
Phosphoproteome Analysis Using Two-Dimensional Electrophoresis Coupled with Chemical Dephosphorylation

Abstract
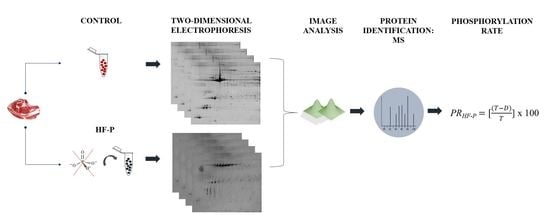
:
1. Introduction
2. Materials and Methods
2.1. Animal Material
2.2. Protein Extraction and Quantification
2.3. Two-Dimensional Electrophoresis (2-DE)
2.4. Detection of Phosphoproteins and Total Proteins
2.5. Image Analysis
2.6. Protein Dephosphorylation
2.7. Mass Spectrometry (MS) Analysis
2.8. Statistical Analysis
3. Results and Discussion
3.1. Phosphoproteome Map: An Overview
3.2. Phosphorylation Level of Proteins
3.3. Efficiency Assessment of the Dephosphorylation Method with HF-P
3.4. Importance of HF-P on the Study of Phosphoproteome at Global Level
3.5. Importance of HF-P in the Meat Industry
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chou, K.C. Progresses in Predicting Post-translational Modification. Int. J. Pept. Res. Ther. 2020, 26, 873–888. [Google Scholar] [CrossRef]
- Veenstra, T.D. Phosphorylation. In Proteomics for Biological Discovery; Veenstra, T.D., Yates, J.R., III, Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2019; pp. 265–289. [Google Scholar]
- Mato, A.; Rodríguez-Vázquez, R.; López-Pedrouso, M.; Bravo, S.; Franco, D.; Zapata, C. The first evidence of global meat phosphoproteome changes in response to pre-slaughter stress. BMC Genom. 2019, 20, 590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Needham, E.J.; Parker, B.L.; Burykin, T.; James, D.E.; Humphrey, S.J. Illuminating the dark phosphoproteome. Sci. Signal. 2019, 12, eaau8645. [Google Scholar] [CrossRef] [Green Version]
- Kaufmann, H.; Bailey, J.E.; Fussenegger, M. Use of antibodies for detection of phosphorylated proteins separated by two-dimensional gel electrophoresis. Proteomics 2001, 1, 194–199. [Google Scholar] [CrossRef]
- Bendt, A.K.; Burkovski, A.; Schaffer, S.; Bott, M.; Farwick, M.; Hermann, T. Towards a phosphoproteome map of Corynebacterium glutamicum. Proteomics 2003, 3, 1637–1646. [Google Scholar] [CrossRef]
- Thingholm, T.E.; Jensen, O.N.; Larsen, M.R. Analytical strategies for phosphoproteomics. Proteomics 2009, 9, 1451–1468. [Google Scholar] [CrossRef] [PubMed]
- Angel, T.E.; Aryal, U.K.; Hengel, S.M.; Baker, E.S.; Kelly, R.T.; Robinson, E.W.; Smith, R.D. Mass spectrometry-based proteomics: Existing capabilities and future directions. Chem. Soc. Rev. 2012, 41, 3912–3928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliveira, B.M.; Coorssen, J.R.; Martins-de-Souza, D. 2DE: The Phoenix of Proteomics. J. Proteomics 2014, 104, 140–150. [Google Scholar] [CrossRef] [PubMed]
- Naryzhny, S. Towards the full realization of 2DE power. Proteomes 2016, 4, 33. [Google Scholar] [CrossRef] [Green Version]
- Lee, P.Y.; Saraygord-Afshari, N.; Low, T.Y. The evolution of two-dimensional gel electrophoresis—From proteomics to emerging alternative applications. J. Chromatogr. A 2020, 1615, 460763. [Google Scholar] [CrossRef]
- Hackett, M. Science, marketing and wishful thinking in quantitative proteomics. Proteomics 2008, 8, 4618–4623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baudin, B. Two-Dimensional Gel Electrophoresis (2-DE). In Gel Electrophoresis-Principles and Basics; Magdelin, S., Ed.; InTech: Rijeka, Croatia, 2012; pp. 137–156. [Google Scholar]
- Zhan, X.; Li, B.; Zhan, X.; Schlüter, H.; Jungblut, P.R.; Coorssen, J.R. Innovating the Concept and Practice of Two-Dimensional Gel Electrophoresis in the Analysis of Proteomes at the Proteoform Level. Proteomes 2019, 7, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodríguez-Vázquez, R.; Mato, A.; López-Pedrouso, M.; Franco, D.; Sentandreu, M.A.; Zapata, C. Measuring quantitative proteomic distance between Spanish beef breeds. Food Chem. 2020, 315, 126293. [Google Scholar] [CrossRef] [PubMed]
- Zhu, K.; Zhao, J.; Lubman, D.M.; Miller, F.R.; Barder, T.J.; Avenue, S.C. Protein pI Shifts due to Posttranslational Modifications in the Separation and Characterization of Proteins. Anal. Chem. 2005, 77, 2745–2755. [Google Scholar] [CrossRef]
- Rabilloud, T. When 2D is not enough, go for an extra dimension. Proteomics 2013, 13, 2065–2068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rogowska-Wrzesinska, A.; Le Bihan, M.; Thaysen-andersen, M.; Roepstorff, P. 2D gels still have a niche in proteomics. J. Proteom. 2013, 88, 4–13. [Google Scholar] [CrossRef] [PubMed]
- Jastorff, A.M.; Turck, C.W. Staining of Two-Dimensional Gels. In Post-Translational Modification of Proteins: Tools for Functional Proteomics; Kannicht, C., Ed.; Humana: New York, NY, USA, 2019; Volume 1934, pp. 21–32. ISBN 9781493990559. [Google Scholar]
- Goodman, T.; Schulenberg, B.; Steinberg, T.H.; Patton, W.F. Detection of phosphoproteins on electroblot membranes using a small-molecule organic fluorophore. Electrophoresis 2004, 25, 2533–2538. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, G.K.; Thelen, J.J. Development of a simplified, economical polyacrylamide gel staining protocol for phosphoproteins. Proteomics 2005, 5, 4684–4688. [Google Scholar] [CrossRef] [PubMed]
- Han, C.; Yang, P. Two Dimensional Gel Electrophoresis-Based Plant Phosphoproteomics. In Phospho-Proteomics: Methods and Protocols; von Stechow, L., Ed.; Springer: New York, NY, USA, 2016; pp. 213–223. ISBN 978-1-4939-3049-4. [Google Scholar]
- Murray, J.; Marusich, M.F.; Capaldi, R.A.; Aggeler, R. Focused proteomics: Monoclonal antibody-based isolation of the oxidative phosphorylation machinery and detection of phosphoproteins using a fluorescent phosphoprotein gel stain. Electrophoresis 2004, 25, 2520–2525. [Google Scholar] [CrossRef]
- Hopper, R.K.; Carroll, S.; Aponte, A.M.; Johnson, D.T.; French, S.; Shen, R.F.; Witzmann, F.A.; Harris, R.A.; Balaban, R.S. Mitochondrial matrix phosphoproteome: Effect of extra mitochondrial calcium. Biochemistry 2006, 45, 2524–2536. [Google Scholar] [CrossRef] [PubMed]
- Marcantonio, M.; Trost, M.; Courcelles, M.; Desjardins, M.; Thibault, P. Combined enzymatic and data mining approaches for comprehensive phosphoproteome analyses: Application to cell signaling events of interferon-γ-stimulated macrophages. Mol. Cell. Proteom. 2008, 7, 645–660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lan, G.Q.; Abdullah, N.; Jalaludin, S.; Ho, Y.W. In vitro and in vivo enzymatic dephosphorylation of phytate in maize-soya bean meal diets for broiler chickens by phytase of Mitsuokella jalaludinii. Anim. Feed Sci. Technol. 2010, 158, 155–164. [Google Scholar] [CrossRef]
- Deracinois, B.; Matéos, A.; Romelard, A.; Boulier, A.; Auger, J.; Baniel, A.; Ravallec, R.; Flahaut, C. Partial-, double-enzymatic dephosphorylation and endogluc hydrolysis as an original approach to enhancing identification of casein phosphopeptides (Cpps) by mass spectrometry. Foods 2021, 10, 2134. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Wang, Y.; Yu, Y.; Hu, J.; Lu, N.; Regenstein, J.M.; Wang, M.; Zhou, P. Effects of enzymatic dephosphorylation on infant in vitro gastrointestinal digestibility of milk protein concentrate. Food Chem. 2016, 197, 891–899. [Google Scholar] [CrossRef] [PubMed]
- Volk, S.P.; Ahn, D.U.; Zeece, M.; Jung, S. Effects of high-pressure processing and enzymatic dephosphorylation on phosvitin properties. J. Sci. Food Agric. 2012, 92, 3095–3098. [Google Scholar] [CrossRef]
- Zeller, M.; König, S. The impact of chromatography and mass spectrometry on the analysis of protein phosphorylation sites. Anal. Bioanal. Chem. 2004, 378, 898–909. [Google Scholar] [CrossRef]
- Schlosser, A.; Pipkom, R.; Bossemeyer, D.; Lehmann, W.D. Analysis of protein phosphorylation by a combination of elastase digestion and neutral loss tandem mass spectrometry. Anal. Chem. 2001, 73, 170–176. [Google Scholar] [CrossRef]
- Kuyama, H.; Toda, C.; Watanabe, M.; Tanaka, K.; Nishimura, O. An efficient chemical method for dephosphorylation of phosphopeptides. Rapid Commun. Mass Spectrom. 2003, 17, 1493–1496. [Google Scholar] [CrossRef]
- Hunter, A.P.; Games, D.E. Chromatographic and mass spectrometric methods for the identification of phosphorylation sites in phosphoproteins. Rapid Commun. Mass Spectrom. 1994, 8, 559–570. [Google Scholar] [CrossRef]
- Kita, K.; Okumura, N.; Takao, T.; Watanabe, M.; Matsubara, T.; Nishimura, O.; Nagai, K. Evidence for phosphorylation of rat liver glucose-regulated protein 58, GRP58/ERp57/ER-60, induced by fasting and leptin. FEBS Lett. 2006, 580, 199–205. [Google Scholar] [CrossRef]
- López-Pedrouso, M.; Alonso, J.; Zapata, C. Evidence for phosphorylation of the major seed storage protein of the common bean and its phosphorylation-dependent degradation during germination. Plant Mol. Biol. 2014, 84, 415–428. [Google Scholar] [CrossRef] [PubMed]
- Bernal, J.; López-Pedrouso, M.; Franco, D.; Bravo, S.; García, L.; Zapata, C. Identification and Mapping of Phosphorylated Isoforms of the Major Storage Protein of Potato Based on Two- Dimensional Electrophoresis. In Advances in Seed Biology; Jimenez-Lopez, J., Ed.; InTech: Rijeka, Croatia, 2017; pp. 65–82. [Google Scholar]
- Bernal, J.; Mouzo, D.; Franco, D.; Garc, L.; Zapata, C. The Major Storage Protein in Potato Tuber Is Mobilized by a Mechanism Dependent on Its Phosphorylation Status. Int. J. Mol. Sci. 2019, 20, 1889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ortea, I.; O’Connor, G.; Maquet, A. Review on proteomics for food authentication. J. Proteom. 2016, 147, 212–225. [Google Scholar] [CrossRef]
- Gallardo, J.M.; Ortea, I.; Carrera, M. Proteomics and its applications for food authentication and food-technology research. TrAC Trends Anal. Chem. 2013, 52, 135–141. [Google Scholar] [CrossRef]
- Afzaal, M.; Saeed, F.; Hussain, M.; Shahid, F.; Siddeeg, A.; Al-Farga, A. Proteomics as a promising biomarker in food authentication, quality and safety: A review. Food Sci. Nutr. 2022, 10, 2333–2346. [Google Scholar] [CrossRef] [PubMed]
- Franco, D.; Mato, A.; Salgado, F.J.; López-Pedrouso, M.; Carrera, M.; Bravo, S.; Parrado, M.; Gallardo, J.M.; Zapata, C. Tackling proteome changes in the Longissimus thoracis bovine muscle in response to pre-slaughter stress. J. Proteom. 2015, 122, 73–85. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez-Vázquez, R.; Pateiro, M.; López-Pedrouso, M.; Gende, A.; Crecente, S.; Serrano, M.P.; González, J.; Lorenzo, J.M.; Zapata, C.; Franco, D. Influence of production system and finishing feeding on meat quality of Rubia Gallega calves. Span. J. Agric. Res. 2020, 18, e0606. [Google Scholar] [CrossRef]
- Görg, A.; Obermaier, C.; Boguth, G.; Harder, A.; Scheibe, B.; Wildgruber, R.; Weiss, W. The current state of two-dimensional electrophoresis with immobilized pH gradients. Electrophoresis 1988, 9, 1037–1053. [Google Scholar] [CrossRef]
- Agrawal, G.K.; Thelen, J.J. A high-resolution two dimensional Gel- and Pro-Q DPS-based proteomics workflow for phosphoprotein identification and quantitative profiling. Methods Mol. Biol. 2009, 527, 3–19. [Google Scholar] [CrossRef]
- Bio-Rad PDQuest 2-D Analysis Software. Available online: https://www.bio-rad.com/en-rs/product/pdquest-2-d-analysis-software?ID=966deb78-2656-437f-b7a4-ab0a9bd45c8d (accessed on 1 September 2022).
- Efron, B. The Jackknife, the Bootstrap, and Other Resampling Plans; Society for Industrial and Applied Mathematics: Philadelphia, PA, USA, 1982. [Google Scholar]
- Steinberg, T.H.; Agnew, B.J.; Gee, K.R.; Leung, W.; Goodman, T.; Hendrickson, J.; Beechem, J.M.; Haugland, R.P.; Patton, W.F. Global quantitative phosphoprotein analysis using Multiplexed Proteomics technology. Proteomics 2003, 3, 1128–1144. [Google Scholar] [CrossRef]
- López-Pedrouso, M.; Bernal, J.; Franco, D.; Zapata, C. Evaluating two-dimensional electrophoresis profiles of the protein phaseolin as markers of genetic differentiation and seed protein quality in common bean (Phaseolus vulgaris L.). J. Agric. Food Chem. 2014, 62, 7200–7208. [Google Scholar] [CrossRef]
- Vincenzetti, S.; Vita, A.; Carpi, F.M.; Micozzi, D.; Polidori, P. Effect of Dephosphorylation on Donkey Milk Caseins. In Trends in Veterinary Sciences: Current Aspects in Veterinary Morphophysiology, Biochemistry, Animal Production, Food Hygiene and Clinical Sciences; Boiti, C., Ferlazzo, A., Gaiti, A., Pugliese, A., Eds.; Springer: Berlin/Heidelberg, Germany, 2013; pp. 21–25. ISBN 978-3-642-36488-4. [Google Scholar]
- Wu, Z.; Tiambeng, T.N.; Cai, W.; Chen, B.; Lin, Z.; Gregorich, Z.R.; Ge, Y. Impact of Phosphorylation on the Mass Spectrometry Quantification of Intact Phosphoproteins. Anal. Chem. 2018, 90, 4935–4939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rath, A.; Glibowicka, M.; Nadeau, V.G.; Chen, G.; Deber, C.M. Detergent binding explains anomalous SDS-PAGE migration of membrane proteins. Proc. Natl. Acad. Sci. USA 2009, 106, 1760–1765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.; Park, Y.; Min, H.; Kim, Y. Determination of protein phosphorylation by polyacrylamide gel electrophoresis. J. Microbiol. 2019, 57, 93–100. [Google Scholar] [CrossRef]
- Miller, I.; Crawford, J.; Gianazza, E. Protein stains for proteomic applications: Which, when, why ? Proteomics 2006, 6, 5385–5408. [Google Scholar] [CrossRef]
- Agrawal, G.K.; Thelen, J.J. Large Scale Identification and Quantitative Profiling of Phosphoproteins Expressed during Seed Filling in Oilseed Rape *. Mol. Cell. Proteom. 2006, 5, 2044–2059. [Google Scholar] [CrossRef] [Green Version]
- Berggren, K.N.; Schulenberg, B.; Lopez, M.F.; Steinberg, T.H.; Bogdanova, A.; Smejkal, G.; Wang, A.; Patton, W.F. An improved formulation of SYPRO Ruby protein gel stain: Comparison with the original formulation and with a ruthenium II tris (bathophenanthroline disulfonate) formulation. Proteomics 2002, 2, 486–498. [Google Scholar] [CrossRef]
- Thornton, K.J.; Chapalamadugu, K.C.; Eldredge, E.M.; Murdoch, G.K. Analysis of Longissimus thoracis Protein Expression Associated with Variation in Carcass Quality Grade and Marbling of Beef Cattle Raised in the Pacific Northwestern United States. J. Agric. Food Chem. 2017, 65, 1434–1442. [Google Scholar] [CrossRef]
- Rosa, A.F.; Moncau, C.T.; Poleti, M.D.; Fonseca, L.D.; Balieiro, J.C.C.; Silva, S.L.E.; Eler, J.P. Proteome changes of beef in Nellore cattle with different genotypes for tenderness. Meat Sci. 2018, 138, 1–9. [Google Scholar] [CrossRef]
- Bjarnadóttir, S.G.; Hollung, K.; Høy, M.; Bendixen, E.; Codrea, M.C.; Veiseth-Kent, E. Changes in protein abundance between tender and tough meat from bovine Longissimus thoracis muscle assessed by isobaric Tag for Relative and Absolute Quantitation (iTRAQ) and 2-dimensional gel electrophoresis analysis. J. Anim. Sci. 2012, 90, 2035–2043. [Google Scholar] [CrossRef]
- De Souza Rodrigues, R.T.; Chizzotti, M.L.; Vital, C.E.; Baracat-Pereira, M.C.; Barros, E.; Busato, K.C.; Gomes, R.A.; Ladeira, M.M.; Da Silva Martins, T. Differences in beef quality between Angus (Bos taurus taurus) and Nellore (Bos taurus indicus) cattle through a proteomic and phosphoproteomic approach. PLoS ONE 2017, 12, e0170294. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Wang, J.; Wang, G.; Ji, Z.; Hou, L.; Liu, Z.; Chao, T. Molecular cloning and mRNA expression analysis of sheep MYL3 and MYL4 genes. Gene 2016, 577, 209–214. [Google Scholar] [CrossRef] [PubMed]
- Su, H.; Iii, C.A.H.; Giddings, M.C. Mycoplasma pneumoniae. BMC Microbiol. 2007, 7, 63. [Google Scholar] [CrossRef] [Green Version]
- Roux, P.P.; Thibault, P. The coming of age of phosphoproteomics-from large data sets to inference of protein functions. Mol. Cell. Proteomics 2013, 12, 3453–3464. [Google Scholar] [CrossRef] [Green Version]
- Kinoshita, E.; Kinoshita-kikuta, E.; Koike, T. Separation and detection of large phosphoproteins using Phos-tag SDS-PAGE. Nat. Protoc. 2009, 4, 1513–1521. [Google Scholar] [CrossRef]
- Shen, C.; Zhang, K.; Gao, N.; Wei, S.; Liu, G.; Chai, Y.; Yang, M. Colorimetric and electrochemical determination of the activity of protein kinase based on retarded particle growth due to binding of phosphorylated peptides to DNA—capped silver nanoclusters. Microchim. Acta 2016, 183, 2933–2939. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, L.; Liang, R.P.; Bai, J.M.; Qiu, J.D. Using graphene quantum dots as photoluminescent probes for protein kinase sensing. Anal. Chem. 2013, 85, 9148–9155. [Google Scholar] [CrossRef] [PubMed]
- Solari, F.A.; Dell’Aica, M.; Sickmann, A.; Zahedi, R.P. Why phosphoproteomics is still a challenge. Mol. BioSyst. 2015, 11, 1487–1493. [Google Scholar] [CrossRef] [Green Version]
- Dowsey, A.W.; Dunn, M.J.; Yang, G.Z. The role of bioinformatics in two-dimensional gel electrophoresis. Proteomics 2003, 3, 1567–1596. [Google Scholar] [CrossRef]
- Ijaz, M.; Li, X.; Zhang, D.; Bai, Y.; Hou, C.; Hussain, Z.; Zheng, X.; Huang, C. Sarcoplasmic and myofibrillar phosphoproteins profile of beef M. Longissimus thoracis with different pHu at different days postmortem. J. Sci. Food Agric. 2022, 102, 2464–2471. [Google Scholar] [CrossRef] [PubMed]
- Erban, T.; Shcherbachenko, E.; Talacko, P.; Harant, K. A single honey proteome dataset for identifying adulteration by foreign amylases and mining various protein markers natural to honey. J. Proteom. 2021, 239, 104157. [Google Scholar] [CrossRef] [PubMed]
- Guarino, C.; De Simone, L.; Santoro, S.; Caira, S.; Lilla, S.; Calabrese, M.G.; Chianese, L.; Addeo, F. The Proteomic Changes in Cynara cardunculus L. var. altilis DC Following the Etiolation Phenomena Using De Novo Sequence Analysis. J. Bot. 2010, 2010, 496893. [Google Scholar] [CrossRef]
- Li, X.; Zhang, D.; Ren, C.; Bai, Y.; Ijaz, M.; Hou, C.; Chen, L. Effects of protein posttranslational modifications on meat quality: A review. Compr. Rev. Food Sci. Food Saf. 2021, 20, 289–331. [Google Scholar] [CrossRef] [PubMed]
- D’Alessandro, A.; Zolla, L. Foodomics to investigate meat tenderness. TrAC Trends Anal. Chem. 2013, 52, 47–53. [Google Scholar] [CrossRef]
- Weng, K.; Li, Y.; Huo, W.; Zhang, Y.; Cao, Z.; Zhang, Y.; Xu, Q.; Chen, G. Comparative phosphoproteomic provides insights into meat quality differences between slow- and fast-growing broilers. Food Chem. 2022, 373, 131408. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.; Li, X.; Li, C. Seasons affect the phosphorylation of pork sarcoplasmic proteins related to meat quality. Anim. Biosci. 2022, 35, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Gu, M.; Wei, Y.; Jiao, Y.; Zhang, D.; Liu, Y. Insights from proteome to phosphorylated proteome: Deciphering different regulatory mechanisms in goat muscles with high- and low-meat quality. Int. J. Food Sci. Technol. 2022, 57, 3532–3543. [Google Scholar] [CrossRef]
- Weng, K.; Huo, W.; Gu, T.; Bao, Q.; Cao, Z.; Zhang, Y.; Zhang, Y.; Xu, Q.; Chen, G. Quantitative phosphoproteomic analysis unveil the effect of marketable ages on meat quality in geese. Food Chem. 2021, 361, 130093. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Spot Code 1 | Protein 2 | Abbrev. | Accession No. (Uniprot) | Mascot Score | Sequence Cov. (%) | No. of Matched Peptides | pI Th/ Obs 3 | Mr Th/ Obs (kDa) 3 | Method of Identification |
|---|---|---|---|---|---|---|---|---|---|
| 1A | -- | -- | -- | -- | -- | -- | --/ | --/ | |
| 2A | Myosin light chain 1/3, skeletal muscle isoform | MYL1 | A0JNJ5 | 170 | 41 | 8 | 4.96/4.87 | 21.0/24.8 | MALDI-TOF and MALDI-TOF/TOF |
| 3A | Myosin light chain 1/3, skeletal muscle isoform | MYL1 | A0JNJ5 | -- | 45 | 14 | 4.96/4.95 | 21.0/24.8 | [56] |
| 4A | Myosin light chain 3 | MYL3 | P85100 | -- | 43 | 4 | 5.00/4.99 | 21.9/25.2 | [41] |
| 5A | -- | -- | -- | -- | -- | -- | --/ | --/ | |
| 1B | Myosin regulatory light chain 2, ventricular/cardiac muscle isoform. | MYL2 | Q3SZE5 | 200 | 63 | 12 | 4.86/4.70 | 18.9/18.8 | MALDI-TOF and MALDI-TOF/TOF |
| 2B | Myosin regulatory light chain 2, ventricular/cardiac muscle isoform. | MYL2 | Q3SZE5 | 166 | 60 | 11 | 4.86/4.75 | 18.9/18.8 | MALDI-TOF and MALDI-TOF/TOF |
| 3B | Myosin regulatory light chain 2, ventricular/cardiac muscle isoform. | MYL2 | Q3SZE5 | 166 | 60 | 13 | 4.86/4.80 | 18.0/18.8 | [56,57] |
| 4B | Myosin regulatory light chain 2, skeletal muscle isoform | MYLPF | Q0P571 | 134 | 20 | 3 | 4.91/4.68 | 19.1/18.5 | MALDI-TOF and MALDI-TOF/TOF |
| 5B | Myosin regulatory light chain 2, fast skeletal muscle isoform | MYLPF | Q0P571 | 126 | 20 | 5 | 4.91/4.73 | 19.1/18.5 | MALDI-TOF and MALDI-TOF/TOF |
| 6B | Myosin regulatory light chain 2, fast skeletal muscle isoform | MYLPF | Q0P571 | 126 | 80 | 22 | 4.91/4.81 | 19.1/18.5 | [57,58] |
| 7B | -- | -- | -- | -- | -- | -- | -- | -- | |
| 1C | Myosin regulatory light chain 2, ventricular/cardiac muscle isoform | MYL2 | Q3SZE5 | 221 | 23 | 9 | 4.86/4.60 | 18.9/15.5 | [41] |
| 2C | MYL1 protein | MYL1 | Q08E10 | 239 | 52 | 9 | 4.73/4.57 | 19.7/15.0 | MALDI-TOF and MALDI-TOF/TOF |
| 3C | MYL1 protein | MYL1 | Q08E10 | 97 | 32 | 5 | 4.73/4.60 | 19.7/15.0 | MALDI-TOF and MALDI-TOF/TOF |
| Spot Code 1 | PR | p-Value 4 | PR Representation 5 | |||
|---|---|---|---|---|---|---|
| HF-P | Pro-Q DPS | |||||
| Mean (±SE) 2 | 95% Bootstrap CI (CL, CU) 3 | Mean (±SE) 2 | 95% Bootstrap CI (CL, CU) 3 | |||
| 1A | 100 ± 0.0 | 100, 100 | 0.0 ± 0.0 | 0.0, 0.0 | <0.05 |  |
| 2A | 87.4 ± 3.7 | 83.2, 94.2 | 27.2 ± 16.6 | 8.9, 59.7 | <0.05 |  |
| 3A | 47.9 ± 8.3 | 38.5, 63.2 | 0.0 ± 0.0 | 0.0, 0.0 | <0.05 |  |
| 4A | 100 ± 0.0 | 100, 100 | 0.0 ± 0.0 | 0.0, 0.0 | <0.05 |  |
| 5A | N/A | N/A | 0.0 ± 0.0 | 0.0, 0.0 | -- | -- |
| 1B | 59.0 ± 2.1 | 56.5, 62.8 | 100 ± 0.0 | 100, 100 | <0.05 |  |
| 2B | 32.5 ± 5.0 | 36.5, 38.5 | 100 ± 0.0 | 100, 100 | <0.05 |  |
| 3B | 22.6 ± 9.4 | 4.7, 36.8 | 0 ± 0.0 | 0, 0 | <0.05 |  |
| 4B | 100 ± 0.0 | 100, 100 | 100 ± 0.0 | 100, 100 | ns |  |
| 5B | 69.6 ± 5.4 | 63.3, 79.5 | 100 ± 0.0 | 100, 100 | <0.05 |  |
| 6B | 63.7 ± 21.4 | 37.5, 89.9 | 51.8 ± 17.7 | 34.0, 87.3 | ns |  |
| 7B | N/A | N/A | -- | -- | -- | -- |
| 1C | 100 ± 0.0 | 100, 100 | 0.0 ± 0.0 | 0.0, 0.0 | <0.05 |  |
| 2C | 100 ± 0.0 | 100, 100 | 0.0 ± 0.0 | 0.0, 0.0 | <0.05 |  |
| 3C | 88.8 ± 3.9 | 81.4, 94.9 | 5.5 ± 5.3 | 0.1, 16.0 | < 0.05 |  |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodríguez-Vázquez, R.; Mouzo, D.; Zapata, C. Phosphoproteome Analysis Using Two-Dimensional Electrophoresis Coupled with Chemical Dephosphorylation. Foods 2022, 11, 3119. https://doi.org/10.3390/foods11193119
Rodríguez-Vázquez R, Mouzo D, Zapata C. Phosphoproteome Analysis Using Two-Dimensional Electrophoresis Coupled with Chemical Dephosphorylation. Foods. 2022; 11(19):3119. https://doi.org/10.3390/foods11193119
Chicago/Turabian StyleRodríguez-Vázquez, Raquel, Daniel Mouzo, and Carlos Zapata. 2022. "Phosphoproteome Analysis Using Two-Dimensional Electrophoresis Coupled with Chemical Dephosphorylation" Foods 11, no. 19: 3119. https://doi.org/10.3390/foods11193119