
Comparative Genomics Analysis Provides New Insights into High Ethanol Tolerance of Lactiplantibacillus pentosus LTJ12, a Novel Strain Isolated from Chinese Baijiu
 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Bacterial Strains
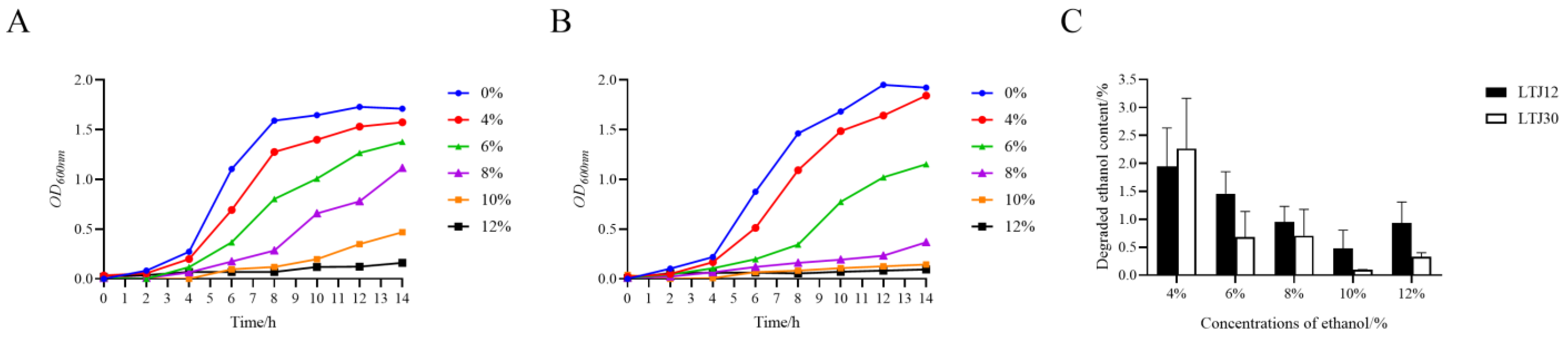
2.2. Determination of Ethanol Degradation Ability and the Effect of Ethanol on the Growth of Strains
2.3. Extraction of Genomic DNA of L. pentosus LTJ12
2.4. Library Construction and Sequencing
2.5. Genome Assembly, Gene Prediction and Annotation
2.6. Accession Number
2.7. Comparative Genomics Analysis
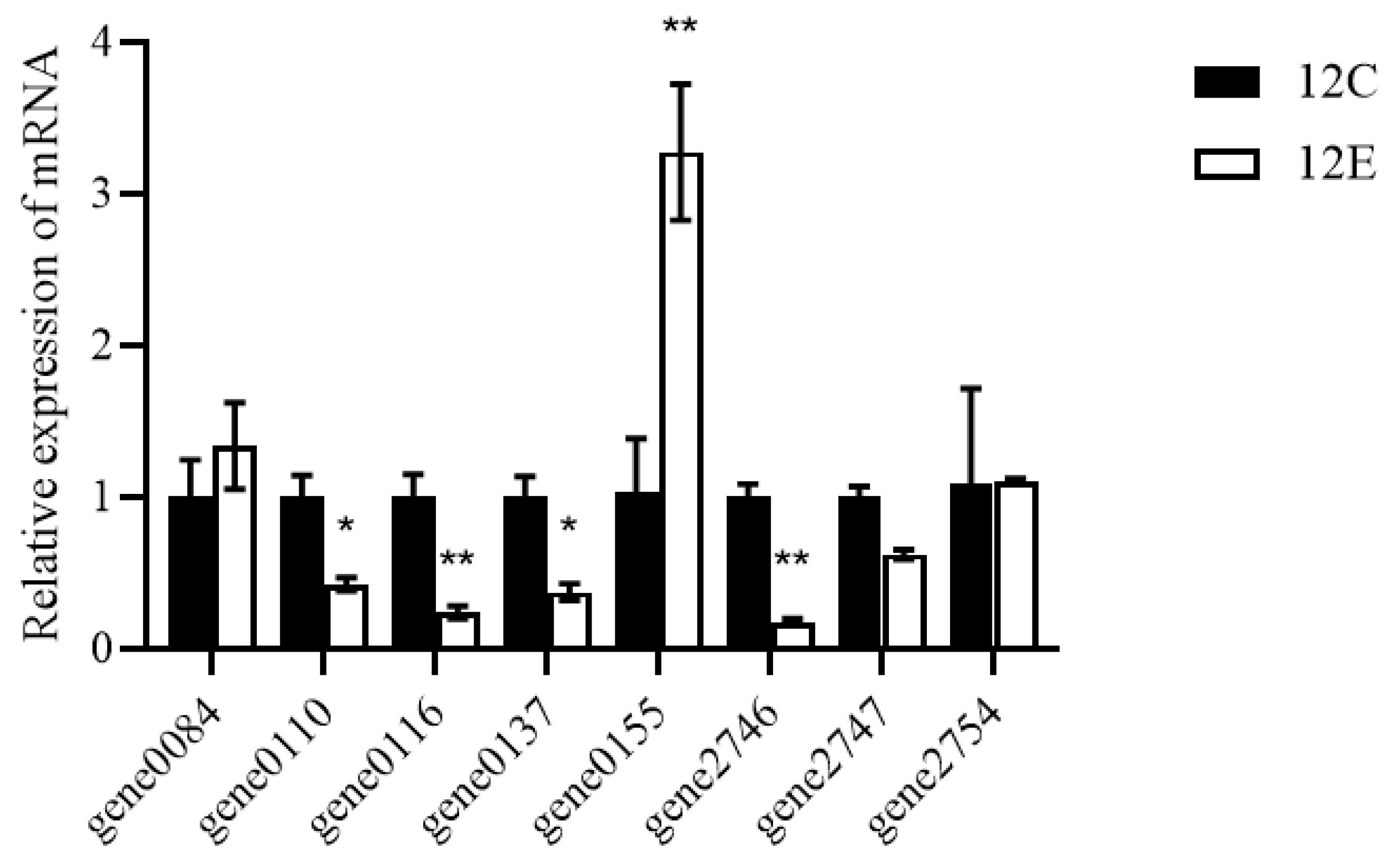
2.8. Quantitative Real-Time PCR
3. Results and Discussion
3.1. LTJ12 Has the Ability to Resist and Degrade Ethanol
3.2. Reclassification of LTJ12 via Genomic Analysis
3.3. Genome Sequencing and Assembly of L. pentosus LTJ12
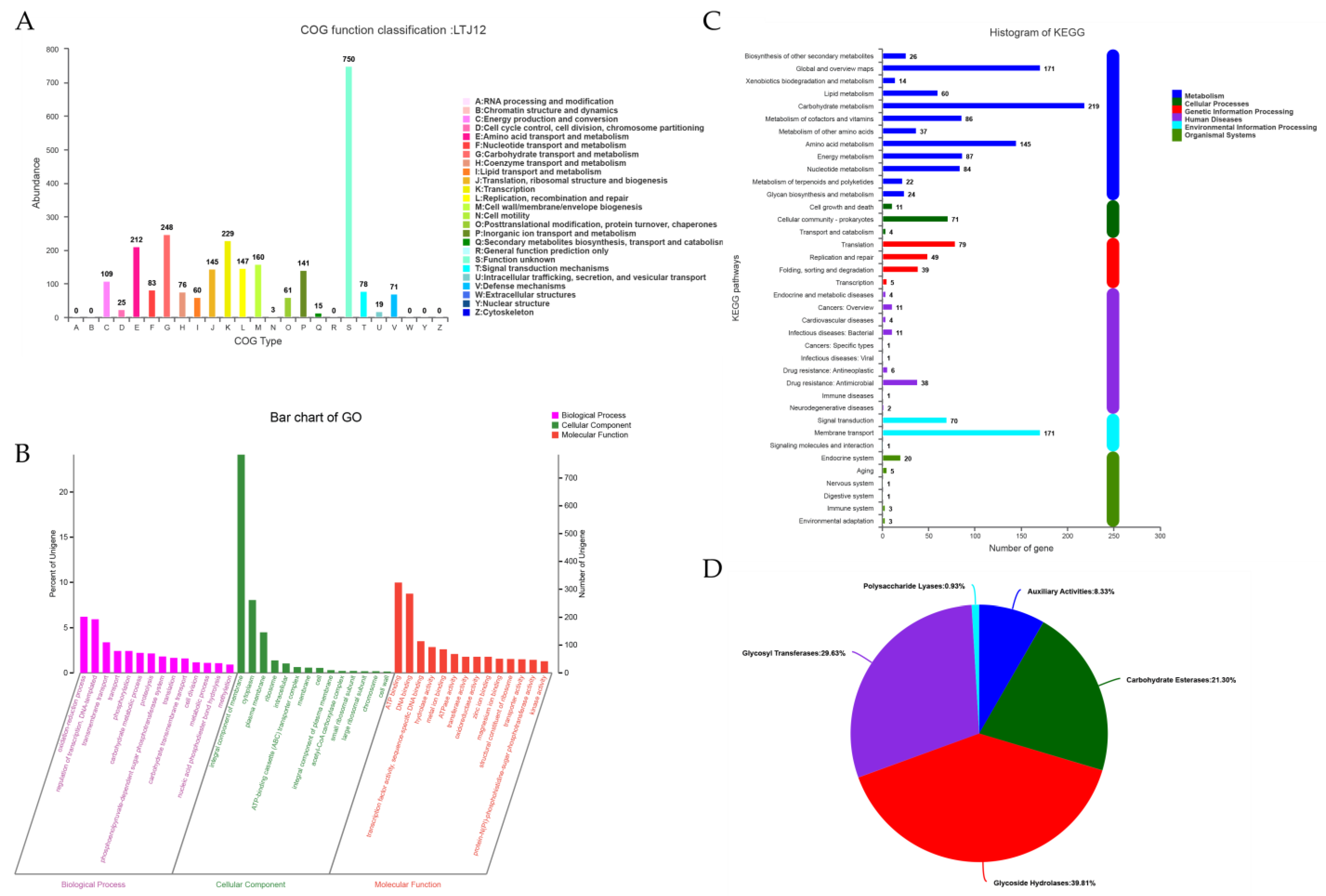
3.4. Functional Annotation of L. pentosus LTJ12
3.5. Carbohydrate-active Enzyme (CAZyme) Annotation
3.6. Alcohol-Related Genes of LTJ12 May Be Involved in the Degradation of Alcohol
3.7. LTJ12 Has Potential Probiotic Properties
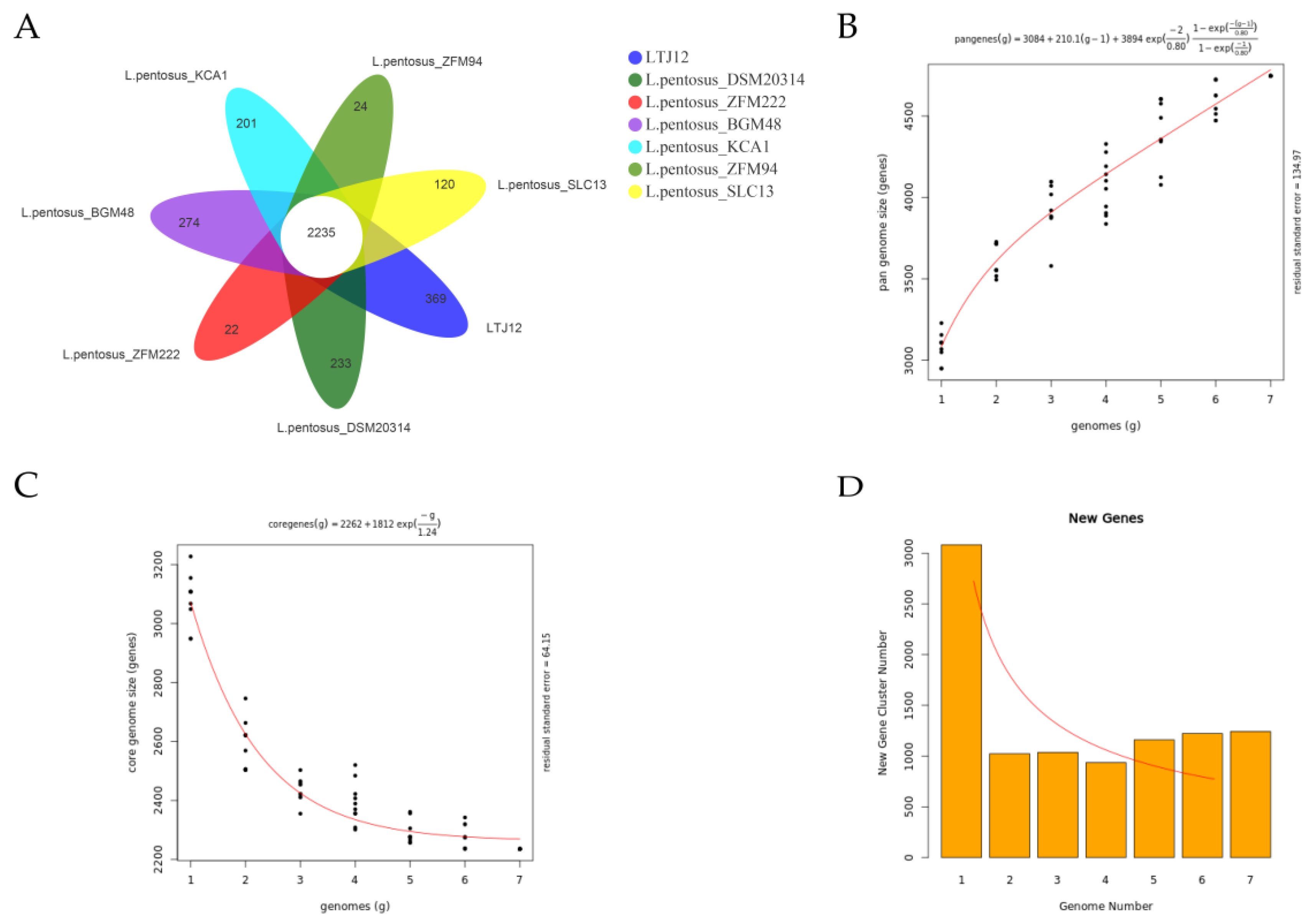
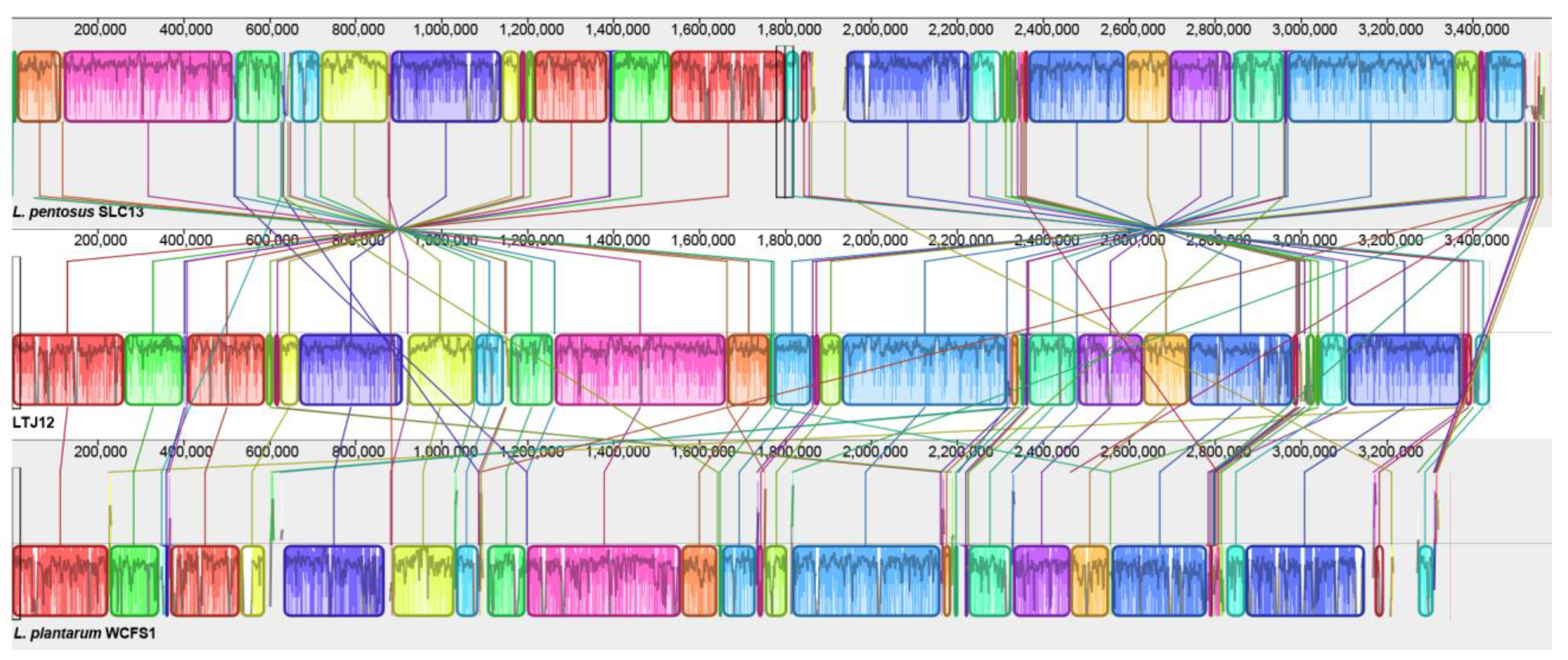
3.8. Comparative Genomic Analysis Suggests That LTJ12 Has More Functional Genes
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pradhan, P.; Tamang, J. Probiotic properties of lactic acid bacteria isolated from traditionally prepared dry starters of the Eastern Himalayas. World J. Microbiol. Biotechnol. 2021, 37, 7. [Google Scholar] [CrossRef] [PubMed]
- Liong, M.T. Safety of probiotics: Translocation and infection. Nutr. Rev. 2008, 66, 192–202. [Google Scholar] [CrossRef] [PubMed]
- Duar, R.; Lin, X.; Zheng, J.; Martino, M.; Grenier, T.; Pérez-Muñoz, M.; Leulier, F.; Gänzle, M.; Walter, J. Lifestyles in transition: Evolution and natural history of the genus Lactobacillus. FEMS Microbiol. Rev. 2017, 41, S27–S48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, J.; Wittouck, S.; Salvetti, E.; Franz, C.; Harris, H.; Mattarelli, P.; O’Toole, P.; Pot, B.; Vandamme, P.; Walter, J.; et al. A taxonomic note on the genus Lactobacillus: Description of 23 novel genera, emended description of the genus Lactobacillus Beijerinck 1901, and union of Lactobacillaceae and Leuconostocaceae. Int. J. Syst. Evol. Microbiol. 2020, 70, 2782–2858. [Google Scholar] [CrossRef]
- Gupta, J.A.; Thapa, S.; Verma, M.; Som, R.; Mukherjee, K.J. Genomics and transcriptomics analysis reveals the mechanism of isobutanol tolerance of a laboratory evolved Lactococcus lactis strain. Sci. Rep. 2020, 10, 10850. [Google Scholar] [CrossRef]
- Chen, X.; Wang, T.; Jin, M.; Tan, Y.; Liu, L.; Liu, L.; Li, C.; Yang, Y.; Du, P. Metabolomics analysis of growth inhibition of Lactobacillus plantarum under ethanol stress. Int. J. Food Sci. Tech. 2020, 55, 3441–3454. [Google Scholar] [CrossRef]
- Schmid, M.; Muri, J.; Melidis, D.; Varadarajan, A.R.; Somerville, V.; Wicki, A.; Moser, A.; Bourqui, M.; Wenzel, C.; Eugster-Meier, E. Comparative genomics of completely sequenced Lactobacillus helveticus genomes provides insights into strain-specific genes and resolves metagenomics data down to the strain level. Front. Microbiol. 2018, 9, 63. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.J.; Wang, R.; Gong, F.M.; Liu, X.F.; Zheng, H.J.; Luo, Y.Y.; Li, X.R. Complete genome sequences and comparative genome analysis of Lactobacillus plantarum strain 5-2 isolated from fermented soybean. Genomics 2015, 106, 404–411. [Google Scholar] [CrossRef]
- Shi, Y.; Zhao, J.; Kellingray, L.; Zhang, H.; Narbad, A.; Zhai, Q.; Chen, W. In vitro and in vivo evaluation of Lactobacillus strains and comparative genomic analysis of Lactobacillus plantarum CGMCC12436 reveal candidates of colonise-related genes. Food Res. Int. 2019, 119, 813–821. [Google Scholar] [CrossRef]
- Pérez-Díaz, I.; Johanningsmeier, S.D.; Anekella, K.; Pagán-Medina, C.; Arroyo-López, F. Genotypic and phenotypic diversity among Lactobacillus plantarum and Lactobacillus pentosus isolated from industrial scale cucumber fermentations. Food Microbiol. 2021, 94, 103652. [Google Scholar] [CrossRef]
- Ye, K.; Li, P.; Gu, Q. Complete genome sequence analysis of a strain Lactobacillus pentosus ZFM94 and its probiotic characteristics. Genomics 2020, 112, 3142–3149. [Google Scholar] [CrossRef]
- Stergiou, O.; Tegopoulos, K.; Kiousi, D.; Tsifintaris, M.; Papageorgiou, A.; Tassou, C.; Chorianopoulos, N.; Kolovos, P.; Galanis, A. Whole-genome sequencing, phylogenetic and genomic analysis of Lactiplantibacillus pentosus L33, a potential probiotic strain isolated from fermented sausages. Front. Microbiol. 2021, 12, 746659. [Google Scholar] [CrossRef]
- Spano, G.; Massa, S. Environmental stress response in wine lactic acid bacteria: Beyond Bacillus subtilis. Crit. Rev. Microbiol. 2006, 32, 77–86. [Google Scholar] [CrossRef]
- Eva, G.A.; Isabel, L.; Ignacio, R.J.; Julio, S.; Eva, F.E.; Myriam, Z.; Marta, D.; Carmen, T.; Fe Rnanda, R.L. High tolerance of wild Lactobacillus plantarum and Oenococcus oeni strains to lyophilisation and stress environmental conditions of acid pH and ethanol. FEMS Microbiol. Lett. 2004, 230, 53–61. [Google Scholar]
- Wang, J.; Lu, C.; Xu, Q.; Li, Z.; Song, Y.; Zhou, S.; Zhang, T.; Luo, X. Bacterial diversity and lactic acid bacteria with high alcohol tolerance in the fermented grains of soy sauce aroma type baijiu in North China. Foods 2022, 11, 1794. [Google Scholar] [CrossRef]
- Koren, S.; Walenz, B.P.; Berlin, K.; Miller, J.R.; Bergman, N.H.; Phillippy, A.M. Canu: Scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017, 27, 722–736. [Google Scholar] [CrossRef] [Green Version]
- Delcher, A.L.; Bratke, K.A.; Powers, E.C.; Salzberg, S.L. Identifying bacterial genes and endosymbiont DNA with Glimmer. Bioinformatics 2007, 23, 673–679. [Google Scholar] [CrossRef] [Green Version]
- Borodovsky, M.; McIninch, J. GENMARK: Parallel gene recognition for both DNA strands. Comput. Chem. 1993, 17, 123–133. [Google Scholar] [CrossRef]
- Chen, Y.; Li, N.; Zhao, S.; Zhang, C.; Qiao, N.; Duan, H.; Xiao, Y.; Yan, B.; Zhao, J.; Tian, F. Integrated phenotypic-genotypic analysis of Latilactobacillus sakei from different niches. Foods 2021, 10, 1717. [Google Scholar] [CrossRef]
- Cosentino, S.; Voldby Larsen, M.; Møller Aarestrup, F.; Lund, O. PathogenFinder--distinguishing friend from foe using bacterial whole genome sequence data. PLoS ONE 2013, 8, e77302. [Google Scholar] [CrossRef]
- Ren, Y.; Yu, G.; Shi, C.; Liu, L.; Guo, Q.; Han, C.; Zhang, D.; Zhang, L.; Liu, B.; Gao, H.; et al. Majorbio Cloud: A one-stop, comprehensive bioinformatic platform for multiomics analyses. iMeta 2022, 1, e12. [Google Scholar] [CrossRef]
- Valeriano, V.; Oh, J.; Bagon, B.; Kim, H.; Kang, D. Comparative genomic analysis of Lactobacillus mucosae LM1 identifies potential niche-specific genes and pathways for gastrointestinal adaptation. Genomics 2019, 111, 24–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Binda, S.; Hill, C.; Johansen, E.; Obis, D.; Ouwehand, A.C. Criteria to qualify microorganisms as "probiotic" in foods and dietary supplements. Front. Microbiol. 2020, 11, 1662. [Google Scholar] [CrossRef] [PubMed]
- Heo, S.; Kim, J.H.; Kwak, M.S.; Jeong, D.W.; Sung, M.H. Functional genomic insights into probiotic Bacillus siamensis strain B28 from traditional Korean fermented kimchi. Foods 2021, 10, 1906. [Google Scholar] [CrossRef] [PubMed]
- Shanahan, F. A commentary on the safety of probiotics. Gastroenterol. Clin. N. Am. 2012, 41, 869–876. [Google Scholar] [CrossRef]
- Ventura, M.; O’Flaherty, S.; Claesson, M.; Turroni, F.; Klaenhammer, T.; van Sinderen, D.; O’Toole, P. Genome-scale analyses of health-promoting bacteria: Probiogenomics. Nat. Rev. Microbiol. 2009, 7, 61–71. [Google Scholar] [CrossRef] [Green Version]
- Goel, A.; Halami, P.M.; Tamang, J.P. Genome analysis of Lactobacillus plantarum isolated from some Indian fermented foods for bacteriocin production and probiotic marker genes. Front. Microbiol. 2020, 11, 40. [Google Scholar] [CrossRef]
- Chokesajjawatee, N.; Santiyanont, P.; Chantarasakha, K.; Kocharin, K.; Visessanguan, W. Safety assessment of a Nham starter culture Lactobacillus plantarum BCC9546 via whole-genome analysis. Sci. Rep. 2020, 10, 10241. [Google Scholar] [CrossRef]
- Beck, B.R.; Park, G.S.; Lee, Y.H.; Im, S.; Jeong, D.Y.; Kang, J. Whole genome analysis of Lactobacillus plantarum strains isolated from Kimchi and determination of probiotic properties to treat mucosal infections by Candida albicans and Gardnerella vaginalis. Front. Microbiol. 2019, 10, 433. [Google Scholar] [CrossRef]
- Lee, S.M.; Donaldson, G.P.; Mikulski, Z.; Boyajian, S.; Ley, K.; Mazmanian, S.K. Bacterial colonization factors control specificity and stability of the gut microbiota. Nature 2013, 501, 426–429. [Google Scholar] [CrossRef] [Green Version]
- Yuan, J.; Wang, B.; Sun, Z.; Bo, X.; Yuan, X.; He, X.; Zhao, H.; Du, X.; Wang, F.; Jiang, Z. Analysis of host-inducing proteome changes in Bifidobacterium longum NCC2705 grown in Vivo. J. Proteome Res. 2008, 7, 375–385. [Google Scholar] [CrossRef]
- Candela, M.; Centanni, M.; Fiori, J.; Biagi, E.; Turroni, S.; Orrico, C.; Bergmann, S.; Hammerschmidt, S.; Brigidi, P. DnaK from Bifidobacterium animalis subsp. lactis is a surface-exposed human plasminogen receptor upregulated in response to bile salts. Microbiology 2010, 156, 1609–1618. [Google Scholar] [CrossRef] [Green Version]
- de Vries, M.C.; Vaughan, E.E.; Kleerebezem, M.; de Vos, W.M. Lactobacillus plantarum-survival, functional and potential probiotic properties in the human intestinal tract. Int. Dairy. J. 2006, 16, 1018–1028. [Google Scholar] [CrossRef]
- Aslim, B.; Onal, D.; Beyatli, Y. Factors influencing autoaggregation and aggregation of Lactobacillus delbrueckii subsp. bulgaricus isolated from handmade yogurt. J. Food Prot. 2007, 70, 223–227. [Google Scholar] [CrossRef]
- Castro-Bravo, N.; Wells, J.M.; Margolles, A.; Ruas-Madiedo, P. Interactions of surface exopolysaccharides from Bifidobacterium and Lactobacillus within the intestinal environment. Front. Microbiol. 2018, 9, 2426. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, P.T.; Nguyen, T.T.; Bui, D.C.; Hong, P.T.; City, M. Exopolysaccharide production by lactic acid bacteria: The manipulation of environmental stresses for industrial applications. AIMS Microbiol. 2020, 6, 451–469. [Google Scholar] [CrossRef]
- Ale, E.; Rojas, M.; Reinheimer, J.; Binetti, A. Lactobacillus fermentum: Could EPS production ability be responsible for functional properties? Food Microbiol. 2020, 90, 103465. [Google Scholar] [CrossRef]
- Medini, D.; Donati, C.; Tettelin, H.; Masignani, V.; Rappuoli, R. The microbial pan-genome. Curr. Opin. Genet. Dev. 2005, 15, 589–594. [Google Scholar] [CrossRef]
- Lei, L.; Ruiyun, W.; Jinlan, Z.; Nan, S.; Pinglan, L. D-Ribose interferes with quorum sensing to inhibit biofilm formation of Lactobacillus paraplantarum L-ZS9. Front. Microbiol. 2017, 8, 1860. [Google Scholar]
- Yin, X.; Salemi, M.R.; Phinney, B.S.; Gotcheva, V.; Angelov, A.; Marco, M.L. Proteomes of Lactobacillus delbrueckii subsp. bulgaricus LBB.B5 incubated in milk at optimal and low temperatures. mSystems 2017, 2, e00027-17. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.; Wang, J.; Li, J.; Liu, Y.; Liu, X.; Li, Z.; Kurni, K.; Deng, Y.; Wang, G.; Ralph, J.D. Prevalence of phase variable epigenetic invertons among host-associated bacteria. Nucleic Acids Res. 2020, 48, 11468–11485. [Google Scholar] [CrossRef] [PubMed]
- Abriouel, H.; Pérez Montoro, B.; Casado Muñoz, M.; Knapp, C.; Gálvez, A.; Benomar, N. In silico genomic insights into aspects of food safety and defense mechanisms of a potentially probiotic Lactobacillus pentosus MP-10 isolated from brines of naturally fermented Aloreña green table olives. PLoS ONE 2017, 12, e0176801. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.L.; Huang, J.Y.; Kao, C.Y.; Fang, T.J. Complete genome sequence of Lactobacillus pentosus SLC13, isolated from mustard pickles, a potential probiotic strain with antimicrobial activity against foodborne pathogenic microorganisms. Gut Pathog. 2018, 10, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Yang, L.; Nan, W.; Lu, J.; Zhang, S.; Ujiroghene, O.; Pang, X.; Lv, J. Whole-genome sequencing and genomic-based acid tolerance mechanisms of Lactobacillus delbrueckii subsp. bulgaricus LJJ. Appl. Microbiol. Biotechnol. 2020, 104, 7631–7642. [Google Scholar] [CrossRef]
- Sun, Z.; Harris, H.; McCann, A.; Guo, C.; Argimón, S.; Zhang, W.; Yang, X.; Jeffery, I.; Cooney, J.; Kagawa, T.; et al. Expanding the biotechnology potential of Lactobacilli through comparative genomics of 213 strains and associated genera. Nat. Commun. 2015, 6, 8322. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene ID | Gene Name | Forward Primer | Reveres Primer | Product Size |
|---|---|---|---|---|
| gene0084 | adh | TACCTTCGCTGAGTTTATC | CTGAACCACCAGGAATAA | 173 |
| gene0110 | - | ACGACGGTCTCACAACGC | AGGCCAAACGAGGCAAGT | 175 |
| gene0116 | - | ACCATCCGACCAGAAGAA | GAGGGTTGCGACTGTGAG | 113 |
| gene0137 | gldA | CACCAACCGCTGGCTTAT | GTCGCCTTCGCTTCAATA | 181 |
| gene0155 | - | GTGAGACTTTTAGCCGTTTG | TTCTTCCGACTGACATCC | 175 |
| gene2746 | dhaT | TATGACTAACGGTGACGAC | ACGAACTTGTAATGGGTG | 156 |
| gene2747 | - | GTGACAACTGTTTGAAAGGGAT | CACCGTATGAGTCGTGAATG | 155 |
| gene2754 | - | GTAATGATTCCAACGACAG | TTGACATCCAACAGAACTAA | 134 |
| - | 16S rRNA | AAGGGTTTCGGCTCGTAAAA | TGCACTCAAGTTTCCCAGTT | 247 |
| Features | Genome |
|---|---|
| Genome Size (bp) | 3,512,307 |
| GC Content (%) | 46.37 |
| Gene (CDS) NO. | 3248 |
| Gene Len (bp) | 2,835,894 |
| Gene Average Len (bp) | 873.12 |
| Gene Density | 0.92 |
| GC Content in Gene Region (%) | 47.59 |
| Gene/Genome (%) | 80.74 |
| Intergenetic Region Len (bp) | 676,413 |
| GC Content in Intergenetic Region (%) | 41.24 |
| Intergenetic Len/Genome (%) | 19.26 |
| tRNA NO. | 73 |
| Type of tRNAs NO. | 21 |
| rRNAs NO. | 16 |
| 16S rRNA | 5 |
| 23S rRNA | 5 |
| 5S rRNA | 6 |
| sRNAs NO. | 38 |
| NR annotation | 3247 |
| Swiss-Prot annotation | 2168 |
| Pfam annotation | 2535 |
| COG annotation | 2593 |
| GO annotation | 2437 |
| KEGG annotation | 1541 |
| Gene ID | Gene Name | Description | COG Description | GO Description | KO Description |
|---|---|---|---|---|---|
| gene0084 | adh | oxidoreductase | alcohol dehydrogenase | oxidation-reduction process; oxidoreductase activity; zinc ion binding | alcohol dehydrogenase [EC:1.1.1.1] |
| gene0110 | - | alcohol dehydrogenase | alcohol dehydrogenase | oxidation-reduction process; oxidoreductase activity; zinc ion binding | - |
| gene0116 | - | oxidoreductase | alcohol dehydrogenase | oxidation-reduction process; oxidoreductase activity; zinc ion binding | - |
| gene0137 | gldA | iron-containing alcohol dehydrogenase | Dehydrogenase | oxidation-reduction process; metal ion binding; glycerol dehydrogenase [NAD+] activity | glycerol dehydrogenase [EC:1.1.1.6] |
| gene0155 | - | alcohol dehydrogenase | - | - | - |
| gene2746 | dhaT | 1,3-propanediol dehydrogenase | alcohol dehydrogenase | oxidation-reduction process; metal ion binding; 1,3-propanediol dehydrogenase activity | 1,3-propanediol dehydrogenase [EC:1.1.1.202] |
| gene2747 | - | aryl-alcohol dehydrogenase | alcohol dehydrogenase | oxidation-reduction process; zinc ion binding; aryl-alcohol dehydrogenase (NAD+) activity | aryl-alcohol dehydrogenase [EC:1.1.1.90] |
| gene2754 | - | alcohol dehydrogenase | alcohol dehydrogenase | oxidation-reduction process; metal ion binding; lactaldehyde reductase activity | - |
| Strains | Accession | Size (Mb) | CDS | GC% | |
|---|---|---|---|---|---|
| L. plantarum | L. plantarum WCFS1 | NC_004567.2 | 3.35 | 3041 | 44.45 |
| L. plantarum strain LY-78 | NZ_CP015308.1 | 3.13 | 2830 | 44.77 | |
| L. plantarum strain JBE245 | NZ_CP014780.1 | 3.26 | 2919 | 44.5 | |
| L. plantarum DOMLa | NZ_CP004406.1 | 3.21 | 2897 | 44.67 | |
| L. plantarum strain LZ206 | NZ_CP015966.1 | 3.26 | 2942 | 44.5 | |
| L. plantarum strain C410L1 | NZ_CP017954.1 | 3.39 | 2996 | 44.42 | |
| L. plantarum subsp. plantarum P-8 | NC_021224.2 | 3.25 | 2951 | 44.55 | |
| L. plantarum strain JBE490 | NZ_CP020861.1 | 3.20 | 2820 | 44.57 | |
| L. pentosus | L. pentosus strain SLC13 | NZ_CP022130.1 | 3.58 | 3084 | 46.41 |
| L. pentosus strain BGM48 | NZ_CP016491.1 | 3.68 | 3236 | 46.13 | |
| L. pentosus KCA1 | NZ_CM001538.1 | 3.43 | 5931 | 46.40 | |
| L. pentosus strain DSM 20314 | NZ_CP032757.1 | 3.67 | 3199 | 46.33 | |
| L. pentosus strain ZFM222 | NZ_CP032654.1 | 3.70 | 3198 | 46.14 | |
| L. pentosus strain ZFM94 | NZ_CP032659.1 | 3.69 | 3179 | 46.19 | |
| L. pentosus strain MS031 | NZ_CP043671.1 | 3.81 | 3303 | 46.10 | |
| L. pentosus strain KZ0310 | NZ_CP044245.1 | 3.87 | 3372 | 46.03 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, J.; Lu, C.; Xu, Q.; Li, Z.; Song, Y.; Zhou, S.; Guo, L.; Zhang, T.; Luo, X. Comparative Genomics Analysis Provides New Insights into High Ethanol Tolerance of Lactiplantibacillus pentosus LTJ12, a Novel Strain Isolated from Chinese Baijiu. Foods 2023, 12, 35. https://doi.org/10.3390/foods12010035
Wang J, Lu C, Xu Q, Li Z, Song Y, Zhou S, Guo L, Zhang T, Luo X. Comparative Genomics Analysis Provides New Insights into High Ethanol Tolerance of Lactiplantibacillus pentosus LTJ12, a Novel Strain Isolated from Chinese Baijiu. Foods. 2023; 12(1):35. https://doi.org/10.3390/foods12010035
Chicago/Turabian StyleWang, Jiali, Chengshun Lu, Qiang Xu, Zhongyuan Li, Yajian Song, Sa Zhou, Le Guo, Tongcun Zhang, and Xuegang Luo. 2023. "Comparative Genomics Analysis Provides New Insights into High Ethanol Tolerance of Lactiplantibacillus pentosus LTJ12, a Novel Strain Isolated from Chinese Baijiu" Foods 12, no. 1: 35. https://doi.org/10.3390/foods12010035