


Analysis of Dynamics and Diversity of Microbial Community during Production of Germinated Brown Rice

Abstract
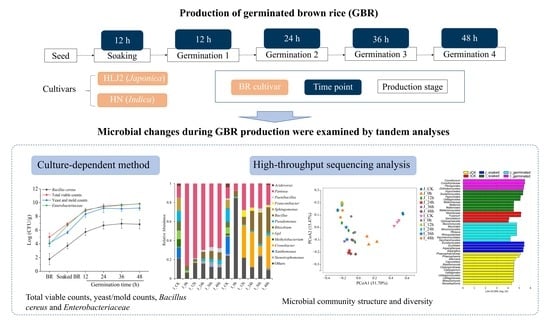
:
1. Introduction
2. Materials and Methods
2.1. Brown Rice Samples and Preparation
2.2. Germination of Brown Rice
2.3. Microbiological Determination
2.4. Microbial Diversity Analysis
2.4.1. DNA Extraction and PCR Amplification
2.4.2. Sequencing and Bioinformatics Processing
2.5. Statistical Analysis
3. Results
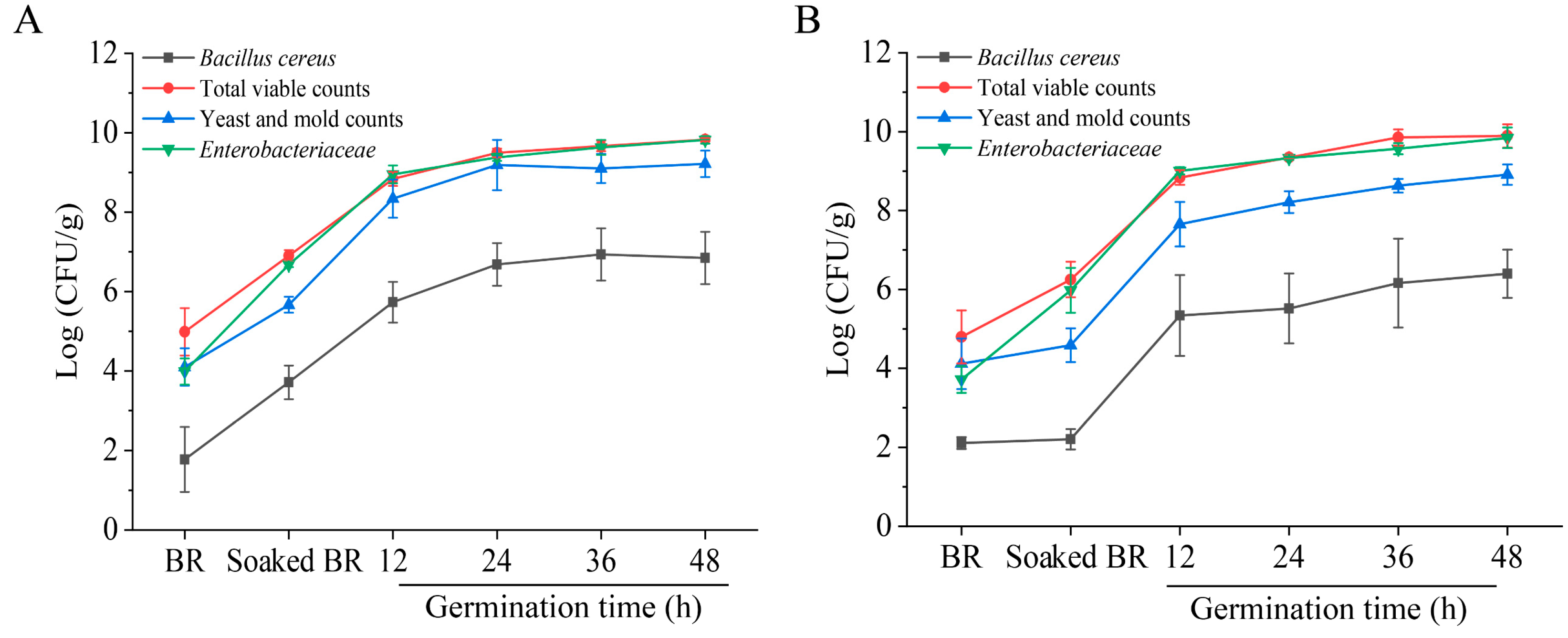
3.1. Natural Microbial Counts of BR Samples
3.2. Microbiological Changes during BR Germination
3.3. Microbiome Analysis
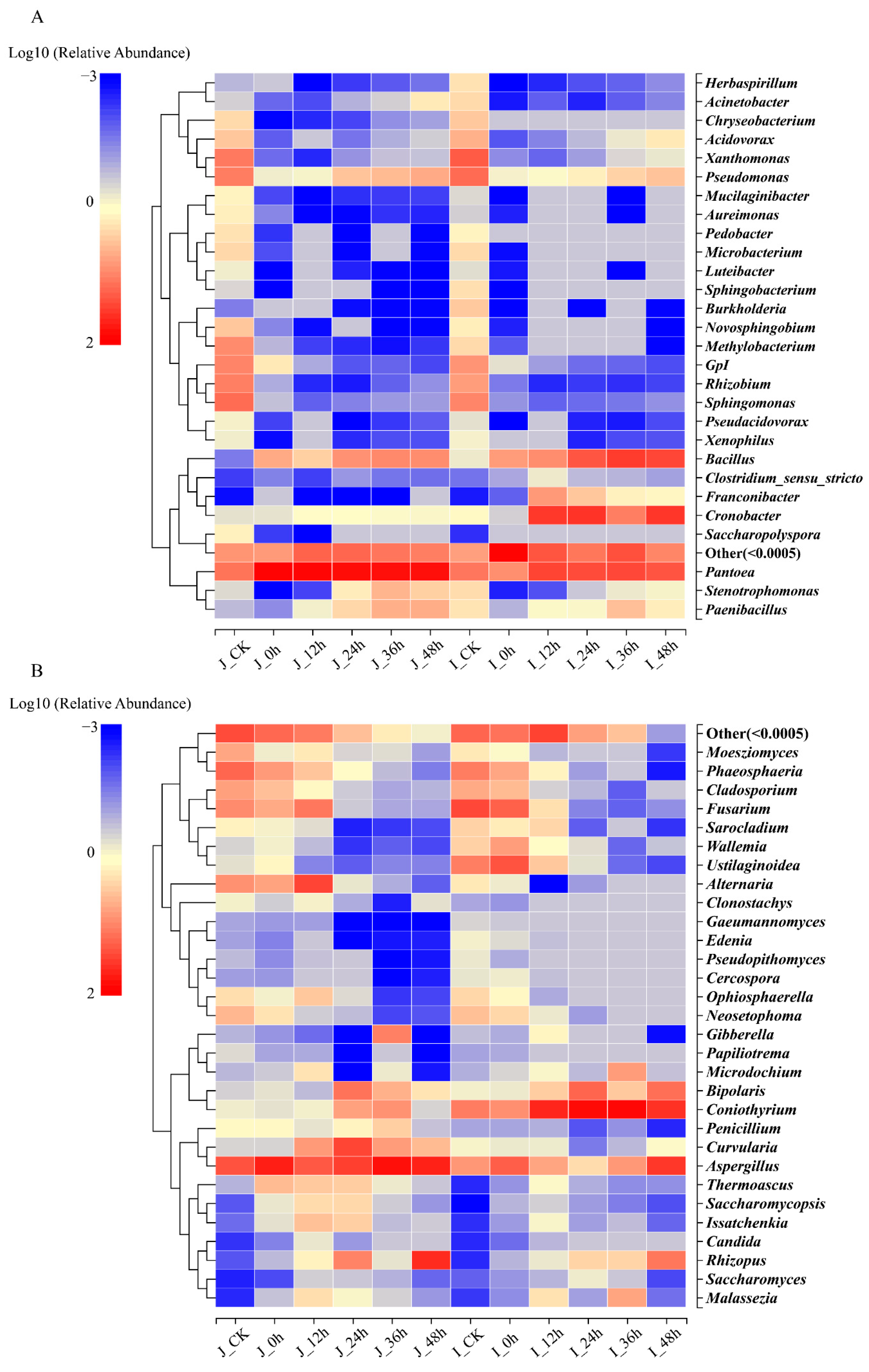
3.3.1. Community Abundance and Diversity
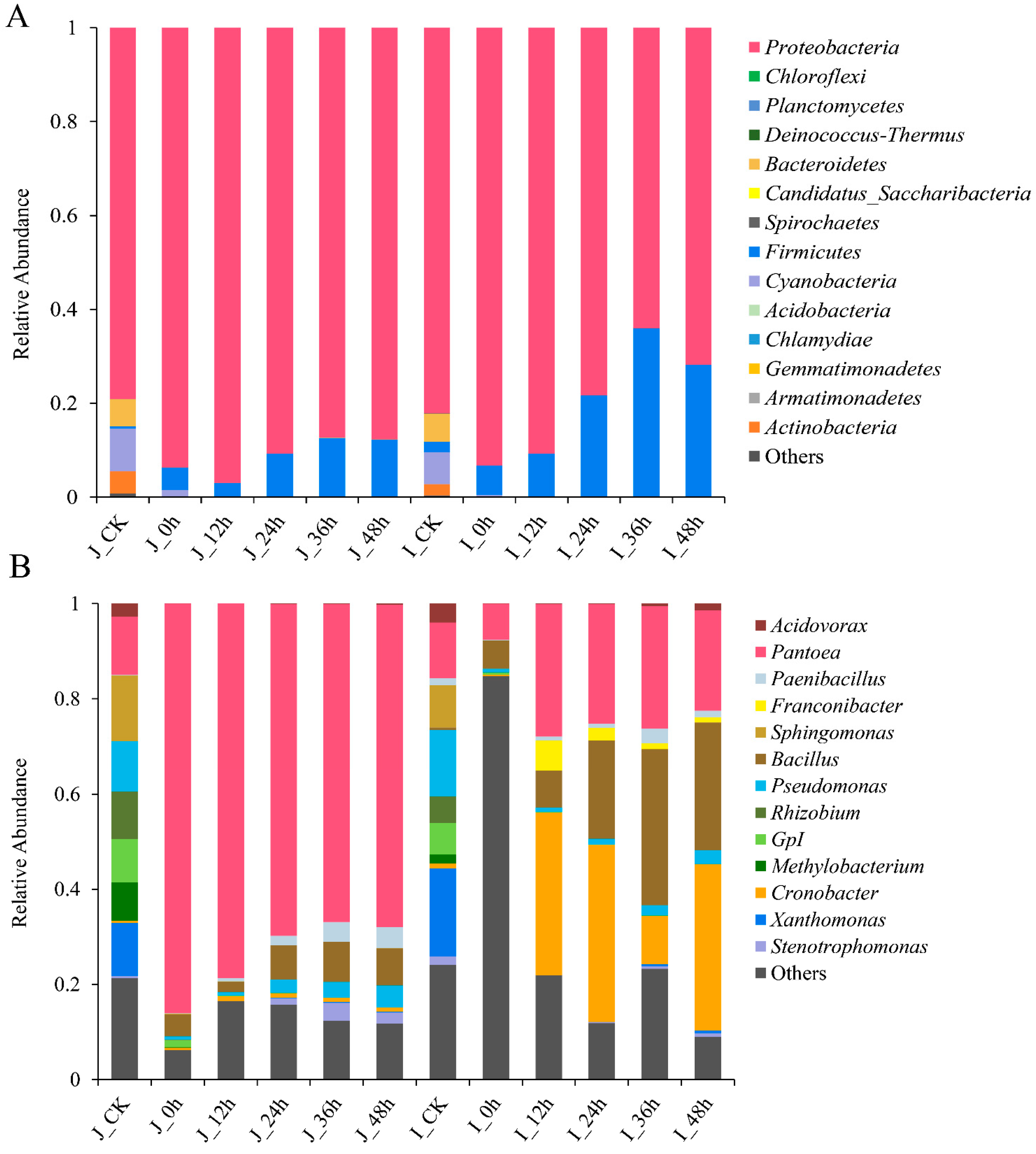
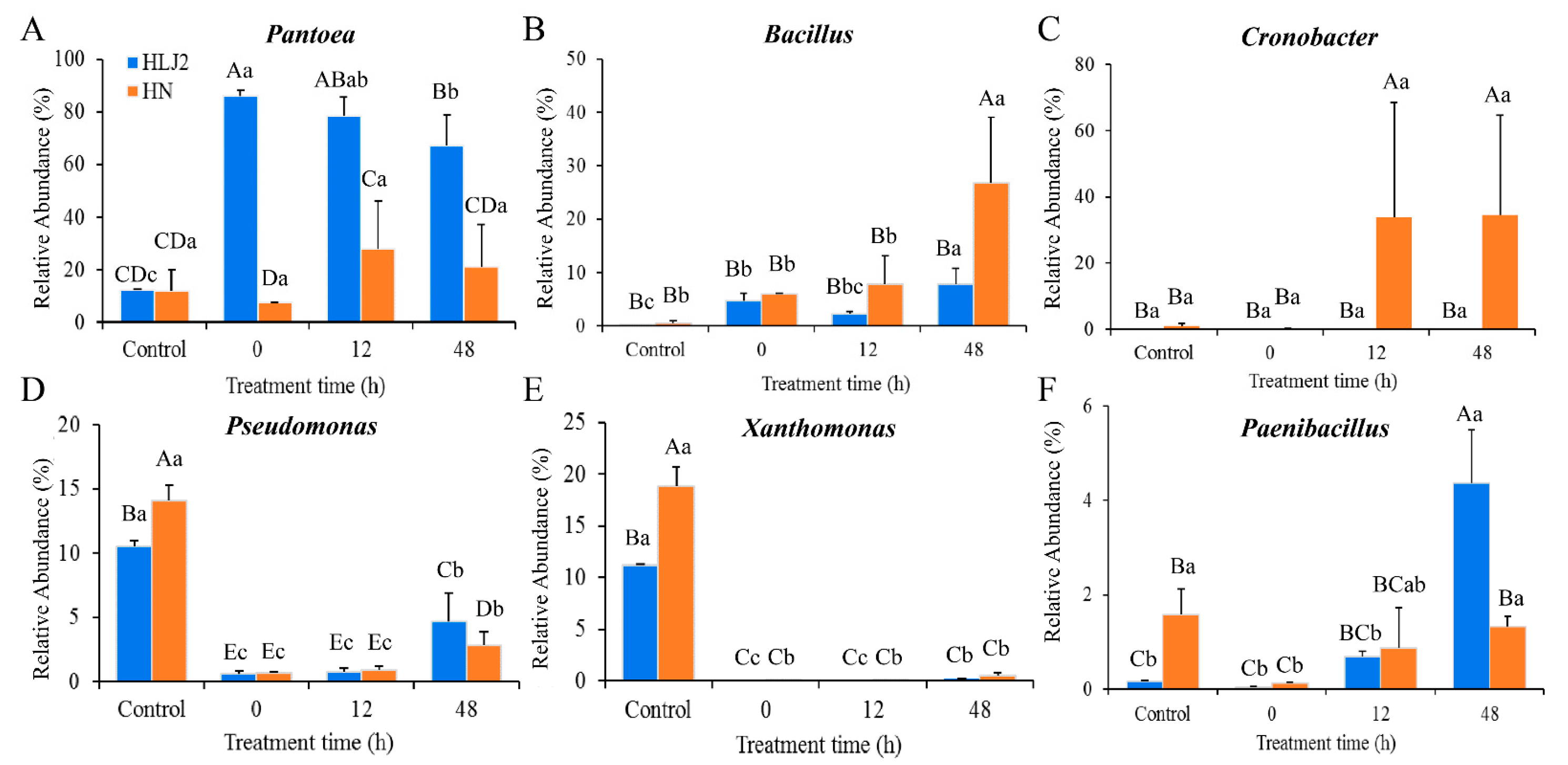
3.3.2. Composition Analysis of Bacterial Community during BR Germination
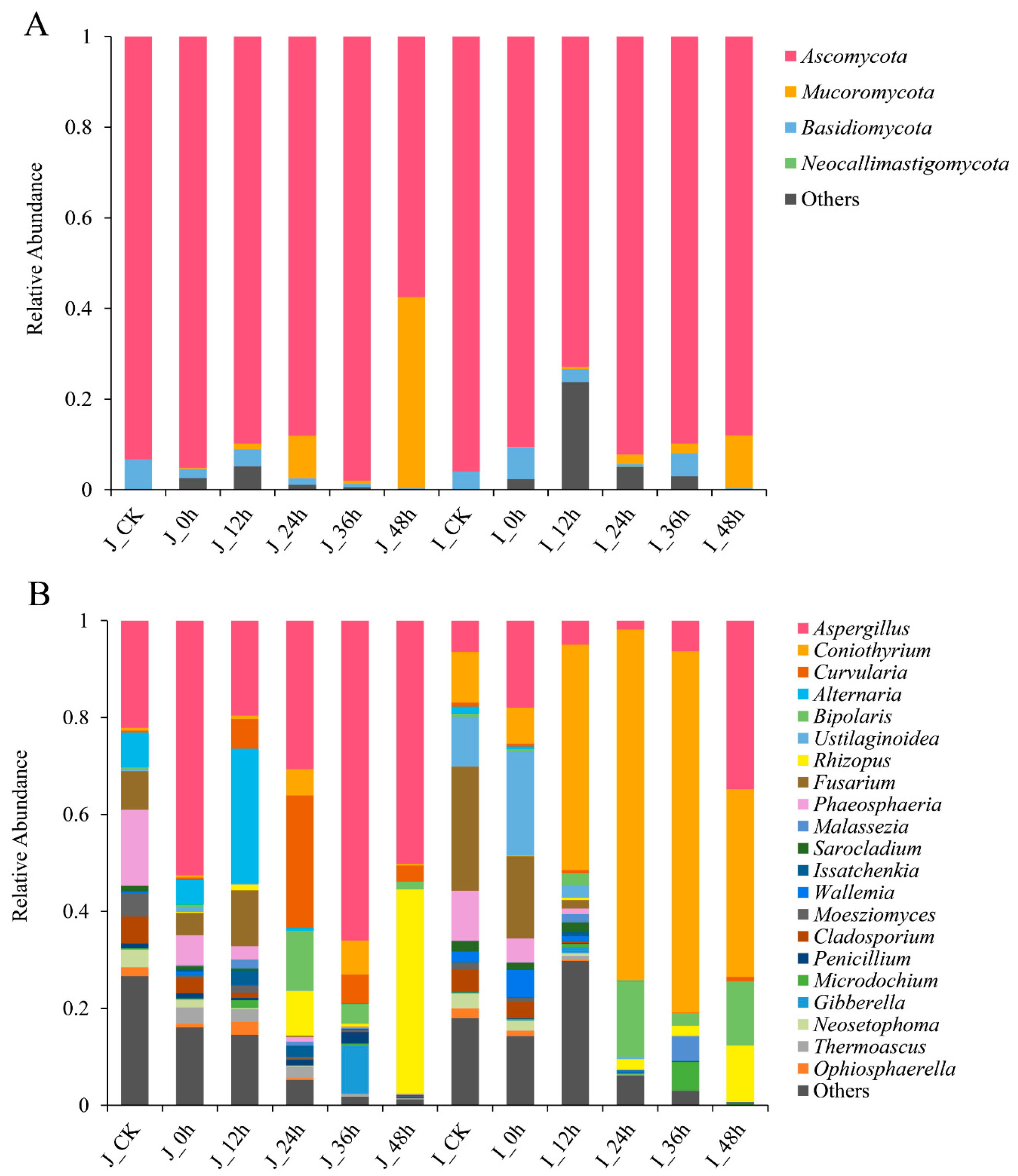
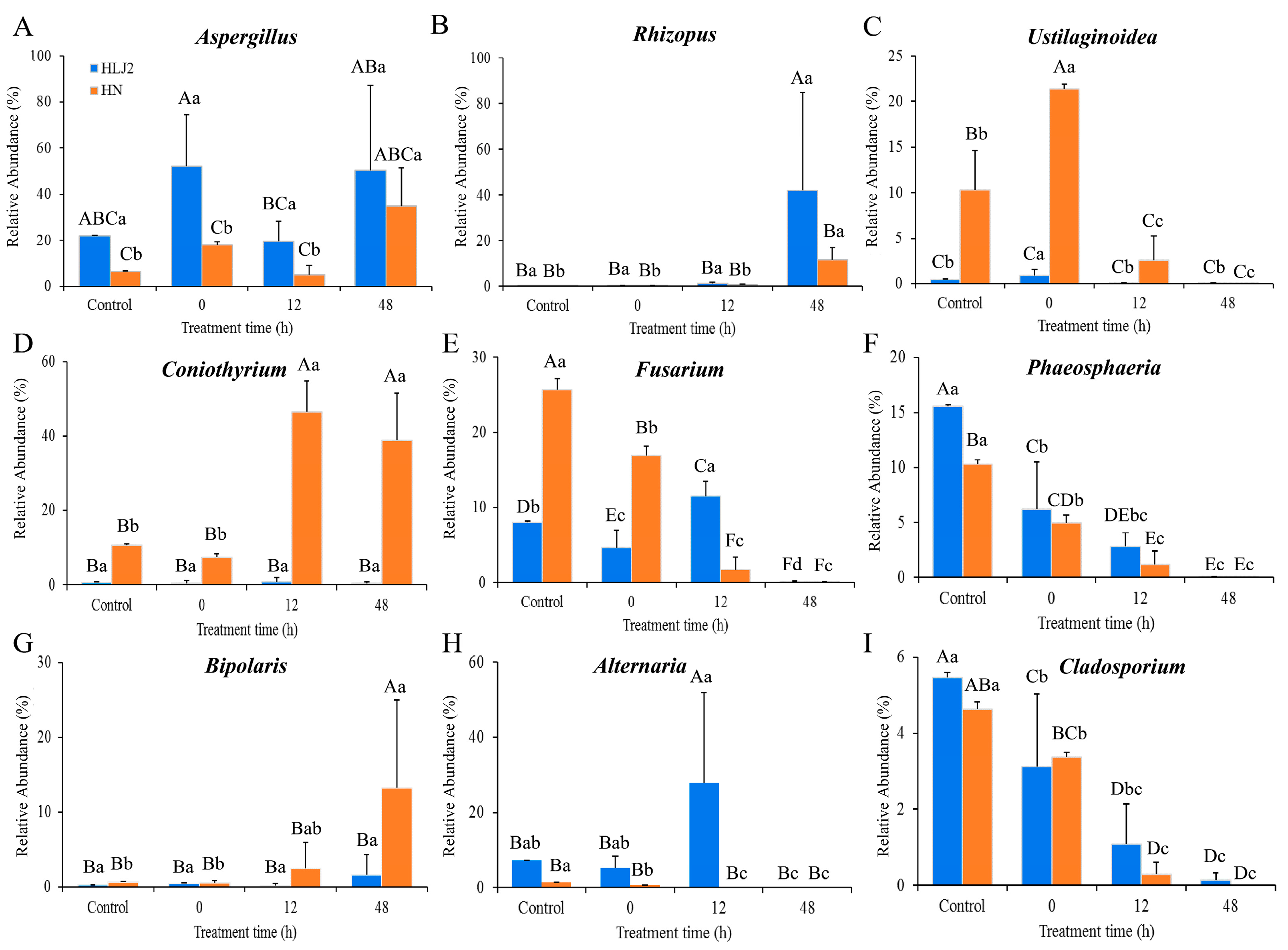
3.3.3. Composition Analysis of Fungal Community during BR Germination
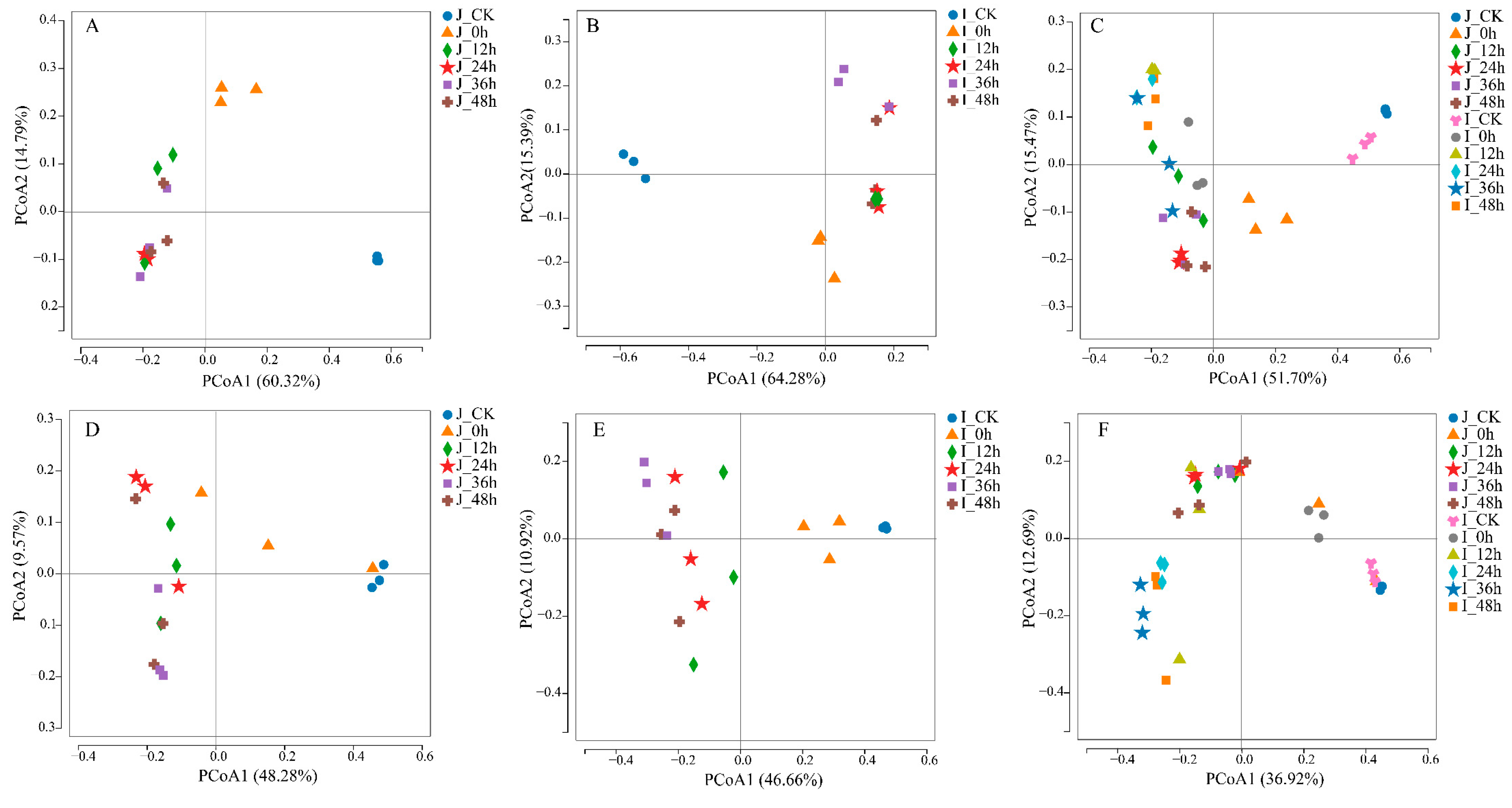
3.3.4. Comparison of Microbial Community Structure between HLJ2 and HN Samples
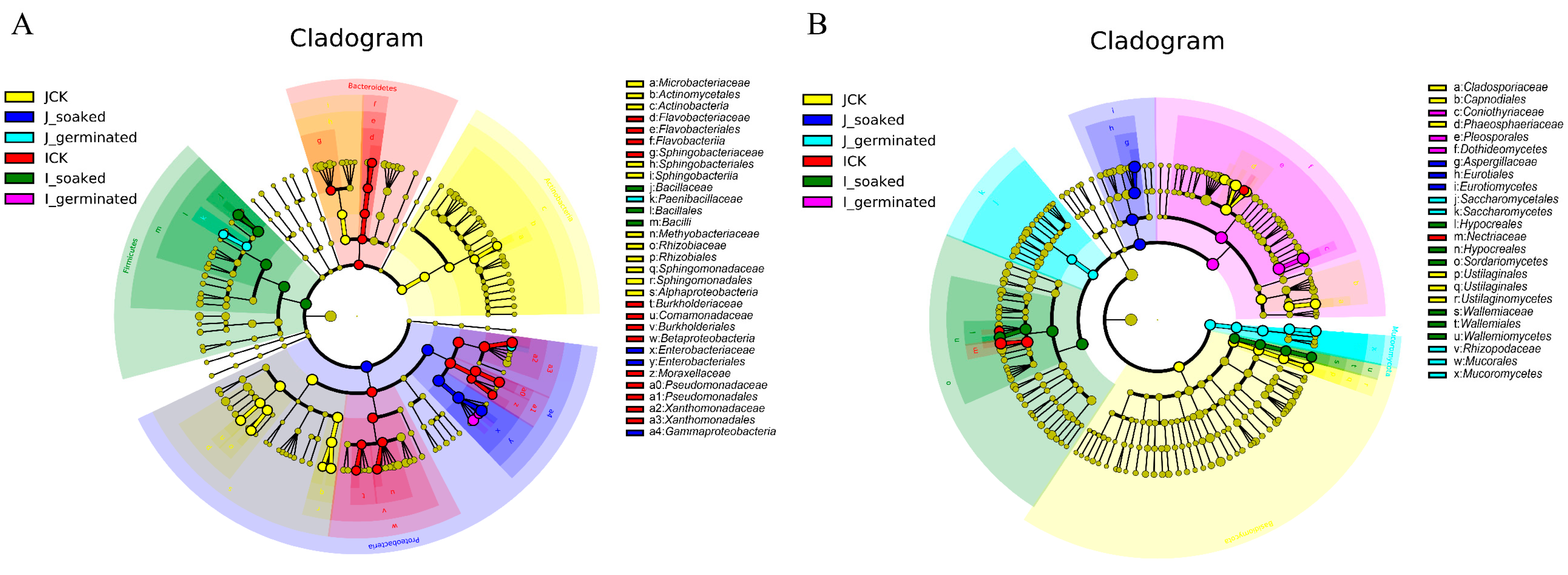
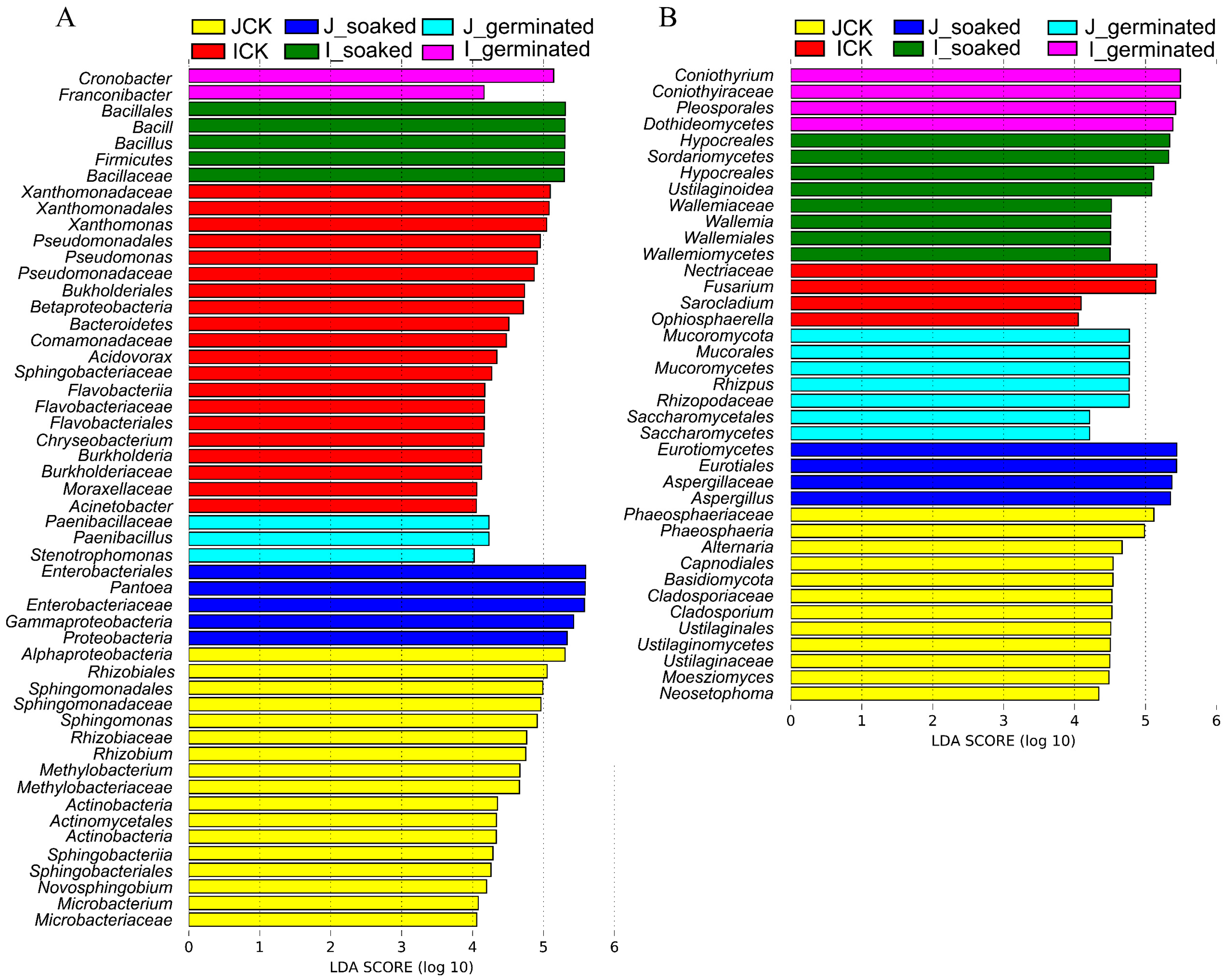
3.3.5. LDA Effect Size (LEfSe) Analysis
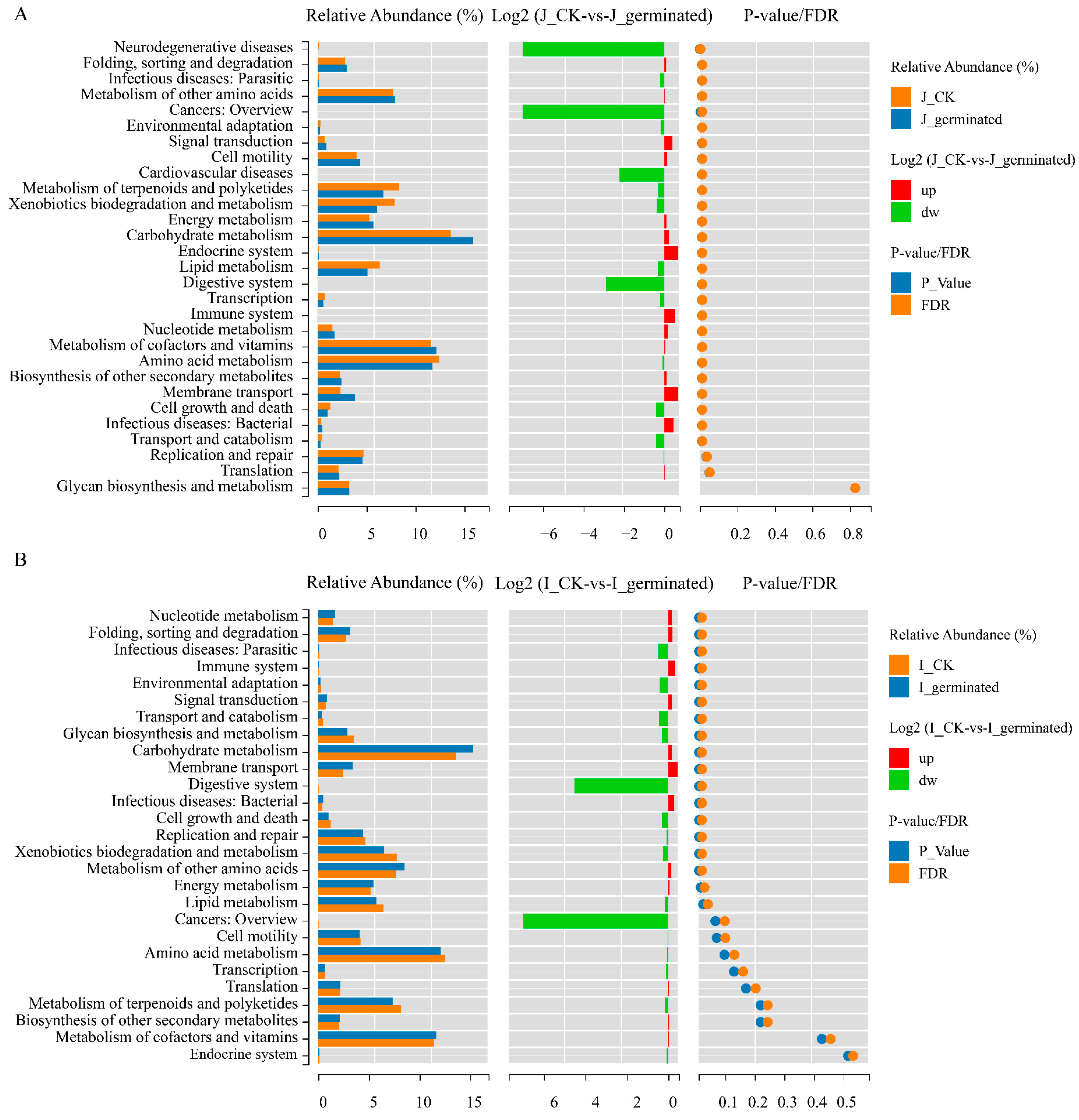
3.3.6. Functional Prediction Analysis of Microbial Community
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Saleh, A.S.M.; Wang, P.; Wang, N.; Yang, L.; Xiao, Z. Brown rice versus white rice: Nutritional quality, potential health benefits, development of food products, and preservation technologies. Compr. Rev. Food Sci. Food Saf. 2019, 18, 1070–1096. [Google Scholar] [PubMed]
- Lemmens, E.; Moroni, A.V.; Pagand, J.; Heirbaut, P.; Ritala, A.; Karlen, Y.; Lê, K.A.; Den Broeck, H.C.; Brouns, F.J.P.H.; Brier, N. Impact of cereal seed sprouting on its nutritional and technological properties: A critical review. Compr. Rev. Food Sci. Food Saf. 2019, 18, 305–328. [Google Scholar] [PubMed]
- Cho, D.H.; Lim, S.T. Germinated brown rice and its bio-functional compounds. Food Chem. 2016, 196, 259–271. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.S.; Kim, B.H.; Kim, M.J.; Han, J.K.; Kum, J.S.; Lee, H.Y. Quantitative microbiological profiles of brown rice and germinated brown rice. Food Sci. Biotechnol. 2012, 21, 1785–1788. [Google Scholar] [CrossRef]
- Lu, Z.H.; Zhang, Y.; Li, L.T.; Curtis, R.B.; Kong, X.L.; Fulcher, R.G.; Zhang, G.; Cao, W. Inhibition of microbial growth and enrichment of gamma-aminobutyric acid during germination of brown rice by electrolyzed oxidizing water. J. Food Protect. 2010, 73, 483–487. [Google Scholar] [CrossRef]
- Bourneow, C.; Toontam, N. Microbiological quality and some bioactive compounds in relation to sensory profiles during germination of brown-purple-pigmented rice. Int. Food Res. J. 2019, 26, 1011–1021. [Google Scholar]
- Miyahira, R.F.; Costa Antunes, A.E. Bacteriological safety of sprouts: A brief review. Int. J. Food Microbiol. 2021, 352, 109266. [Google Scholar] [CrossRef]
- Ostlie, H.M.; Porcellato, D.; Kvam, G.; Wicklund, T. Investigation of the microbiota associated with ungerminated and germinated Norwegian barley cultivars with focus on lactic acid bacteria. Int. J. Food Microbiol. 2021, 341, 109059. [Google Scholar] [CrossRef]
- Los, A.; Ziuzina, D.; Boehm, D.; Cullen, P.J.; Bourke, P. The potential of atmospheric air cold plasma for control of bacterial contaminants relevant to cereal grain production. Innov. Food Sci. Emerg. Technol. 2017, 44, 36–45. [Google Scholar] [CrossRef]
- Laca, A.; Mousia, Z.; Diaz, M.; Webb, C.; Pandiella, S.S. Distribution of microbial contamination within cereal grains. J. Food Eng. 2006, 72, 332–338. [Google Scholar] [CrossRef]
- Piernas, V.; Guiraud, J.P. Microbial hazards related to rice sprouting. Int. J. Food Sci. Technol. 1997, 32, 33–39. [Google Scholar] [CrossRef]
- Dechet, A.M.; Herman, K.M.; Chen Parker, C.; Taormina, P.; Johanson, J.; Tauxe, R.V.; Mahon, B.E. Outbreaks caused by sprouts, United States, 1998–2010: Lessons learned and solutions needed. Foodborne Pathog. Dis. 2014, 11, 635–644. [Google Scholar]
- Callejón, R.M.; Rodriguez-Naranjo, M.I.; Ubeda, C.; Hornedo-Ortega, R.; GarciaParrilla, M.C.; Troncoso, A.M. Reported foodborne outbreaks due to fresh produce in the United States and European union: Trends and causes. Foodborne Pathog. Dis. 2015, 12, 32–38. [Google Scholar] [CrossRef]
- Villeneuve, S.; Power, K.A.; Guévremont, E.; Mondor, M.; Tsao, R.; Wanasundara, J.P.D.; Mercier, S. Effect of a short-time germination process on the nutrient composition, microbial counts and bread-making potential of whole flaxseed. J. Food Process. Pres. 2015, 39, 1574–1586. [Google Scholar] [CrossRef]
- Cai, H.; Zhang, T.; Zhang, Q.; Luo, J.; Cai, C.; Mao, J. Microbial diversity and chemical analysis of the starters used in traditional Chinese sweet rice wine. Food Microbiol. 2018, 73, 319–326. [Google Scholar] [CrossRef]
- Zhao, G.; Li, J.; Zheng, F.; Yao, Y. The fermentation properties and microbial diversity of soy sauce fermented by germinated soybean. J. Sci. Food Agric. 2021, 101, 2920–2929. [Google Scholar] [CrossRef] [PubMed]
- Keshri, J.; Krouptiski, Y.; Abu-Fani, L.; Achmon, Y.; Bauer, T.S.; Zarka, O.; Maler, I.; Pinto, R.; Saldinger, S.S. Dynamics of bacterial communities in alfalfa and mung bean sprouts during refrigerated conditions. Food Microbiol. 2019, 84, 103261. [Google Scholar]
- Los, A.; Ziuzina, D.; Boehm, D.; Bourke, P. Effects of cold plasma on wheat grain microbiome and antimicrobial efficacy against challenge pathogens and their resistance. Int. J. Food Microbiol. 2020, 335, 108889. [Google Scholar] [CrossRef] [PubMed]
- Solanki, M.K.; Abdelfattah, A.; Britzi, M.; Zakin, V.; Wisniewski, M.; Droby, S.; Sionov, E. Shifts in the composition of the microbiota of stored wheat grains in response to fumigation. Front. Microbiol. 2019, 10, 1098. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Ban, G.H.; Hong, Y.W.; Jang, M.J.; Kim, S.A. Microbiome shifts in sprouts (alfalfa, radish, and rapeseed) during production from seed to sprout using 16S rRNA microbiome sequencing. Food Res. Int. 2022, 152, 110896. [Google Scholar]
- Moongngarm, A.; Saetung, N. Comparison of chemical compositions and bioactive compounds of germinated rough rice and brown rice. Food Chem. 2010, 122, 782–788. [Google Scholar]
- Magoc, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [PubMed]
- Edgar, R.C. UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 2013, 10, 996–998. [Google Scholar] [CrossRef]
- Edgar, R.C.; Haas, B.J.; Clemente, J.C.; Quince, C.; Knight, R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 2011, 27, 2194–2200. [Google Scholar] [PubMed]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Pena, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [Green Version]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [PubMed]
- Landry, K.S.; Sela, D.A.; Mclandsborough, L. Influence of sprouting environment on the microbiota of sprouts. J. Food Saf. 2017, 38, e12380. [Google Scholar]
- Liu, H.K.; Li, Z.H.; Zhang, X.W.; Liu, Y.P.; Zhao, X.Y. The effects of ultrasound on the growth, nutritional quality and microbiological quality of sprouts. Trends Food Sci. Technol. 2021, 111, 92–300. [Google Scholar]
- Landry, K.S.; Micheli, S.; Mcclements, D.J.; Mclandsborough, L. Effectiveness of a spontaneous carvacrol nanoemulsion against Salmonella enterica Enteritidis and Escherichia coli O157:H7 on contaminated broccoli and radish seeds. Food Microbiol. 2015, 51, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Peñas, E.; Gómez, R.; Frías, J.; Vidal-Valverde, C. Effects of combined treatments of high pressure, temperature and antimicrobial products on germination of mung bean seeds and microbial quality of sprouts. Food Control 2010, 21, 82–88. [Google Scholar]
- Park, H.; Puligundla, P.; Mok, C. Cold plasma decontamination of brown rice: Impact on biochemical and sensory qualities of their corresponding seedlings and aqueous tea infusions. LWT Food Sci. Technol. 2020, 131, 109508. [Google Scholar]
- Kim, B.; Bang, J.; Kim, H.; Kim, Y.; Kim, B.S.; Beuchat, L.R.; Ryu, J.H. Bacillus cereus and Bacillus thuringiensis spores in Korean rice: Prevalence and toxin production as affected by production area and degree of milling. Food Microbiol. 2014, 42, 89–94. [Google Scholar] [CrossRef]
- Mir, S.A.; Farooq, S.; Shah, M.A.; Sofi, S.A.; Khaneghah, A.M. An overview of sprouts nutritional properties, pathogens and decontamination technologies. LWT Food Sci. Technol. 2021, 141, 110900. [Google Scholar]
- Li, X.; Cai, G.; Wu, D.; Zhang, M.; Lin, C.; Lu, J. Microbial community dynamics of Dan’er barley grain during the industrial malting process. Food Microbiol. 2018, 76, 110–116. [Google Scholar] [PubMed]
- Gloria, T.C.; Sophie, B.; Olivier, B.; Clémence, G.; Martial, B.; Marie-Agnès, J.; Matthieu, B. Functional microbial features driving community assembly during seed germination and emergence. Front. Plant Sci. 2018, 9, 902. [Google Scholar]
- Huang, Y.; Kuang, Z.; Wang, W.; Cao, L. Exploring potential bacterial and fungal biocontrol agents transmitted from seeds to sprouts of wheat. Biol. Control 2016, 98, 27–33. [Google Scholar] [CrossRef]
- Bziuk, N.; Maccario, L.; Straube, B.; Wehner, G.; Sorensen, S.J.; Schikora, A.; Small, K. The treasure inside barley seeds: Microbial diversity and plant beneficial bacteria. Environ. Microbiome 2021, 16, 20. [Google Scholar] [PubMed]
- Fierer, N.; Bradford, M.A.; Jackson, R.B. Toward an ecological classification of soil bacteria. Ecology 2007, 88, 1354–1364. [Google Scholar] [CrossRef] [PubMed]
- He, H.D.; Li, W.C.; Yu, R.Q.; Ye, Z.H. Illumina-based analysis of bulk and rhizosphere soil bacterial communities in paddy fields under mixed heavy metal contamination. Pedosphere 2017, 27, 569–578. [Google Scholar]
- Walterson, A.M.; Stavrinides, J. Pantoea: Insights into a highly versatile and diverse genus within the Enterobacteriaceae. FEMS Microbiol. Rev. 2015, 39, 968–984. [Google Scholar] [PubMed]
- Leff, J.W.; Noah, F.; Gabriele, B. Bacterial communities associated with the surfaces of fresh fruits and vegetables. PLoS ONE 2013, 8, e59310. [Google Scholar] [CrossRef] [PubMed]
- Logan, N.A. Bacillus and relatives in foodborne illness. J. Appl. Microbiol. 2011, 112, 417–429. [Google Scholar]
- Chenu, J.W.; Cox, J.M. Cronobacter (‘Enterobacter sakazakii’): Current status and future prospects. Lett. Appl. Microbiol. 2010, 49, 153–159. [Google Scholar]
- Berthold-Pluta, A.; Garbowska, M.; Stefańska, I.; Pluta, A. Microbiological quality of selected ready-to-eat leaf vegetables, sprouts and non-pasteurized fresh fruit-vegetable juices including the presence of Cronobacter spp. Food Microbiol. 2017, 65, 221–230. [Google Scholar] [PubMed]
- Healy, B.; Cooney, S.; O’Brien, S.; Iversen, C.; Whyte, P.; Nally, J.; Callanan, J.J.; Fanning, S. Cronobacter (Enterobacter sakazakii): An opportunistic foodborne pathogen. Foodborne Pathog. Dis. 2010, 7, 339–350. [Google Scholar]
- He, Z.; Chen, H.; Wang, X.; Lin, X.; Ji, C.; Li, S.; Liang, H. Effects of different temperatures on bacterial diversity and volatile flavor compounds during the fermentation of suancai, a traditional fermented vegetable food from northeastern China. LWT Food Sci. Technol. 2020, 118, 108773. [Google Scholar]
- Zhang, J.; Zhang, C.; Wu, W.; Lv, X.; Xin, X.; Liu, D.; Hu, H.; Guo, S. Correlation of the bacterial communities with umami components, and chemical characteristics in Zhejiang xuecai and fermented brine. Food Res. Int. 2021, 140, 109986. [Google Scholar] [CrossRef]
- Jiang, L.; Jeong, J.C.; Lee, J.S.; Park, J.M.; Yang, J.W.; Lee, M.H.; Choi, S.H.; Kim, C.Y.; Kim, D.H.; Kim, S.W.; et al. Potential of Pantoea dispersa as an effective biocontrol agent for black rot in sweet potato. Sci. Rep. 2019, 9, 16354. [Google Scholar]
- Singha, K.M.; Singh, B.; Pandey, P. Host specific endophytic microbiome diversity and associated functions in three varieties of scented black rice are dependent on growth stage. Sci. Rep. 2021, 11, 12259. [Google Scholar] [PubMed]
- Huang, Y.; Zhang, M.; Deng, Z.; Cao, L. Evaluation of probiotic diversity from soybean (Glycine max) seeds and sprouts using illumina-based sequencing method. Probiotics Antimicrob. Proteins 2018, 10, 293–298. [Google Scholar] [CrossRef] [PubMed]
- Park, H.S.; Jun, S.C.; Han, K.H.; Hong, S.B.; Yu, J.H. Diversity, application, and synthetic biology of industrially important Aspergillus Fungi. Adv. Appl. Microbiol. 2017, 100, 161–202. [Google Scholar] [PubMed]
- Perrone, G.; Susca, A.; Cozzi, G.; Ehrlich, K.; Varga, J.; Frisvad, J.C.; Meijer, M.; Noonim, P.; Mahakamchanakul, W.; Samson, R.A. Biodiversity of Aspergillus species in some important agricultural products. Stud. Mycol. 2007, 59, 53–66. [Google Scholar] [PubMed]
- Xie, M.; An, F.; Wu, J.; Liu, Y.; Shi, H.; Wu, R. Meta-omics reveal microbial assortments and key enzymes in bean sauce mash, a traditional fermented soybean product. J. Sci. Food Agric. 2019, 99, 6522–6534. [Google Scholar] [CrossRef]
- Justé, A.; Malfliet, S.; Lenaerts, M.; Cooman, L.D.; Lievens, B. Microflora during malting of barley: Overview and impact on malt quality. Brew. Sci. 2011, 64, 22–31. [Google Scholar]
- Gagkaeva, T.Y.; Gavrilova, O.P.; Orina, A.S.; Blinova, E.V.; Loskutov, I.G. Response of wild Avena species to fungal infection of grain. Crop. J. 2017, 5, 499–508. [Google Scholar]
- El-Demerdash, A. Chemical diversity and biological activities of Phaeosphaeria fungi genus: A systematic review. J. Fungi 2018, 4, 130. [Google Scholar]
- Zheng, X.; Xu, X.; Ma, Y.; Zhu, L.; Xiao, J.; Deng, L.; Shi, X.; Wang, B. Diversity and potential function of bacterial communities during milk fermentation of Kazak artisanal cheese. Process. Biochem. 2021, 111, 191–200. [Google Scholar] [CrossRef]
- Wang, Y.; Cai, W.; Wang, W.; Shu, N.; Zhang, Z.; Hou, Q.; Shan, C.; Guo, Z. Analysis of microbial diversity and functional differences in different types of high-temperature Daqu. Food Sci. Nutr. 2021, 9, 1003–1016. [Google Scholar]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | Cultivars | Producing Regions | Moisture Content (% w.b.) | Viable Counts (log10 CFU/g) | |||
|---|---|---|---|---|---|---|---|
| TVC | Molds and Yeasts | Bacillus cereus | Enterobacteriaceae | ||||
| AH1 | Japonica | Anhui | 12.21 ± 0.32 | 5.45 ± 0.20 abcd | 4.11 ± 0.16 c | <1.3 | 3.38 ± 0.34 e |
| AH2 | Japonica | Anhui | 11.99 ± 0.10 | 5.11 ± 0.24 d | 4.22 ± 0.15 bc | 1.60 ± 0.49 e | 3.91 ± 0.33 d |
| GD | Indica | Guangdong | 12.12 ± 0.35 | 5.79 ± 0.05 a | 5.33 ± 0.28 a | 2.45 ± 0.17 ab | 4.76 ± 0.25 b |
| GX | Japonica | Guangxi | 14.00 ± 0.33 | 4.10 ± 0.11 f | 5.20 ± 0.14 a | 1.76 ± 0.11 d e | 3.09 ± 0.09 e |
| HB | Indica | Hubei | 11.89 ± 0.22 | 4.55 ± 0.10 e | 4.22 ± 0.09 bc | 2.59 ± 0.16 a | 3.17 ± 0.06 e |
| HLJ1 | Indica | Heilongjiang | 11.61 ± 0.91 | 4.41 ± 0.09 ef | 4.16 ± 0.45 bc | <1.3 | 3.86 ± 0.52 d |
| HLJ2 | Japonica | Heilongjiang | 11.42 ± 0.66 | 5.32 ± 0.28 bcd | 4.60 ± 0.45 abc | 2.05 ± 0.34 bcd | 3.98 ± 0.33 cd |
| HN | Indica | Hunan | 12.53 ± 0.24 | 5.18 ± 0.02 cd | 4.65 ± 0.16 abc | 2.30 ± 0.32 abc | 3.94 ± 0.35 d |
| JS | Japonica | Jiangsu | 12.75 ± 0.29 | 5.44 ± 0.29 abcd | 5.00 ± 0.26 a | 1.54 ± 0.34 e | 4.37 ± 0.08 c |
| JX | Japonica | Jiangxi | 13.55 ± 0.14 | 5.11 ± 0.19 d | 4.86±0.51 ab | <1.3 | 3.33 ± 0.24 e |
| LN1 | Japonica | Liaoning | 11.84 ± 0.05 | 5.86 ± 0.13 a | 5.14±0.39 a | 1.99 ± 0.24 dce | 5.22 ± 0.23 a |
| LN2 | Japonica | Liaoning | 12.43 ± 0.05 | 5.60 ± 0.01 abc | 3.93±0.03 c | ND * | 3.34 ± 0.26 e |
| SC | Japonica | Sichuan | 11.67 ± 0.25 | 5.65 ± 0.22 ab | 4.98±0.26 a | 1.61 ± 0.32 e | 4.13 ± 0.09 cd |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, G.; Xu, J.; Xu, Y.; Li, R.; Wang, S. Analysis of Dynamics and Diversity of Microbial Community during Production of Germinated Brown Rice. Foods 2023, 12, 755. https://doi.org/10.3390/foods12040755
Yang G, Xu J, Xu Y, Li R, Wang S. Analysis of Dynamics and Diversity of Microbial Community during Production of Germinated Brown Rice. Foods. 2023; 12(4):755. https://doi.org/10.3390/foods12040755
Chicago/Turabian StyleYang, Gaoji, Juanjuan Xu, Yuanmei Xu, Rui Li, and Shaojin Wang. 2023. "Analysis of Dynamics and Diversity of Microbial Community during Production of Germinated Brown Rice" Foods 12, no. 4: 755. https://doi.org/10.3390/foods12040755