; Casini, I.; Caproni, R.; Orban, E. Trimethylamine as a freshness indicator for seafood stored in ice: Analysis by GC-FID of four species caught in the Tyrrhenian Sea. Ital. J. Food Sci. 2018, 30, 522–534. [Google Scholar] [CrossRef]
- Chan, S.T.; Yao, M.W.Y.; Wong, Y.C.; Wong, T.; Mok, C.S.; Sin, D.W.M. Evaluation of chemical indicators for monitoring freshness of food and determination of volatile amines in fish by headspace solid-phase microextraction and gas chromatography-mass spectrometry. Eur. Food Res. Technol. 2006, 224, 67–74. [Google Scholar] [CrossRef]
- Wang, X.; Rogers, K.M.; Li, Y.; Yang, S.; Chen, L.; Zhou, J. Untargeted and Targeted Discrimination of Honey Collected by Apis cerana and Apis mellifera Based on Volatiles Using HS-GC-IMS and HS-SPME-GC-MS. J. Agric. Food Chem. 2019. [Google Scholar] [CrossRef] [PubMed]
- Perestrelo, R.; Silva, C.; Silva, P.; Medina, S.; Câmara, J.S. Differentiation of fresh and processed fruit juices using volatile composition. Molecules 2019, 24, 974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sung, J.; Suh, J.H.; Chambers, A.H.; Crane, J.; Wang, Y. Relationship between Sensory Attributes and Chemical Composition of Different Mango Cultivars. J. Agric. Food Chem. 2019, 67, 5177–5188. [Google Scholar] [CrossRef]
- Silva, P.; Silva, C.L.; Perestrelo, R.; Nunes, F.M.; Câmara, J.S. A useful strategy based on chromatographic data combined with quality-by-design approach for food analysis applications. The case study of furanic derivatives in sugarcane honey. J. Chromatogr. A 2017, 1520, 117–126. [Google Scholar] [CrossRef]
- Debrus, B.; Guillarme, D.; Rudaz, S. Improved quality-by-design compliant methodology for method development in reversed-phase liquid chromatography. J. Pharm. Biomed. Anal. 2013, 84, 215–223. [Google Scholar] [CrossRef]
- Orlandini, S.; Pinzauti, S.; Furlanetto, S. Application of quality by design to the development of analytical separation methods. Anal. Bioanal. Chem. 2013, 405, 443–450. [Google Scholar] [CrossRef]
- Vogt, F.G.; Kord, A.S. Development of Quality-By-Design Analytical Methods. J. Pharm. Sci. 2011, 100, 797–812. [Google Scholar] [CrossRef]
- Tome, T.; Žigart, N.; Časar, Z.; Obreza, A. Development and Optimization of Liquid Chromatography Analytical Methods by Using AQbD Principles: Overview and Recent Advances. Org. Process Res. Dev. 2019, 23, 1784–1802. [Google Scholar] [CrossRef] [Green Version]
- Mohammed, A.Q.; Sunkari, P.K.; Srinivas, P.; Roy, A.K. Quality by Design in Action 1: Controlling Critical Quality Attributes of an Active Pharmaceutical Ingredient. Org. Process Res. Dev. 2015, 19, 1634–1644. [Google Scholar] [CrossRef]
- Muteki, K.; Morgado, J.E.; Reid, G.L.; Wang, J.; Xue, G.; Riley, F.W.; Harwood, J.W.; Fortin, D.T.; Miller, I.J. Quantitative structure retention relationship models in an analytical quality by design framework: Simultaneously accounting for compound properties, mobile-phase conditions, and stationary-phase properties. Ind. Eng. Chem. Res. 2013, 52, 12269–12284. [Google Scholar] [CrossRef]
- Dehaut, A.; Duthen, S.; Grard, T.; Krzewinski, F.; N’Guessan, A.; Brisabois, A.; Duflos, G. Development of an SPME-GC-MS method for the specific quantification of dimethylamine and trimethylamine: Use of a new ratio for the freshness monitoring of cod fillets. J. Sci. Food Agric. 2016, 96, 3787–3794. [Google Scholar] [CrossRef] [PubMed]
- Freitas, J.; Vaz-Pires, P.; Câmara, J.S. Freshness assessment and shelf life prediction for seriola dumerili from aquaculture based on the quality index method. Molecules 2019, 24, 3530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, M.; Ellison, S.L.R.; Wood, R. Harmonized guidelines for single-laboratory validation of methods of analysis (IUPAC Technical Report). Pure Appl. Chem. 2002, 74, 835–855. [Google Scholar] [CrossRef]
- Horwitz, W. AOAC Guidelines for Single Laboratory Validation of Chemical Methods for Dietary Supplements and Botanicals; AOAC International: Rockville, MD, USA, 2002; Volume 1219. [Google Scholar]
- Silva, P.; Freitas, J.; Silva, C.L.; Perestrelo, R.; Nunes, F.M.; Câmara, J.S. Establishment of authenticity and typicality of sugarcane honey based on volatile profile and multivariate analysis. Food Control 2017, 73, 1176–1188. [Google Scholar] [CrossRef]
- Alasalvar, C.; Taylor, K.D.A.; Shahidi, F. Comparative quality assessment of cultured and wild sea bream (Sparus aurata) stored in ice. J. Agric. Food Chem. 2002, 50, 2039–2045. [Google Scholar] [CrossRef]
- Gonçalves, A.A.; de Lima, J.T.A.X.; de Paula, F.E.R. Development of Quality Index Method (QIM) scheme for spiny lobster (Panulirus argus, Latreille, 1804) stored in ice. Food Control 2015, 47, 237–245. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Li, J.; Wang, J.; Lv, W. Applying Different Methods To Evaluate the Freshness of Large Yellow Croacker (Pseudosciaena crocea) Fillets during Chilled Storage. J. Agric. Food Chem. 2012, 60, 11387–11394. [Google Scholar] [CrossRef]
- Triqui, R.; Bouchriti, N. Freshness Assessments of Moroccan Sardine (Sardina pilchardus): Comparison of Overall Sensory Changes to. J. Agric. Food Chem. 2003, 51, 7540–7546. [Google Scholar] [CrossRef]
- Sant’Ana, L.; Soares, S.; Vaz-Pires, P. Development of a quality index method (QIM) sensory scheme and study of shelf life of ice-stored blackspot seabream (Pagellus bogaraveo). LWT—Food Sci. Technol. 2011, 44, 2253–2259. [Google Scholar] [CrossRef] [Green Version]
- Distell User Manual Distell Fish Freshness Meter. Available online: www.distell.com (accessed on 18 June 2020).
- Huidobro, A.; Pastor, A.; Tejada, M. Quality Index Method Developed for Raw Gilthead Seabream (Sparus aurata). J. Food Sci. 2000, 65, 1202–1205. [Google Scholar] [CrossRef]
- Wzorek, B.; Mochalski, P.; Śliwka, I.; Amann, A. Application of GC-MS with a SPME and thermal desorption technique for determination of dimethylamine and trimethylamine in gaseous samples for medical diagnostic purposes. J. Breath Res. 2010. [Google Scholar] [CrossRef] [PubMed]
- Barbosa-Pereira, L.; Otero-Pazos, P.; Rodríguez-Bernaldo De Quirós, A.; Sendón, R.; Vecino, X.; Cruz, J.M.; Romero-Rodríguez, M.A.; Estévez, N.; Maroto, J.; Paseiro-Losada, P. SPME-GC method for the determination of volatile amines as indices of freshness in fish samples. Ital. J. Food Sci. 2012, 24, 211–214. [Google Scholar]
- Kataoka, H.; Lord, H.L.; Pawliszyn, J. Applications of solid-phase microextraction in food analysis. J. Chromatogr. A 2000, 880, 35–62. [Google Scholar] [CrossRef]
- Pawliszyn, J. Applications of Solid Phase Microextraction, 1st ed.; Royal Society of Chemistry: Cambridge, UK, 1999; ISBN 0854045252. [Google Scholar]
- Shirey, R.E. SPME Commercial Devices and Fibre Coatings; Elsevier Inc.: Waltham, MA, USA, 2012; ISBN 9780124160170. [Google Scholar]
- Araujo, P. Key aspects of analytical method validation and linearity evaluation. J. Chromatogr. B 2009, 877, 2224–2234. [Google Scholar] [CrossRef] [PubMed]
- Ochrem, A.; Zapletal, P.; Maj, D.; Gil, Z.; Zychlińska-Buczek, J. Changes in physical and dielectrical properties of carp meat (Cyprinus carpio) during cold storage. J. Food Process Eng. 2014, 37, 177–184. [Google Scholar] [CrossRef]
- Vaz-Pires, P.; Araújo, I.; Kirby, R.M. Physical measurement of the quality of fresh scad (Trachurus trachurus) and rainbow trout (Oncorhynchus mykiss) during ice storage using the RT Freshmeter. Int. J. Food Sci. Technol. 1995, 30, 799–805. [Google Scholar] [CrossRef]
- Vaz-Pires, P.; Seixas, P.; Mota, M.; Lapa-Guimarães, J.; Pickova, J.; Lindo, A.; Silva, T. Sensory, microbiological, physical and chemical properties of cuttlefish (Sepia officinalis) and broadtail shortfin squid (Illex coindetii) stored in ice. LWT—Food Sci. Technol. 2008, 41, 1655–1664. [Google Scholar] [CrossRef] [Green Version]
- Agüeria, D.; Sanzano, P.; Vaz-Pires, P.; Rodríguez, E.; Yeannes, M.I. Development of Quality Index Method Scheme for Common Carp (Cyprinus carpio) Stored in Ice: Shelf life Assessment by Physicochemical, Microbiological, and Sensory Quality Indices. J. Aquat. Food Prod. Technol. 2016, 25, 708–723. [Google Scholar] [CrossRef]
- Billar dos Santos, A.P.; Kushida, M.M.; Viegas, E.M.M.; Lapa-Guimarães, J. Development of Quality Index Method (QIM) scheme for Acoupa weakfish (Cynoscion acoupa). LWT—Food Sci. Technol. 2014, 57, 267–275. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | SPME Fibers | Amines | |||
|---|---|---|---|---|---|
| DVB/CAR/PDMS | CAR/PDMS | PDMS/DVB | TMA | DMA | |
| pKa | - | - | - | 9.80 | 10.37 |
| Molecular Weight | 40–275 | 30–225 | 50–300 | 59.11 | 45.08 |
| Polarity | Bipolar | Bipolar | Bipolar | Polar | Polar |
| Thickness (µm) | 30–55 | 75 | 65 | - | - |
| Core type | Stabflex | Fused silica | Fused silica | - | - |
| Operating temperature | 230–270 | 250–320 | 200–270 | - | - |
| Extraction mechanism | Adsorbent | Adsorbent | Adsorbent | - | - |
| Method | CMP 1 | LSL 2 | USL 3 | CpK 4 |
|---|---|---|---|---|
| HS-SPME | TPA 5 | 0.05 | 0.17 | 1.49 |
| IP 6 | 0.03 | 0.18 | 1.47 | |
| GC-MS | TPA | 0.03 | 0.18 | 1.47 |
| IP | 0.02 | 0.20 | 1.49 | |
| Resolution | 0.04 | 0.17 | 1.38 | |
| TF 7 | 0.02 | 0.19 | 1.36 | |
| SF 8 | 0.05 | 0.17 | 1.37 |
| TMA | DMA | |
|---|---|---|
| Ion (m/z) | 58 | 44 |
| Retention time (min) | 4.12 | 4.23 |
| Concentration range (µg/mL) | 2-20 | 2-20 |
| Regression equation | Y = 11143x2 + 2 × 107 − 1 × 107 | Y = 11153x2 + 2 × 107 − 3 × 107 |
| R2 | 0.9504 | 0.9989 |
| Linearity test (Ftheo/Fexp) | 3.0 1 | 15.4 1 |
| LOD (µg/mL) 5 | 0.4 | 0.6 |
| LOQ (µg/mL) 6 | 1.3 | 2.0 |
| Matrix effect (%) | 105 | 109 |
| Repeatability (%RSD 7) | 8.2 2 6.0 3 9.6 4 | 8.3 2 11.2 3 7.4 3 |
| Intermediate Precision (%RSD 7) | 8.4 2 7.1 3 9.7 4 | 9.6 2 13.6 3 8.8 4 |
| Recovery (%) | 93 2 98 3 99 4 | 90 3 93 2 95 4 |
| Method | Rejection Criteria | Value at Rejection | Estimated Rejection Day |
|---|---|---|---|
| QIM | Off odors | 13 | 7–8 |
| Torrymeter | Off odorsSlope change | 8 | 8–9 |
| Microbiology (TVC) | Log cfu/cm2 = 7–9 | 8.0 | 8–9 |
| Microbiology (H2S) | Log cfu/cm2 = 7–9 | 7.5 | 9–10 |
| TMA analysis | 12 mg/100 gr | 12.5 | 9–10 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Freitas, J.; Silva, P.; Vaz-Pires, P.; Câmara, J.S. A Systematic AQbD Approach for Optimization of the Most Influential Experimental Parameters on Analysis of Fish Spoilage-Related Volatile Amines. Foods 2020, 9, 1321. https://doi.org/10.3390/foods9091321
Freitas J, Silva P, Vaz-Pires P, Câmara JS. A Systematic AQbD Approach for Optimization of the Most Influential Experimental Parameters on Analysis of Fish Spoilage-Related Volatile Amines. Foods. 2020; 9(9):1321. https://doi.org/10.3390/foods9091321
Chicago/Turabian StyleFreitas, Jorge, Pedro Silva, Paulo Vaz-Pires, and José S. Câmara. 2020. "A Systematic AQbD Approach for Optimization of the Most Influential Experimental Parameters on Analysis of Fish Spoilage-Related Volatile Amines" Foods 9, no. 9: 1321. https://doi.org/10.3390/foods9091321