
Simulated Fermentation of Strong-Flavor Baijiu through Functional Microbial Combination to Realize the Stable Synthesis of Important Flavor Chemicals

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Preparation and Collection
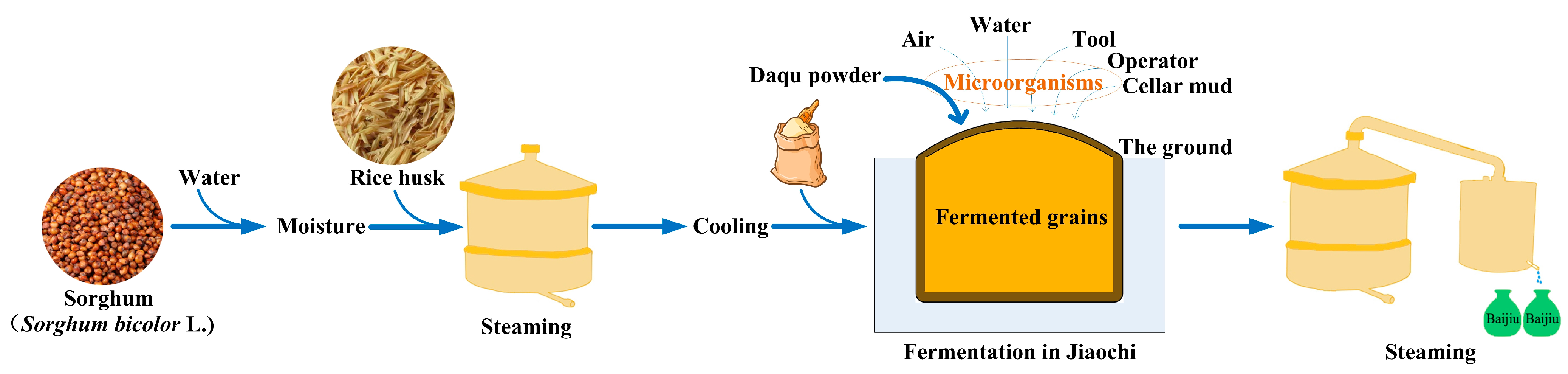
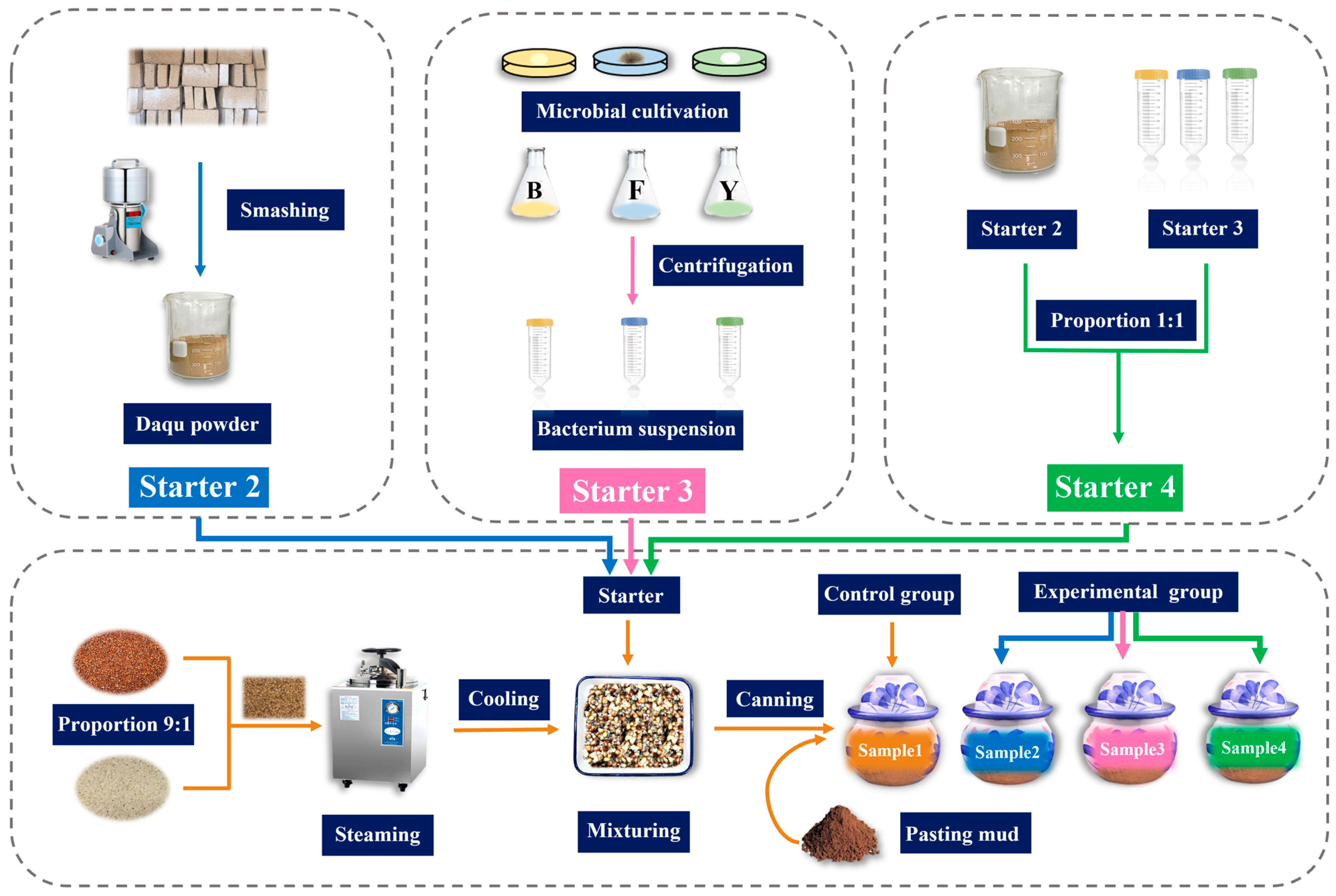
2.1.1. Sample Preparation
2.1.2. Sample Collection
2.2. Determination of Physicochemical Factors
2.3. Analysis of Volatile Flavor Compounds in Fermented Grains
2.4. Microbial Community Analysis
2.5. Data Analysis
3. Results and Discussion
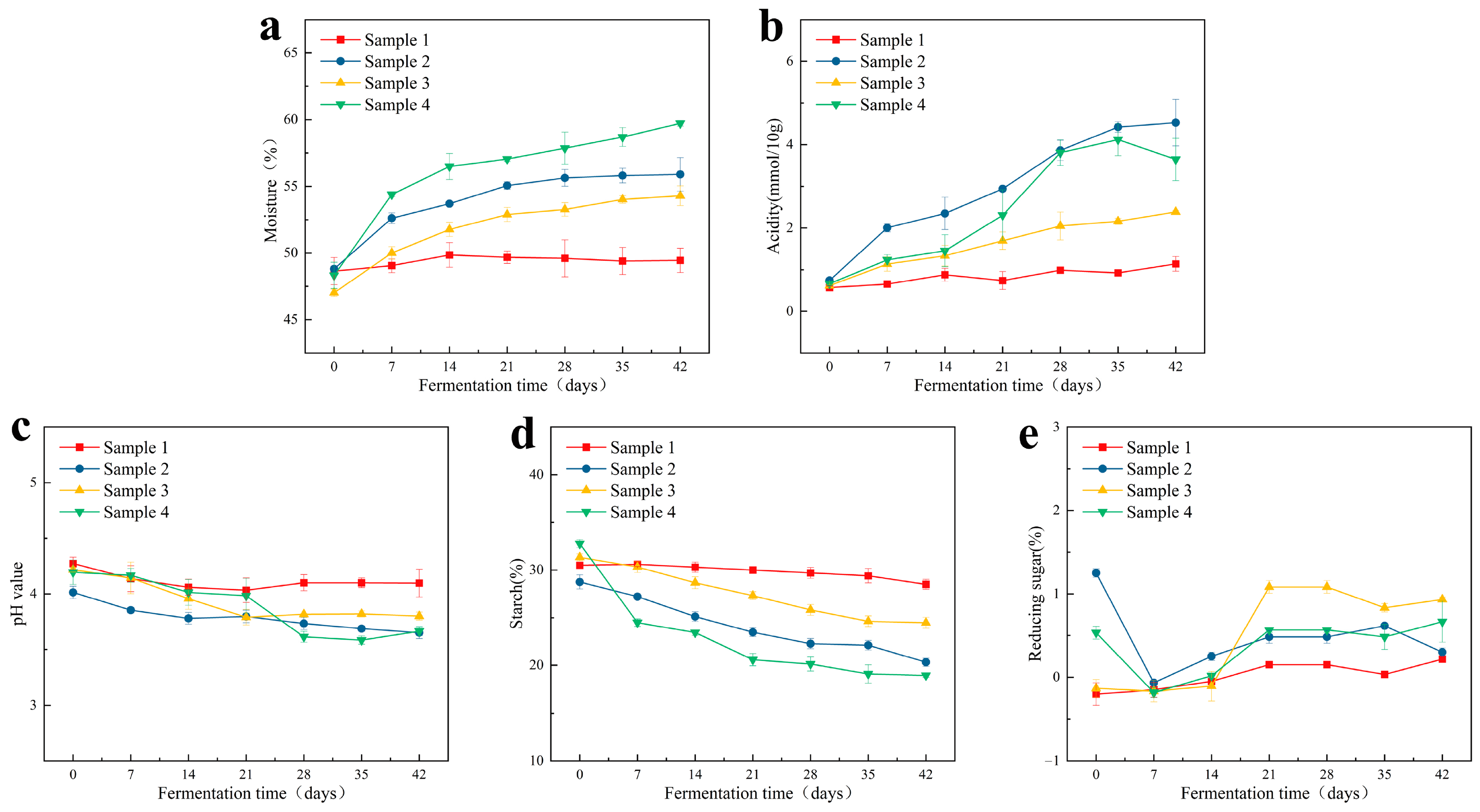
3.1. Analysis of Physicochemical Factors
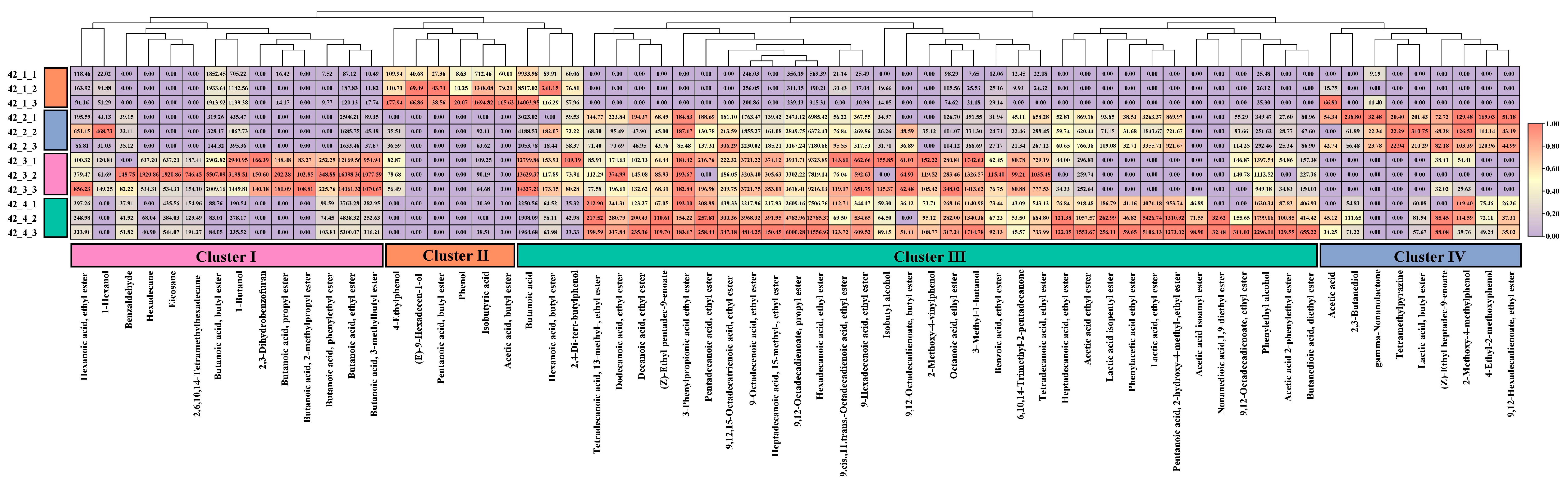
3.2. Analysis of Flavor Compounds
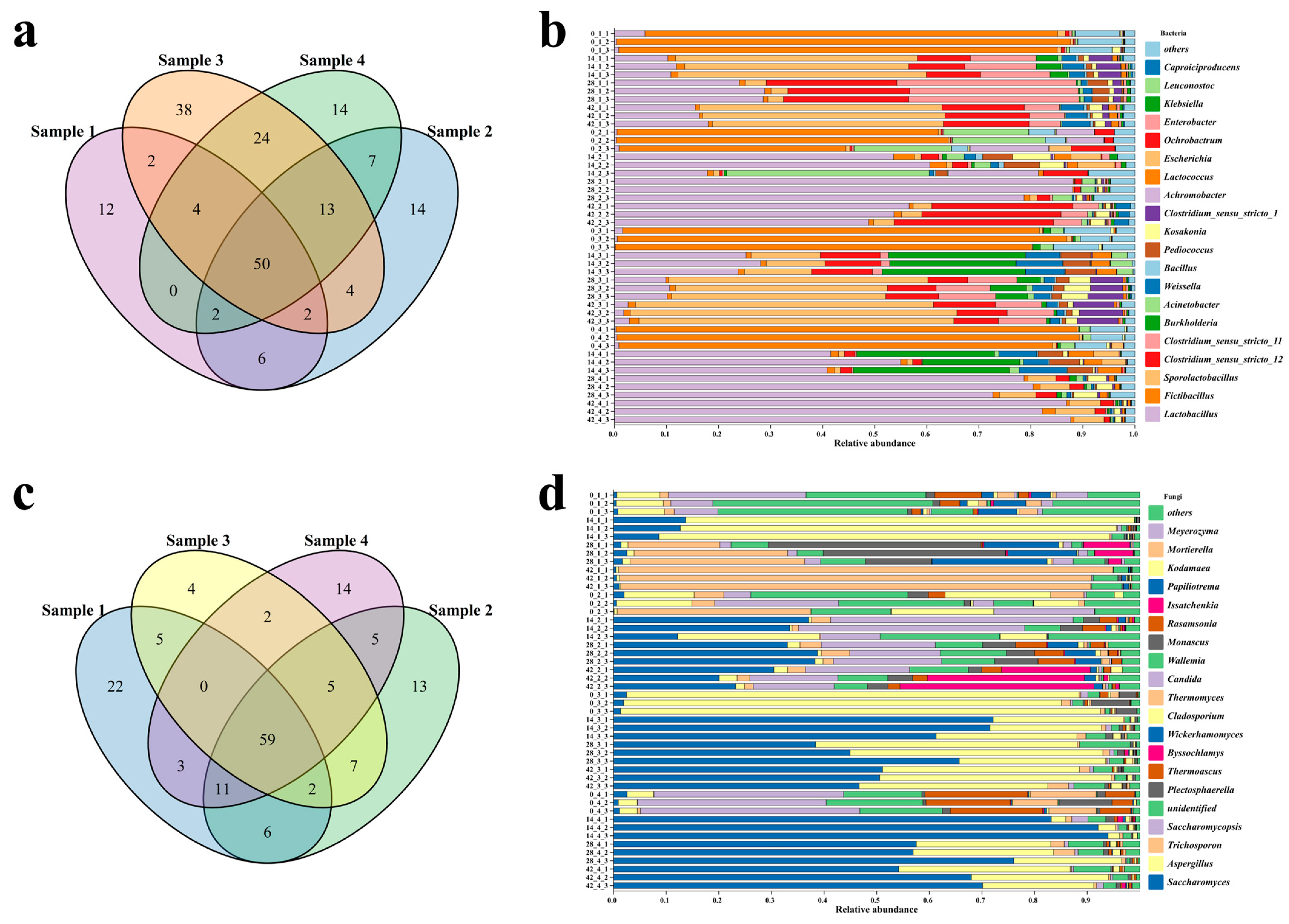
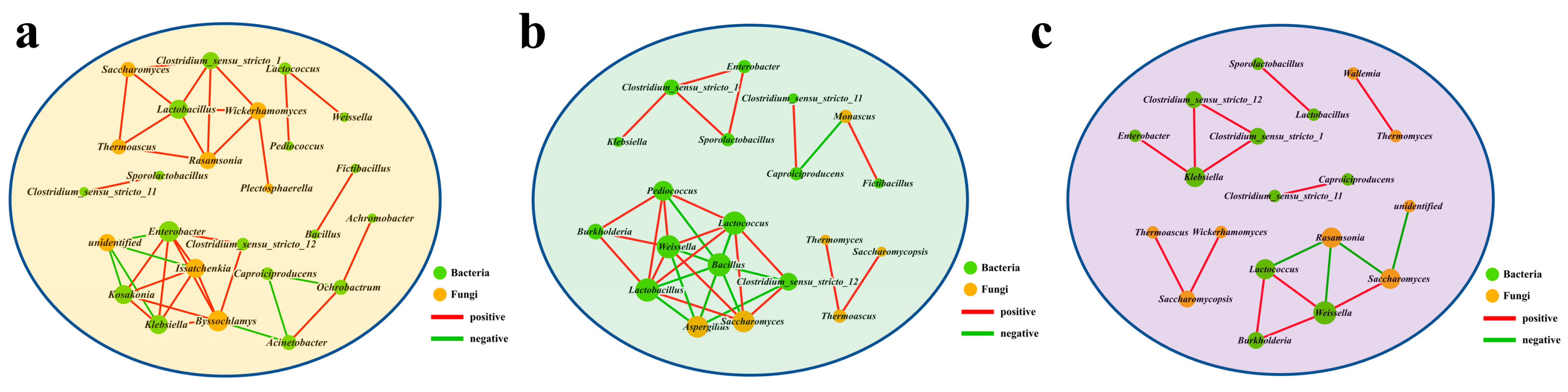
3.3. Succession of Microbial Communities and Microbial Interactions
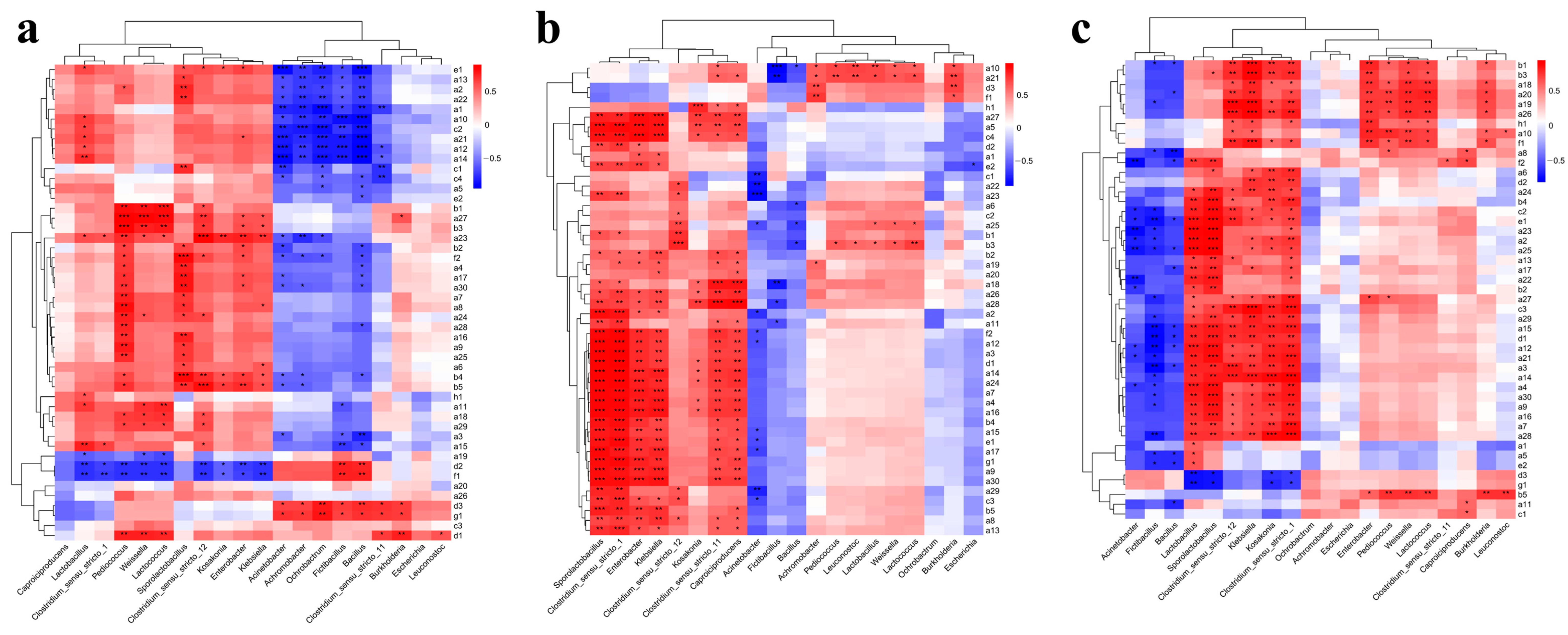
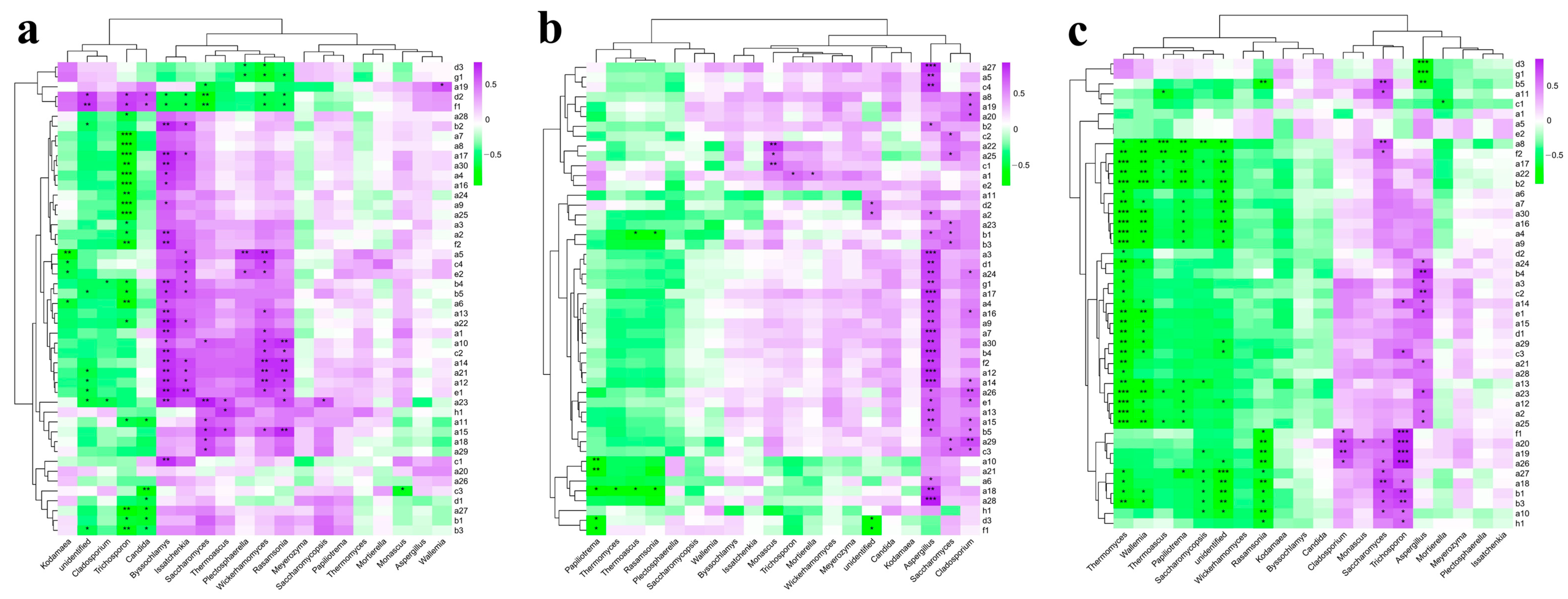
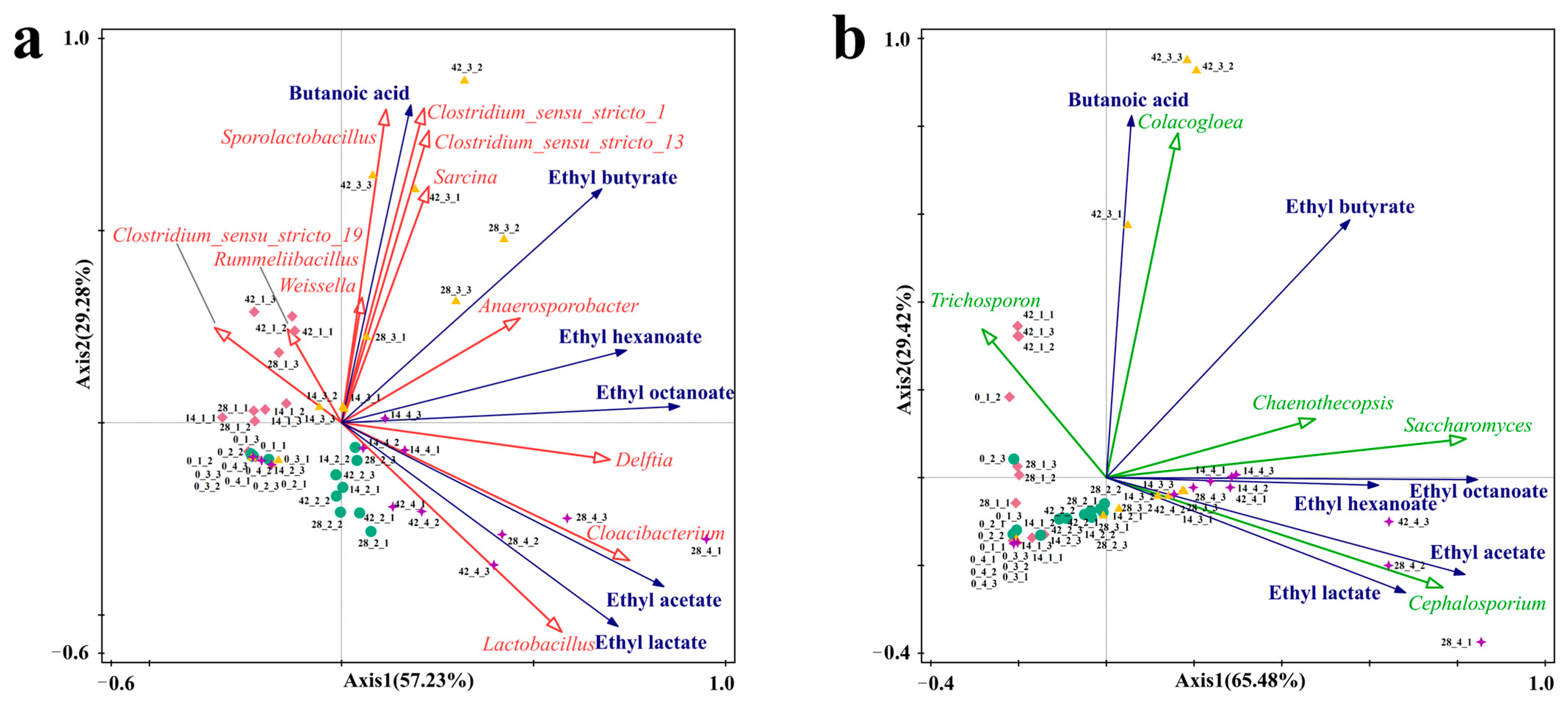
3.4. Correlation Analysis of Flavor Compounds and Microorganisms
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Xu, Y.Q.; Sun, B.G.; Fan, G.S.; Teng, C.; Xiong, K.; Zhu, Y.P.; Li, J.L.; Li, X.T. The brewing process and microbial diversity of strong flavour Chinese spirits: A review. J. Inst. Brew. 2017, 123, 5–12. [Google Scholar] [CrossRef]
- Xu, Y.Q.; Zhao, J.R.; Liu, X.; Zhang, C.S.; Zhao, Z.G.; Li, X.T.; Sun, B.G. Flavor mystery of Chinese traditional fermented baijiu: The great contribution of ester compounds. Food Chem. 2022, 369, 130920. [Google Scholar] [CrossRef]
- Chai, L.J.; Qian, W.; Zhong, X.Z.; Zhang, X.J.; Lu, Z.M.; Zhang, S.Y.; Wang, S.T.; Shen, C.H.; Shi, J.S.; Xu, Z.H. Mining the factors driving the evolution of the pit mud microbiome under the impact of long-term production of strong-flavor Baijiu. Appl. Environ. Microbiol. 2021, 87, AEM0088521. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.Q.; Huang, H.Q.; Lu, H.Y.; Wu, M.Q.; Lin, M.W.; Zhang, C.S.; Zhao, Z.G.; Li, W.W.; Zhang, C.N.; Li, X.T.; et al. Characterization of an Aspergillus niger for efficient fatty acid ethyl ester synthesis in aqueous phase and the molecular mechanism. Front Microbiol. 2022, 12, 820380. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.Q.; Minhazul, K.; Wang, X.C.; Liu, X.; Li, X.T.; Meng, Q.; Li, H.H.; Zhang, C.N.; Sun, X.T.; Sun, B.G. Biodegradation of phthalate esters by Paracoccus kondratievae BJQ0001 isolated from Jiuqu (Baijiu fermentation starter) and identification of the ester bond hydrolysis enzyme. Environ. Pollut. 2020, 263, 114506. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.Q.; Wang, X.C.; Liu, X.; Li, X.T.; Zhang, C.N.; Li, W.W.; Sun, X.T.; Wang, W.H.; Sun, B.G. Discovery and development of a novel short-chain fatty acid ester synthetic biocatalyst under aqueous phase from Monascus purpureus isolated from Baijiu. Food Chem. 2021, 338, 128025. [Google Scholar] [CrossRef] [PubMed]
- Lai, J.H.; Huang, H.Q.; Lin, M.W.; Xu, Y.Q.; Li, X.T.; Sun, B.G. Enzyme catalyzes ester bond synthesis and hydrolysis: The key step for sustainable usage of plastics. Front. Microbiol. 2023, 13, 1113705. [Google Scholar] [CrossRef]
- Zheng, X.W.; Tabrizi, M.R.; Nout, M.J.R.; Han, B.Z. Daqu-a traditional Chinese liquor fermentation starter. J. Inst. Brew 2011, 117, 82–90. [Google Scholar] [CrossRef]
- Li, P.; Lin, W.F.; Liu, X.; Wang, X.W.; Luo, L.X. Environmental factors affecting microbiota dynamics during traditional solid-state fermentation of Chinese Daqu starter. Front. Microbiol. 2016, 7, 1237. [Google Scholar] [CrossRef]
- Wang, S.W.; Wang, Q.H.; Zhai, L.P.; Liu, J.; Yu, Z.D.; Zheng, M.M.; Wang, F. Genetic diversity among the microorganisms in Daqu used for beidacang liquor as revealed by RAPD analyses. In Proceedings of the 2nd International Conference on Applied Biotechnology (ICAB), Tianjin, China, 28–30 November 2014; pp. 99–108. [Google Scholar]
- Ma, S.Y.; Shang, Z.C.; Chen, J.; Shen, Y.J.; Li, Z.J.; Huang, D.; Luo, H.B. Differences in structure, volatile metabolites, and functions of microbial communities in nongxiangxing daqu from different production areas. LWT-Food Sci. Technol. 2022, 166, 113784. [Google Scholar] [CrossRef]
- Sowards, J.W.; Williamson, C.H.D.; Weeks, T.S.; McColskey, J.D.; Spear, J.R. The effect of Acetobacter sp and a sulfate-reducing bacterial consortium from ethanol fuel environments on fatigue crack propagation in pipeline and storage tank steels. Corrosion Sci. 2014, 79, 128–138. [Google Scholar] [CrossRef]
- Wu, Q.; Zhu, Y.; Fang, C.; Wijffels, R.H.; Xu, Y. Can we control microbiota in spontaneous food fermentation? Chinese liquor as a case example. Trends Food Sci. Technol. 2021, 110, 321–331. [Google Scholar] [CrossRef]
- Jin, G.Y.; Zhu, Y.; Xu, Y. Mystery behind Chinese liquor fermentation. Trends Food Sci. Technol. 2017, 63, 18–28. [Google Scholar] [CrossRef]
- Liu, M.K.; Zhao, K.; Tang, Y.M.; Ren, D.Q.; Yao, W.C.; Tian, X.H.; Zhang, X.Y.; Yi, B.; Deng, B. Analysis of Clostridium cluster I community diversity in pit mud used in manufacture of Chinese Luzhou-flavor liquor. Food Sci. Biotechnol. 2015, 24, 995–1000. [Google Scholar] [CrossRef]
- Wang, Y.S.; Li, B.; Dong, H.; Huang, X.D.; Chen, R.Y.; Chen, X.J.; Yang, L.J.; Peng, B.; Xie, G.P.; Cheng, W.; et al. Complete genome sequence of Clostridium kluyveri JZZ applied in Chinese strong-flavor liquor production. Curr. Microbiol. 2018, 75, 1429–1433. [Google Scholar] [CrossRef]
- Zhao, J.R.; Xu, Y.Q.; Lu, H.Y.; Zhao, D.; Zheng, J.; Lin, M.W. Molecular mechanism of LIP05 derived from Monascus purpureus YJX-8 for synthesizing fatty acid ethyl esters under aqueous phase. Front. Microbiol. 2023, 13, 1107104. [Google Scholar] [CrossRef]
- Wolfe, B.E.; Button, J.E.; Santarelli, M.; Dutton, R.J. Cheese rind communities provide tractable systems for in situ and in vitro studies of microbial diversity. Cell 2014, 158, 422–433. [Google Scholar] [CrossRef]
- Wolfe, B.E.; Dutton, R.J. Fermented foods as experimentally tractable microbial ecosystems. Cell 2015, 161, 49–55. [Google Scholar] [CrossRef]
- Huang, Z.R.; Hong, J.L.; Xu, J.X.; Li, L.; Guo, W.L.; Pan, Y.Y.; Chen, S.J.; Bai, W.D.; Rao, P.F.; Ni, L.; et al. Exploring core functional microbiota responsible for the production of volatile flavour during the traditional brewing of Wuyi Hong Qu glutinous rice wine. Food Microbiol. 2018, 76, 487–496. [Google Scholar] [CrossRef]
- Gu, Y.; Zhu, X.J.; Lin, F.; Shen, C.H.; Li, Y.; Ao, L.; Fan, W.L.; Ren, C.; Xu, Y. Caproicibacterium amylolyticum gen. nov., sp. nov., a novel member of the family Oscillospiraceae isolated from pit clay used for making Chinese strong aroma-type liquor. Int. J. Syst. Evol. Microbiol. 2021, 71, 4789. [Google Scholar] [CrossRef]
- Wang, H.L.; Gu, Y.; Zhao, D.; Qiao, Z.W.; Zheng, J.; Gao, J.J.; Ren, C.; Xu, Y. Caproicibacterium lactatifermentans sp. nov., isolated from pit clay used for the production of Chinese strong aroma-type liquor. Int. J. Syst. Evol. Microbiol. 2022, 72, 005206. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.Q.; Wu, M.Q.; Niu, J.L.; Huang, H.Q.; Nie, Z.; Fu, Z.L.; Zhang, C.S.; Zhao, Z.G.; Lu, H.Y.; Li, X.T.; et al. Clostridium btbubcensis BJN0001, a potentially new species isolated from the cellar mud of Chinese strong-flavor baijiu, produces ethanol, acetic acid and butyric acid from glucose. 3 Biotech 2022, 12, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.X.; Wang, L.; Wang, H.Y.; Yang, F.; Chen, L.Q.; Hao, F.; Lv, X.B.; Du, H.; Xu, Y. Effects of initial temperature on microbial community succession rate and volatile flavors during Baijiu fermentation process. Food Res. Int. 2021, 141, 109887. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.; Zhong, H.; Zhao, D.; Du, H.; Xu, Y. Succession rate of microbial community causes flavor difference in strong-aroma Baijiu making process. Int. J. Food Microbiol. 2019, 311, 108350. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.H.; Xu, Y.Q.; Huang, H.Q.; Pang, Z.M.; Fu, Z.L.; Niu, J.L.; Zhang, C.N.; Li, W.W.; Li, X.T.; Sun, B.G. Correlation between microbial communities and flavor compounds during the fifth and sixth rounds of sauce-flavor baijiu fermentation. Food Res. Int. 2021, 150, 110741. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.Q.; Wu, M.Q.; Niu, J.L.; Lin, M.W.; Zhu, H.; Wang, K.; Li, X.T.; Sun, B.G. Characteristics and correlation of the microbial communities and flavor compounds during the first three rounds of fermentation in Chinese sauce-flavor Baijiu. Foods 2023, 12, 207. [Google Scholar] [CrossRef]
- Xu, S.S.; Zhang, M.Z.; Xu, B.Y.; Liu, L.H.; Sun, W.; Mu, D.D.; Wu, X.F.; Li, X.J. Microbial communities and flavor formation in the fermentation of Chinese strong-flavor Baijiu produced from old and new Zaopei. Food Res. Int. 2022, 156, 111162. [Google Scholar] [CrossRef]
- Hu, X.L.; Tian, R.J.; Wang, K.L.; Cao, Z.H.; Yan, P.X.; Li, F.Q.; Li, X.S.; Li, S.L.; He, P.X. The prokaryotic community, physicochemical properties and flavors dynamics and their correlations in fermented grains for Chinese strong-flavor Baijiu production. Food Res. Int. 2021, 148, 110626. [Google Scholar] [CrossRef]
- Shi, X.S.; Zhao, S.M.; Chen, S.X.; Han, X.L.; Yang, Q.; Zhang, L.; Xia, X.; Tu, J.M.; Hu, Y.L. Tetramethylpyrazine in Chinese baijiu: Presence, analysis, formation, and regulation. Front. Nutr. 2022, 9, 1004435. [Google Scholar] [CrossRef]
- Kutanovas, S.; Stankeviciute, J.; Urbelis, G.; Tauraite, D.; Rutkiene, R.; Meskys, R. Identification and characterization of a Tetramethylpyrazine catabolic pathway in Rhodococcus jostii TMP1. Appl. Environ. Microbiol. 2013, 79, 3649–3657. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.J.; Liu, G.Y.; Li, A.J.; Liang, C.C.; Ren, C.; Xu, Y. Domination of pit mud microbes in the formation of diverse flavour compounds during Chinese strong aroma-type Baijiu fermentation. LWT-Food Sci. Technol. 2021, 137, 110442. [Google Scholar] [CrossRef]
- Wang, X.L.; Song, X.B.; Zhu, L.; Geng, X.J.; Zheng, F.P.; Zhao, Q.Z.; Sun, X.T.; Zhao, D.R.; Feng, S.B.; Zhao, M.; et al. Unraveling the acetals as ageing markers of Chinese Highland Qingke Baijiu using comprehensive two-dimensional gas chromatography-time-of-flight mass spectrometry combined with metabolomics approach. Food Qual. Saf. 2021, 5, fyab014. [Google Scholar] [CrossRef]
- Yan, Q.; Zhang, K.Z.; Zou, W.; Hou, Y.C. Three main flavour types of Chinese Baijiu: Characteristics, research, and perspectives. J. Inst. Brew 2021, 127, 317–326. [Google Scholar] [CrossRef]
- Xia, Y.A.; Liu, Y.Q.; Wang, J.; Shuang, Q. Assessment of key aroma compounds in fresh jujube brandy by GC-O-MS and odor activity value. J. Food Process. Preserv. 2020, 44, e14494. [Google Scholar] [CrossRef]
- Layeghifard, M.; Hwang, D.M.; Guttman, D.S. Disentangling interactions in the microbiome: A network perspective. Trends Microbiol. 2017, 25, 217–228. [Google Scholar] [CrossRef]
- Braga, R.M.; Dourado, M.N.; Araujo, W.L. Microbial interactions: Ecology in a molecular perspective. Braz. J. Microbiol. 2016, 47, 86–98. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.A.; Lee, M.K.; Park, T.H.; Rhee, M.S. A combined intervention using fermented ethanol and supercritical carbon dioxide to control Bacillus cereus and Bacillus subtilis in rice. Food Control 2013, 32, 93–98. [Google Scholar] [CrossRef]
- Araque, I.; Bordons, A.; Reguant, C. Effect of ethanol and low pH on citrulline and ornithine excretion and arc gene expression by strains of Lactobacillus brevis and Pediococcus pentosaceus. Food Microbiol. 2013, 33, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.F.; Wu, C.D.; Huang, J.; Zhou, R.Q. Interphase microbial community characteristics in the fermentation cellar of Chinese Luzhou-flavor liquor determined by PLFA and DGGE profiles. Food Res. Int. 2015, 72, 16–24. [Google Scholar] [CrossRef]
- Du, H.; Ji, M.; Xing, M.Y.; Wang, X.S.; Xu, Y. The effects of dynamic bacterial succession on the flavor metabolites during Baijiu fermentation. Food Res. Int. 2021, 140, 109860. [Google Scholar] [CrossRef]
- Liu, M.K.; Tang, Y.M.; Zhao, K.; Liu, Y.; Guo, X.J.; Tian, X.H.; Ren, D.Q.; Yao, W.C. Contrasting bacterial community structure in artificial pit mud-starter cultures of different qualities: A complex biological mixture for Chinese strong-flavor Baijiu production. 3 Biotech 2019, 9, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.L.; Wang, K.L.; Chen, M.G.; Fan, J.H.; Han, S.N.; Hou, J.G.; Chi, L.; Liu, Y.P.; Wei, T. Profiling the composition and metabolic activities of microbial community in fermented grain for the Chinese strong-flavor Baijiu production by using the metatranscriptome, high-throughput 16S rRNA and ITS gene sequencings. Food Res. Int. 2020, 138, 109765. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Chen, S.J.; Wang, J.H.; Zhang, C.Y.; Shi, Y.; Guo, X.W.; Chen, Y.F.; Xiao, D.G. Genetic engineering to alter carbon flux for various higher alcohol productions by Saccharomyces cerevisiae for Chinese Baijiu fermentation. Appl. Microbiol. Biotechnol. 2018, 102, 1783–1795. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.L.; Sun, B.G. Effect of fermentation processing on the flavor of Baijiu. J. Agric. Food Chem. 2018, 66, 5425–5432. [Google Scholar] [CrossRef]
- Zou, W.; Ye, G.B.; Zhang, K.Z. Diversity, function, and application of Clostridium in Chinese strong flavor Baijiu ecosystem: A Review. J. Food Sci. 2018, 83, 1193–1199. [Google Scholar] [CrossRef]
- Liu, M.K.; Tang, Y.M.; Zhao, K.; Liu, Y.; Guo, X.J.; Ren, D.Q.; Yao, W.C.; Tian, X.H.; Gu, Y.F.; Yi, B.; et al. Determination of the fungal community of pit mud in fermentation cellars for Chinese strong-flavor liquor, using DGGE and Illumina MiSeq sequencing. Food Res. Int. 2017, 91, 80–87. [Google Scholar] [CrossRef]
- He, G.Q.; Huang, J.; Wu, C.D.; Jin, Y.; Zhou, R.Q. Bioturbation effect of fortified Daqu on microbial community and flavor metabolite in Chinese strong-flavor liquor brewing microecosystem. Food Res. Int. 2020, 129, 108851. [Google Scholar] [CrossRef]
- Song, Z.W.; Du, H.; Zhang, Y.; Xu, Y. Unraveling core functional microbiota in traditional solid-state fermentation by high-throughput amplicons and metatranscriptomics sequencing. Front. Microbiol. 2017, 8, 1294. [Google Scholar] [CrossRef]
- Du, R.B.; Wu, Q.; Xu, Y. Chinese liquor fermentation: Identification of key flavor-producing Lactobacillus spp. by quantitative profiling with indigenous internal standards. Appl. Environ. Microbiol. 2020, 86. [Google Scholar] [CrossRef]
- Yuan, S.Q.; Jin, Z.Y.; Ali, A.; Wang, C.J.; Liu, J. Caproic acid-producing bacteria in Chinese Baijiu brewing. Front. Microbiol. 2022, 13, 883142. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Microorganism | Strain | Number |
|---|---|---|
| Fungi | Aspergillus niger | CGMCC 3.4309 |
| Monascus purpureus | YJX-8 | |
| Yeast | Saccharomyces cerevisiae | 21-9 |
| Saccharomyces cerevisiae | 22-1 | |
| Saccharomyces cerevisiae | 22-2 | |
| Bacteria | Burkholderia sp. | BJQ0010 |
| Ligilactobacillus acidipiscis | JN-10-1-2 | |
| Clostridium beijerinckii | BCCB2-7-1 | |
| Clostridium tyrobutyricum | Gm-2-1 | |
| Clostridium butyricum | JG-2-1 | |
| Caproiciproducens sp. | BCJD-1 | |
| Clostridium kluyveri | BJN0002 |
| Sample Number | The Date of Samples | Sample Number for Sequencing in Triplicate |
|---|---|---|
| 0_1 | The 0 day of fermented grains of sample 1 | 1_0_1, 1_0_2, 1_0_3 |
| 0_2 | The 0 day of fermented grains of sample 2 | 2_0_1, 2_0_2, 2_0_3 |
| 0_3 | The 0 day of fermented grains of sample 3 | 3_0_1, 3_0_2, 3_0_3 |
| 0_4 | The 0 day of fermented grains of sample 4 | 4_0_1, 4_0_2, 4_0_3 |
| 14_1 | The 14th day of fermented grains of sample 1 | 1_14_1, 1_14_2, 1_14_3 |
| 14_2 | The 14th day of fermented grains of sample 2 | 2_14_1, 2_14_2, 2_14_3 |
| 14_3 | The 14th day of fermented grains of sample 3 | 3_14_1, 3_14_2, 3_14_3 |
| 14_4 | The 14th day of fermented grains of sample 4 | 4_14_1, 4_14_2, 4_14_3 |
| 28_1 | The 28th day of fermented grains of sample 1 | 1_28_1, 1_28_2, 1_28_3 |
| 28_2 | The 28th day of fermented grains of sample 2 | 2_28_1, 2_28_2, 2_28_3 |
| 28_3 | The 28th day of fermented grains of sample 3 | 3_28_1, 3_28_2, 3_28_3 |
| 28_4 | The 28th day of fermented grains of sample 4 | 4_28_1, 4_28_2, 4_28_3 |
| 42_1 | The 42nd day of fermented grains of sample 1 | 1_42_1, 1_42_2, 1_42_3 |
| 42_2 | The 42nd day of fermented grains of sample 2 | 2_42_1, 2_42_2, 2_42_3 |
| 42_3 | The 42nd day of fermented grains of sample 3 | 3_42_1, 3_42_2, 3_42_3 |
| 42_4 | The 42nd day of fermented grains of sample 4 | 4_42_1, 4_42_2, 4_42_3 |
| Number | Flavor Compounds | Number | Flavor Compounds | Number | Flavor Compounds |
|---|---|---|---|---|---|
| a1 | (Z)-Ethyl heptadec-9-enoate | a17 | Heptadecanoic acid, 15-methyl-, ethyl ester | b3 | 3-Methyl-1-butanol |
| a2 | (Z)-Ethyl pentadec-9-enoate | a18 | Decanoic acid, ethyl ester | b4 | 1-Butanol |
| a3 | 3-Phenylpropionic acid ethyl ester | a19 | Hexanoic acid, butyl ester | b5 | 1-Hexanol |
| a4 | 9,12,15-Octadecatrienoic acid, ethyl ester | a20 | Hexanoic acid, ethyl ester | c1 | 2,4-Di-tert-butylphenol |
| a5 | 9,12-Octadecadienoate, butyl ester | a21 | Lactic acid, ethyl ester | c2 | 2-Methoxy-4-methylphenol |
| a6 | 9,12-Octadecadienoate, ethyl ester | a22 | Heptadecanoic acid, ethyl ester | c3 | 2-Methoxy-4-vinylphenol |
| a7 | 9,12-Octadecadienoate, propyl ester | a23 | Tetradecanoic acid, 13-methyl-, ethyl ester | c4 | 4-Ethylphenol |
| a8 | 9.cis.,11. trans.-Octadecadienoic acid, ethyl ester | a24 | Tetradecanoic acid, ethyl ester | d1 | Eicosane |
| a9 | 9-Hexadecenoic acid, ethyl ester | a25 | Pentadecanoic acid, ethyl ester | d2 | Hexadecane |
| a10 | gamma-Nonanolactone | a26 | Octanoic acid, ethyl ester | d3 | Pentylcyclopropane |
| a11 | Benzoic acid, ethyl ester | a27 | Acetic acid 2-phenylethyl ester | e1 | Butanoic acid |
| a12 | Butanedioic acid, diethyl ester | a28 | Acetic acid ethyl ester | e2 | Isobutyric acid |
| a13 | Butanoic acid, butyl ester | a29 | Dodecanoic acid, ethyl ester | f1 | 2-Heptanone |
| a14 | Butanoic acid, ethyl ester | a30 | Hexadecanoic acid, ethyl ester | f2 | 6,10,14-Trimethyl-2-pentadecanone |
| a15 | Butanoic acid, 3-methylbutyl ester | b1 | Phenylethyl alcohol | g1 | 2,3-Dihydrobenzofuran |
| a16 | 9-Octadecenoic acid, ethyl ester | b2 | Isobutyl alcohol | h1 | Benzaldehyde |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, Y.; Wu, M.; Zhao, D.; Zheng, J.; Dai, M.; Li, X.; Li, W.; Zhang, C.; Sun, B. Simulated Fermentation of Strong-Flavor Baijiu through Functional Microbial Combination to Realize the Stable Synthesis of Important Flavor Chemicals. Foods 2023, 12, 644. https://doi.org/10.3390/foods12030644
Xu Y, Wu M, Zhao D, Zheng J, Dai M, Li X, Li W, Zhang C, Sun B. Simulated Fermentation of Strong-Flavor Baijiu through Functional Microbial Combination to Realize the Stable Synthesis of Important Flavor Chemicals. Foods. 2023; 12(3):644. https://doi.org/10.3390/foods12030644
Chicago/Turabian StyleXu, Youqiang, Mengqin Wu, Dong Zhao, Jia Zheng, Mengqi Dai, Xiuting Li, Weiwei Li, Chengnan Zhang, and Baoguo Sun. 2023. "Simulated Fermentation of Strong-Flavor Baijiu through Functional Microbial Combination to Realize the Stable Synthesis of Important Flavor Chemicals" Foods 12, no. 3: 644. https://doi.org/10.3390/foods12030644