

Improving the Thermostability of Serine Protease PB92 from Bacillus alcalophilus via Site-Directed Mutagenesis Based on Semi-Rational Design
Abstract
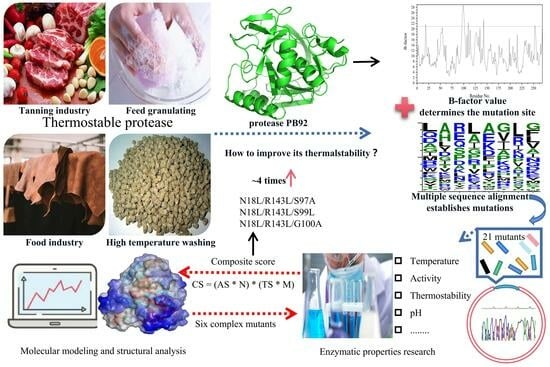
:
1. Introduction
2. Results
2.1. Prediction of Mutagenesis Sites Based on B-Factor Analysis
2.2. Construction and Characterization of Mutant Protease PB92
2.3. Identification of Composite Mutants
2.4. Characterization and Properties of Composite Mutants
2.5. Structural Analysis of Improved Thermostability for Mutants
3. Discussion
4. Materials and Methods
4.1. Strains and Chemicals
4.2. Selection of the Target Residues for Improving the Thermostability of Protease PB92
4.3. Site-Directed Mutagenesis Strategy
4.4. Expression, Purification, and Activity Assay of Protease PB92
4.5. Enzymatic Characterization of Protease PB92
4.6. Identification and Implementation of Complex Mutants
4.7. Molecular Modeling and Structural Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Pessela, B.C.; Okamoto, D.N.; Cabral, H. Editorial: Microbial proteases: Biochemical studies, immobilization and biotechnological application. Front. Microbiol. 2023, 14, 1126989. [Google Scholar] [CrossRef] [PubMed]
- Matkawala, F.; Nighojkar, S.; Kumar, A.; Nighojkar, A. Microbial alkaline serine proteases: Production, properties and applications. World J. Microbiol. Biotechnol. 2021, 37, 63. [Google Scholar] [CrossRef] [PubMed]
- Siezen, R.J.; de Vos, W.M.; Leunissen, J.A.; Dijkstra, B.W. Homology modelling and protein engineering strategy of subtilases, the family of subtilisin-like serine proteinases. Protein Eng. 1991, 4, 719–737. [Google Scholar] [CrossRef]
- van der Laan, J.M.; Teplyakov, A.V.; Kelders, H.; Kalk, K.H.; Misset, O.; Mulleners, L.J.; Dijkstra, B.W. Crystal structure of the high-alkaline serine protease PB92 from Bacillus alcalophilus. Protein Eng. 1992, 5, 401–411. [Google Scholar] [CrossRef]
- Martin, J.R.; Mulder, F.A.; Karimi-Nejad, Y.; van der Zwan, J.; Mariani, M.; Schipper, D.; Boelens, R. The solution structure of serine protease PB92 from Bacillus alcalophilus presents a rigid fold with a flexible substrate-binding site. Structure 1997, 5, 521–532. [Google Scholar] [CrossRef] [PubMed]
- Keshapaga, U.R.; Jathoth, K.; Singh, S.S.; Gogada, R.; Burgula, S. Characterization of high-yield Bacillus subtilis cysteine protease for diverse industrial applications. Braz. J. Microbiol. 2023, 54, 739–752. [Google Scholar] [CrossRef]
- Li, Q.; Yi, Y.; Marek, P.; Iverson, B.L. Commercial proteases: Present and future. FEBS Lett. 2023, 587, 1155–1163. [Google Scholar] [CrossRef] [PubMed]
- Barzkar, N.; Homaei, A.; Hemmati, R.; Patel, S. Thermostable marine microbial proteases for industrial applications: Scopes and risks. Extremophiles 2018, 22, 335–346. [Google Scholar] [CrossRef]
- Zhang, Y.; He, S.D.; Simpson, B.K. Enzymes in food bioprocessing-novel food enzymes, applications, and related techniques. Curr. Opin. Food Sci. 2018, 19, 30–35. [Google Scholar] [CrossRef]
- Frappier, V.; Najmanovich, R. Vibrational entropy differences between mesophile and thermophile proteins and their use in protein engineering. Protein Sci. 2015, 24, 474–483. [Google Scholar] [CrossRef]
- Goldenzweig, A.; Goldsmith, M.; Hill, A.E.; Gertman, O.; Laurino, P.; Ashani, Y.; Dym, O.; Unger, T.; Albeck, S.; Prilusky, J.; et al. Automated structure- and sequence-based design of proteins for high bacterial expression and stability. Mol. Cell 2016, 63, 337–346. [Google Scholar] [CrossRef]
- Nisthal, A.; Wang, C.Y.; Ary, M.L.; Mayo, S.L. Protein stability engineering insights revealed by domain-wide comprehensive mutagenesis. Proc. Natl. Acad. Sci. USA 2019, 116, 16367–16377. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Huang, X.Q.; Li, Q.; Zhu, Y.S. Computational design of variants for cephalosporin C acylase from Pseudomonas strain N176 with improved stability and activity. Appl. Microbiol. Biotechnol. 2017, 101, 621–632. [Google Scholar] [CrossRef] [PubMed]
- Hettiaratchi, M.H.; O’Meara, M.J.; O’Meara, T.R.; Pickering, A.J.; Letko-Khait, N.; Shoichet, M.S. Reengineering biocatalysts: Computational redesign of chondroitinase ABC improves efficacy and stability. Sci. Adv. 2020, 6, eabc6378. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.Y.; Zhang, J.; Li, C.; Wang, T.F.; Qin, H.M. Biochemical and structural characterization of a novel thermophilic and acidophilic β-mannanase from Aspergillus calidoustus. Enzym. Microb. Technol. 2021, 150, 109891. [Google Scholar] [CrossRef] [PubMed]
- Nezhad, N.G.; Rahman, R.N.Z.R.A.R.; Normi, Y.M.; Oslan, S.N.O.; Shariff, F.M.; Leow, T.C. Recent advances in simultaneous thermostability-activity improvement of industrial enzymes through structure modification. Int. J. Biol. Macromol. 2023, 232, 123440. [Google Scholar] [CrossRef]
- Zhao, J.J.; Yan, W.Y.; Yang, Y. DeepTP: A deep learning model for thermophilic protein prediction. Int. J. Mol. Sci. 2023, 24, 2217. [Google Scholar] [CrossRef]
- Bhatia, S.K.; Vivek, N.; Kumar, V.; Chandel, N.; Thakur, M.; Kumar, D.; Yang, Y.H.; Pugazendhi, A.; Kumar, G. Molecular biology interventions for activity improvement and production of industrial enzymes. Bioresource Technol. 2021, 324, 124596. [Google Scholar] [CrossRef]
- Gao, L.; Guo, Q.; Xu, R.N.; Dong, H.F.; Zhou, C.C.; Yu, Z.H.; Zhang, Z.K.; Wang, L.X.; Chen, X.Y.; Wu, X. Loop optimization of Trichoderma reesei endoglucanases for balancing the activity–stability trade-off through cross-strategy between machine learning and the B-factor analysis. GCB Bioenergy 2023, 15, 128–142. [Google Scholar] [CrossRef]
- Szilágyi, A.; Závodszky, P. Structural differences between mesophilic, moderately thermophilic and extremely thermophilic protein subunits: Results of a comprehensive survey. Structure 2000, 8, 493–504. [Google Scholar] [CrossRef]
- Han, Z.L.; Han, S.Y.; Zheng, S.P.; Lin, Y. Enhancing thermostability of a Rhizomucor miehei lipase by engineering a disulfide bond and displaying on the yeast cell surface. Appl. Microbiol. Biotechnol. 2009, 85, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Wang, X.Z.; Deng, R.Q.; Wang, J.W.; Zhou, H.B. Discrimination of thermostable and thermophilic lipases using support vector machines. Protein Pept. Lett. 2011, 18, 707–717. [Google Scholar] [CrossRef]
- Zhu, F.C.; Zhuang, Y.; Wu, B.; Li, J.H.; He, B.F. Rational substitution of surface acidic residues for enhancing the thermostability of thermolysin. Appl. Biochem. Biotechnol. 2016, 178, 725–738. [Google Scholar] [CrossRef] [PubMed]
- Miao, H.B.; Zhe, Y.Y.; Xiang, X.; Cao, Y.; Han, N.Y.; Wu, Q.; Huang, Z.X. Enhanced extracellular expression of a Ca2+- and Mg2+-dependent hyperthermostable protease EA1 in Bacillus subtilis via systematic screening of optimal signal peptides. J. Agric. Food Chem. 2022, 70, 15830–15839. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.T.; Liu, Q.; Qu, G.; Feng, Y.; Reetz, M.T. Utility of B-factors in protein science: Interpreting rigidity, flexibility, and internal motion and engineering thermostability. Chem. Rev. 2019, 119, 1626–1665. [Google Scholar] [CrossRef]
- Jiang, Z.B.; Zhang, C.B.; Tang, M.Y.; Xu, B.; Wang, L.L.; Qian, W.; He, J.D.; Zhao, Z.H.; Wu, Q.; Mu, Y.L.; et al. Improving the thermostability of Rhizopus chinensis lipase through site-directed mutagenesis based on B-factor analysis. Front. Microbiol. 2020, 11, 346. [Google Scholar] [CrossRef]
- Zong, Z.Y.; Gao, L.; Cai, W.S.; Yu, L.; Cui, C.; Chen, S.L.; Zhang, D.Y. Computer-assisted rational modifications to improve the thermostability of β-Glucosidase from Penicillium piceum H16. Bioenergy Res. 2015, 8, 1384–1390. [Google Scholar] [CrossRef]
- Pace, C.N.; Fu, H.L.; Fryar, K.L.; Landua, J.; Trevino, S.T.; Shirley, B.A.; Hendricks, M.M.; Iimura, S.; Gajiwala, K.; Scholtz, J.M.; et al. Contribution of hydrophobic interactions to protein stability. J. Mol. Biol. 2011, 408, 514–528. [Google Scholar] [CrossRef]
- Klaewkla, M.; Pichyangkura, R.; Charoenwongpaiboon, T.; Wangpaiboon, K.; Chunsrivirot, S. Computational design of oligosaccharide producing levansucrase from Bacillus licheniformis RN-01 to improve its thermostability for production of levan-type fructooligosaccharides from sucrose. Int. J. Biol. Macromol. 2020, 160, 252–263. [Google Scholar] [CrossRef]
- Xu, Z.; Cen, Y.K.; Zou, S.P.; Xue, Y.P.; Zheng, Y.G. Recent advances in the improvement of enzyme thermostability by structure modification. Crit. Rev. Biotechnol. 2020, 40, 83–98. [Google Scholar] [CrossRef]
- Chi, H.B.; Wang, Y.L.; Xia, B.J.; Zhou, Y.W.; Lu, Z.X.; Lu, F.X.; Zhu, P. Enhanced thermostability and molecular insights for L-asparaginase from Bacillus licheniformis via structure- and computation-based rational design. J. Agric. Food Chem. 2022, 70, 14499–14509. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.B.; Qiao, C.C.; Li, H.B.; Li, L.J.; Xiao, A.F.; Ni, H.; Jiang, Z.D. Improvement thermostability of Pseudoalteromonas carrageenovora arylsulfatase by rational design. Int. J. Biol. Macromol. 2018, 108, 953–959. [Google Scholar] [CrossRef] [PubMed]
- Long, S.Q.; Zhang, X.; Rao, Z.M.; Chen, K.Y.; Xu, M.J.; Yang, T.W.; Yang, S.T. Amino acid residues adjacent to the catalytic cavity of tetramer L-asparaginase II contribute significantly to its catalytic efficiency and thermostability. Enzym. Microb. Technol. 2016, 82, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Cheng, M.; Huang, Z.L.; Zhang, W.L.; Kim, B.G.; Mu, W.M. Thermostability engineering of an inulin fructotransferase for the biosynthesis of difructose anhydride I. Enzym. Microb. Technol. 2022, 160, 110097. [Google Scholar] [CrossRef]
- Su, B.M.; Xu, X.Q.; Yan, R.X.; Xie, Y.; Lin, J. Mutagenesis on the surface of a β-agarase from Vibrio sp. ZC-1 increased its thermo-stability. Enzym. Microb. Technol. 2019, 12, 22–31. [Google Scholar] [CrossRef]
- Miller, S.R. An appraisal of the enzyme stability-activity trade-off. Evolution 2017, 71, 1876–1887. [Google Scholar] [CrossRef]
- Ban, X.F.; Liu, Y.; Zhang, Y.Z.; Gu, Z.B.; Li, C.M.; Cheng, L.; Hong, Y.; Dhoble, A.S.; Li, Z.F. Thermostabilization of a thermophilic 1,4-alpha-glucan branching enzyme through C-terminal truncation. Int. J. Biol. Macromol. 2018, 107, 1510–1518. [Google Scholar] [CrossRef]
- Li, J.L.; Jiang, L.Y.; Cao, X.; Wu, Y.F.; Lu, F.P.; Liu, F.F.; Li, Y.; Liu, Y.H. Improving the activity and stability of Bacillus clausii alkaline protease using directed evolution and molecular dynamics simulation. Enzym. Microb. Technol. 2021, 147, 109787. [Google Scholar] [CrossRef]
- Miao, H.B.; Jiang, R.; Han, N.Y.; Ma, Y.; Wu, Q.; Mu, Y.L.; Huang, Z.X. Enhanced extracellular expression of α-amylase DL3-4-1 in Bacillus subtilis via systematic screening of optimal signal peptides. Process Biochem. 2021, 108, 176–184. [Google Scholar] [CrossRef]
- Cherqui, A.; Pereira, A.; Simões, N. Purification and characterization of two distinct metalloproteases secreted by the entomopathogenic bacterium Photorhabdus sp. strain Az29. Appl. Environ. Microbiol. 2004, 70, 3831–3838. [Google Scholar] [CrossRef]
- Li, Y.Q.; Middaugh, C.R.; Fang, J.W. A novel scoring function for discriminating hyperthermophilic and mesophilic proteins with application to predicting relative thermostability of protein mutants. BMC Bioinform. 2010, 11, 62. [Google Scholar] [CrossRef]
- Eisenhaber, F.; Lijnzaad, P.; Argos, P.; Sander, C.; Scharf, M. The double cubic lattice method: Efficient approaches to numerical integration of surface area and volume and to dot surface contouring of molecular assemblies. J. Comput. Chem. 1995, 16, 273–284. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protease | Relative Activity (%) b | Topt (°C) c | Protease | Relative Activity (%) b | Topt (°C) c |
|---|---|---|---|---|---|
| Wild type d | 100 | 55 | G100E | 7.88 | 60 |
| N18L | 72.30 | 60 | G100A | 93.82 | 55 |
| N18Q | 0.23 | ND | S101I | 3.26 | ND |
| S97A | 90.89 | 55 | S101G | 90.29 | 55 |
| S97H | 98.52 | 55 | S101V | 12.69 | 65 |
| G98R | 19.95 | 55 | E110L | 31.28 | 60 |
| G98E | 6.19 | 55 | E110R | 0.12 | ND |
| G98Q | 3.45 | ND | R143F | 0.02 | ND |
| S99L | 96.66 | 60 | R143L | 75.73 | 60 |
| S99A | 111.01 | 60 | R143G | 98.24 | 60 |
| G100K | 0.15 | ND | R143V | 78.53 | 55 |
| Protease | t1/2 (min) a | Increase Times | Protease | t1/2 (min) a | Increase Times |
|---|---|---|---|---|---|
| Wild type b | 23.14 | 1.00 | G100E | 548.80 | 23.72 |
| N18L | 46.63 | 2.02 | G100A | 47.92 | 2.07 |
| S97A | 41.25 | 1.78 | S101G | 28.18 | 1.22 |
| S97H | 26.16 | 1.13 | S101V | 190.44 | 8.23 |
| G98R | 230.71 | 9.97 | E110L | 36.94 | 1.60 |
| G98E | 731.39 | 31.61 | R143V | 49.81 | 2.15 |
| S99L | 38.37 | 1.66 | R143L | 62.96 | 2.72 |
| S99A | 32.40 | 1.40 | R143G | 28.14 | 1.65 |
| Protease | Thermostability Score a | Activity Score b | Composite Score c | Protease | Thermostability Score a | Activity Score b | Composite Score c |
|---|---|---|---|---|---|---|---|
| Wild type d | 100 | 100 | 10,000.00 | G100E | 237.17 | 7.88 | 18,691.55 |
| N18L | 201.51 | 72.3 | 14,570.10 | G100A | 207.09 | 93.82 | 19,429.07 |
| S97A | 178.26 | 90.89 | 16,202.84 | S101G | 121.79 | 94.23 | 10,996.84 |
| S97H | 113.47 | 98.52 | 11,178.42 | S101V | 822.99 | 12.69 | 10,445.30 |
| G98R | 997.02 | 19.95 | 19,892.83 | E110L | 159.64 | 31.28 | 4993.94 |
| G98E | 316.07 | 6.19 | 19,552.51 | R143V | 215.25 | 78.53 | 16,903.36 |
| S99L | 165.82 | 96.66 | 16,027.04 | R143L | 272.07 | 75.73 | 20,604.41 |
| S99A | 140.02 | 111.01 | 15,543.38 | R143G | 164.83 | 98.24 | 16,192.72 |
| Protease | t1/2 (min) a | Increase Times | Composite Score b |
|---|---|---|---|
| Wild type c | 23.14 | 1.00 | 10,000 |
| M2 | 49.00 | 2.12 | 16,141.99 |
| M3-1 | 105.42 | 4.55 | 34,207.01 |
| M3-2 | 232.45 | 10.04 | 6692.22 |
| M3-3 | 90.87 | 3.93 | 29,739.52 |
| M3-4 | 91.42 | 3.95 | 30,752.94 |
| M6 | 224.62 | 9.71 | 3544.60 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miao, H.; Xiang, X.; Han, N.; Wu, Q.; Huang, Z. Improving the Thermostability of Serine Protease PB92 from Bacillus alcalophilus via Site-Directed Mutagenesis Based on Semi-Rational Design. Foods 2023, 12, 3081. https://doi.org/10.3390/foods12163081
Miao H, Xiang X, Han N, Wu Q, Huang Z. Improving the Thermostability of Serine Protease PB92 from Bacillus alcalophilus via Site-Directed Mutagenesis Based on Semi-Rational Design. Foods. 2023; 12(16):3081. https://doi.org/10.3390/foods12163081
Chicago/Turabian StyleMiao, Huabiao, Xia Xiang, Nanyu Han, Qian Wu, and Zunxi Huang. 2023. "Improving the Thermostability of Serine Protease PB92 from Bacillus alcalophilus via Site-Directed Mutagenesis Based on Semi-Rational Design" Foods 12, no. 16: 3081. https://doi.org/10.3390/foods12163081