
Synthesis of New Organoselenium-Based Succinanilic and Maleanilic Derivatives and In Silico Studies as Possible SARS-CoV-2 Main Protease Inhibitors

 ,
,  and
and 
Abstract
:1. Introduction
2. Results and Discussion
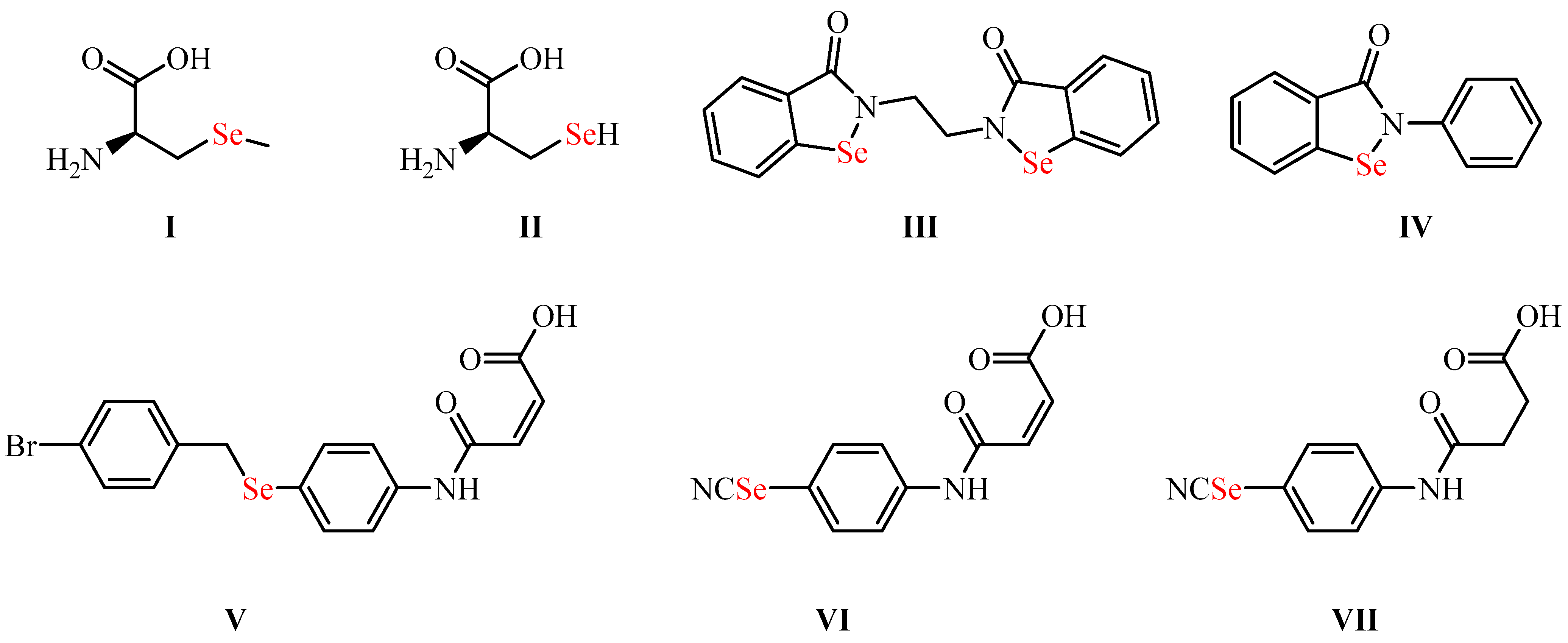
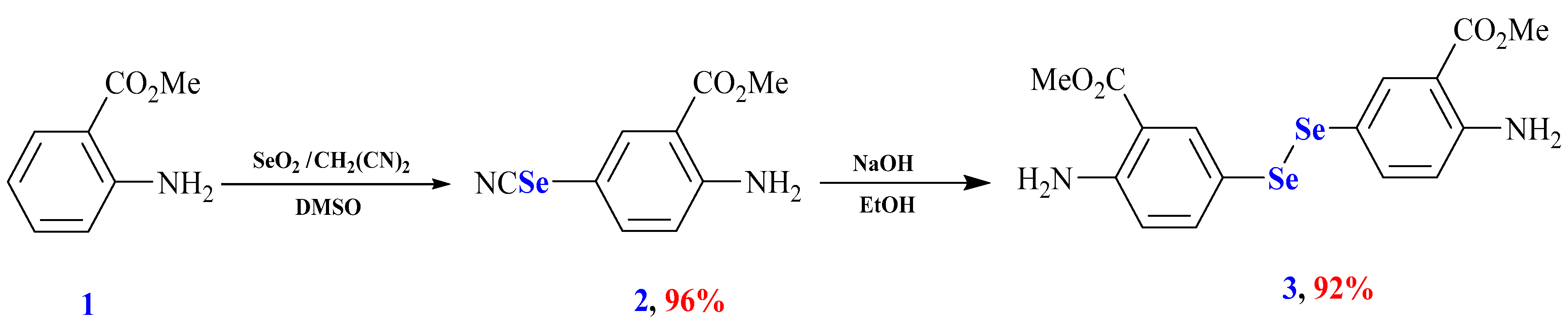
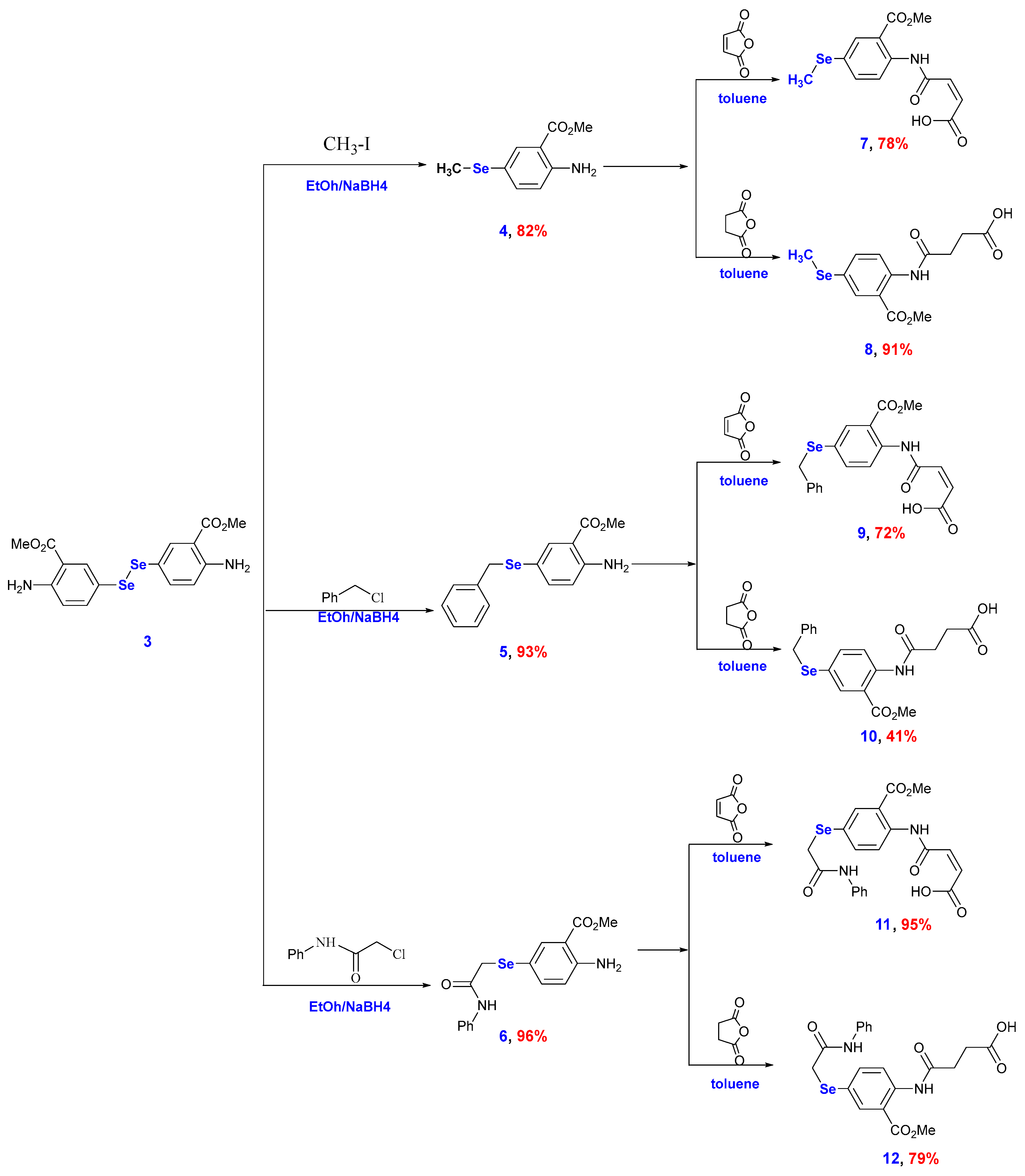
2.1. Chemistry
2.2. DFT Calculations
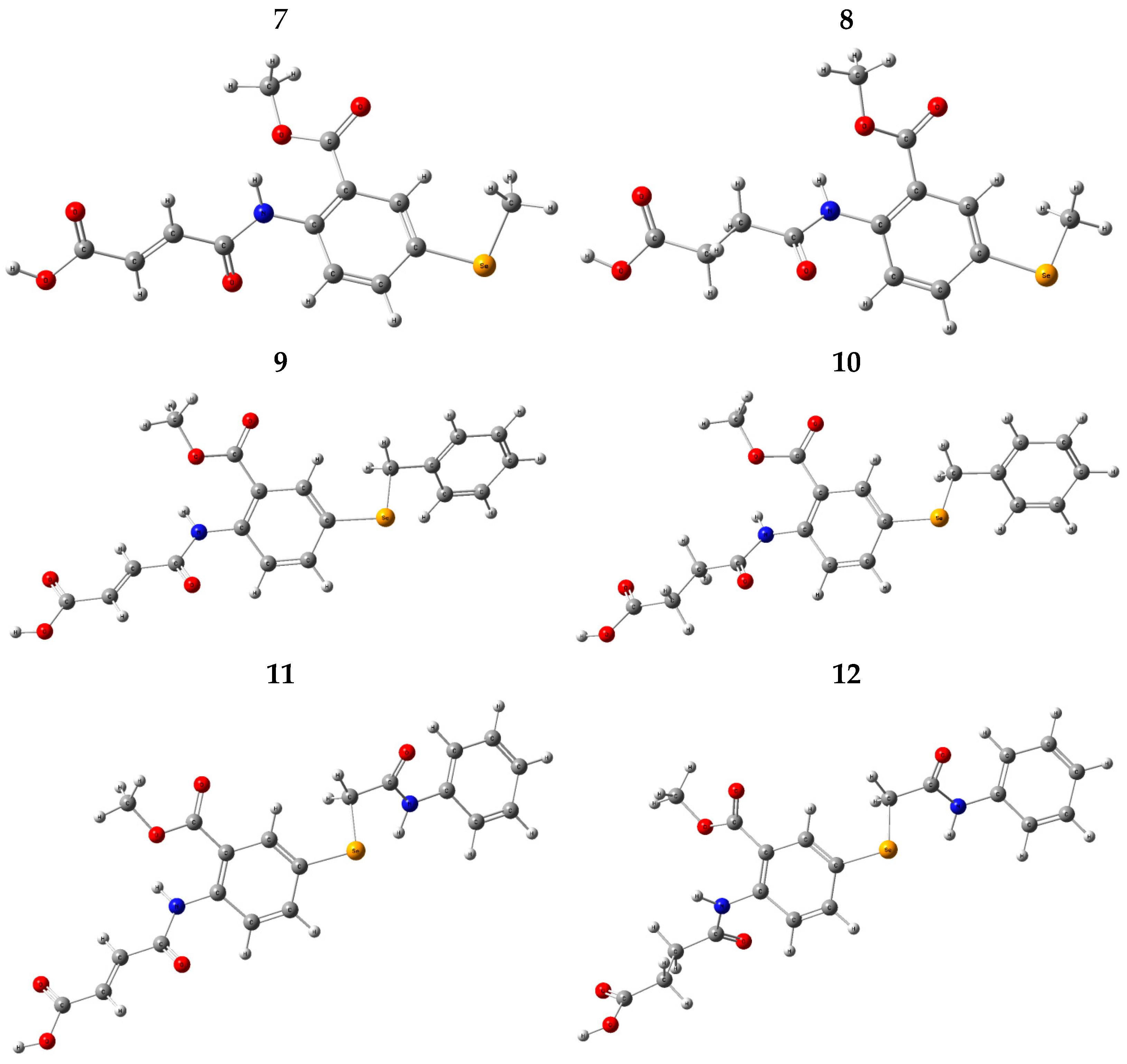
2.2.1. Conformational Analysis
2.2.2. Geometry Optimization
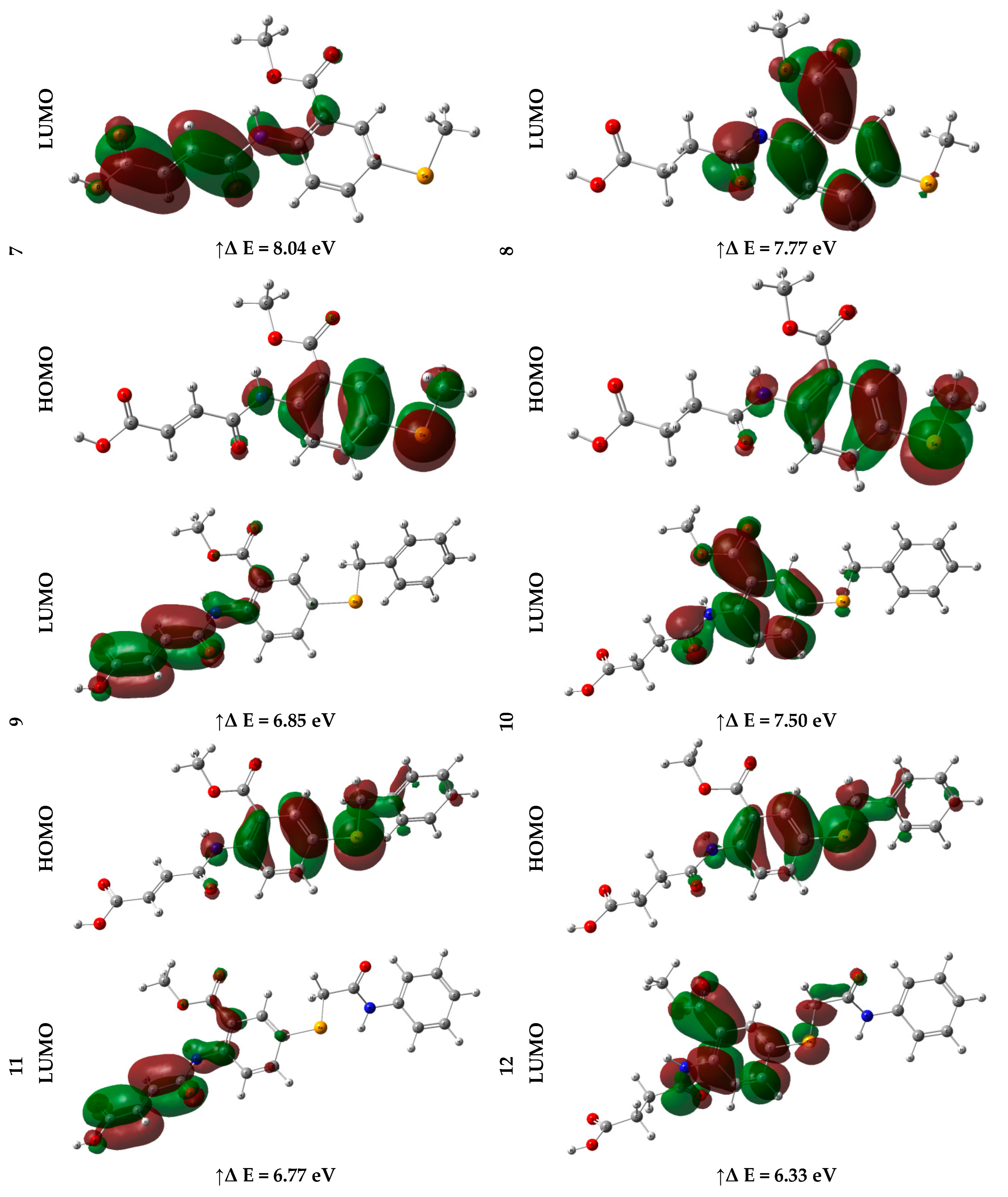
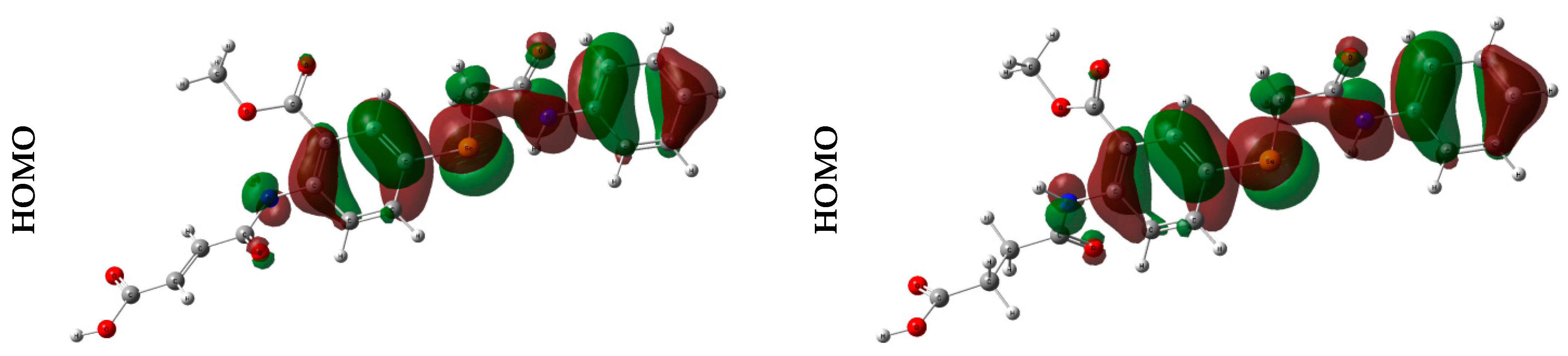
2.2.3. Frontier Molecular Orbital (FMO) Analysis
2.2.4. Global Reactivity
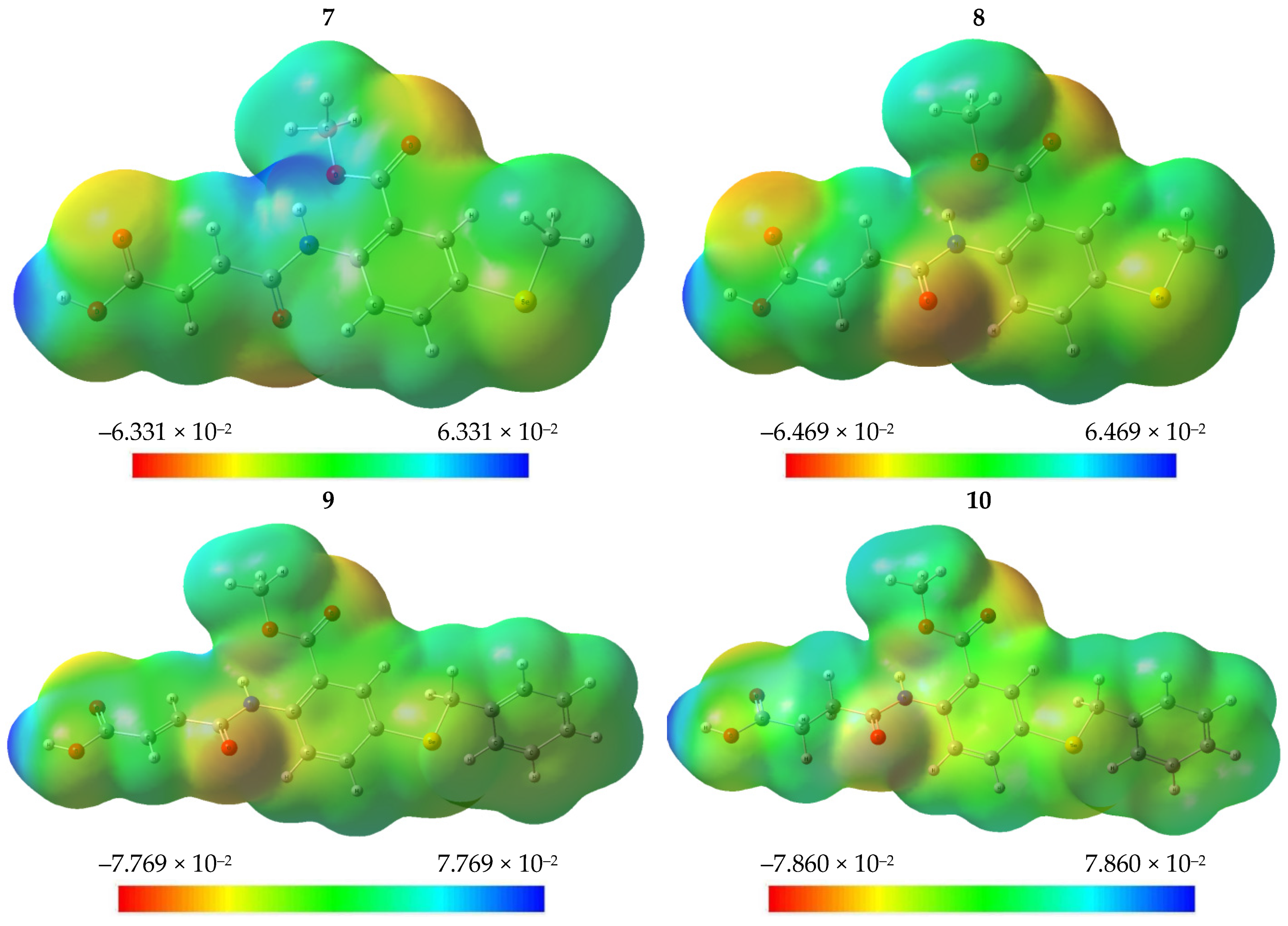
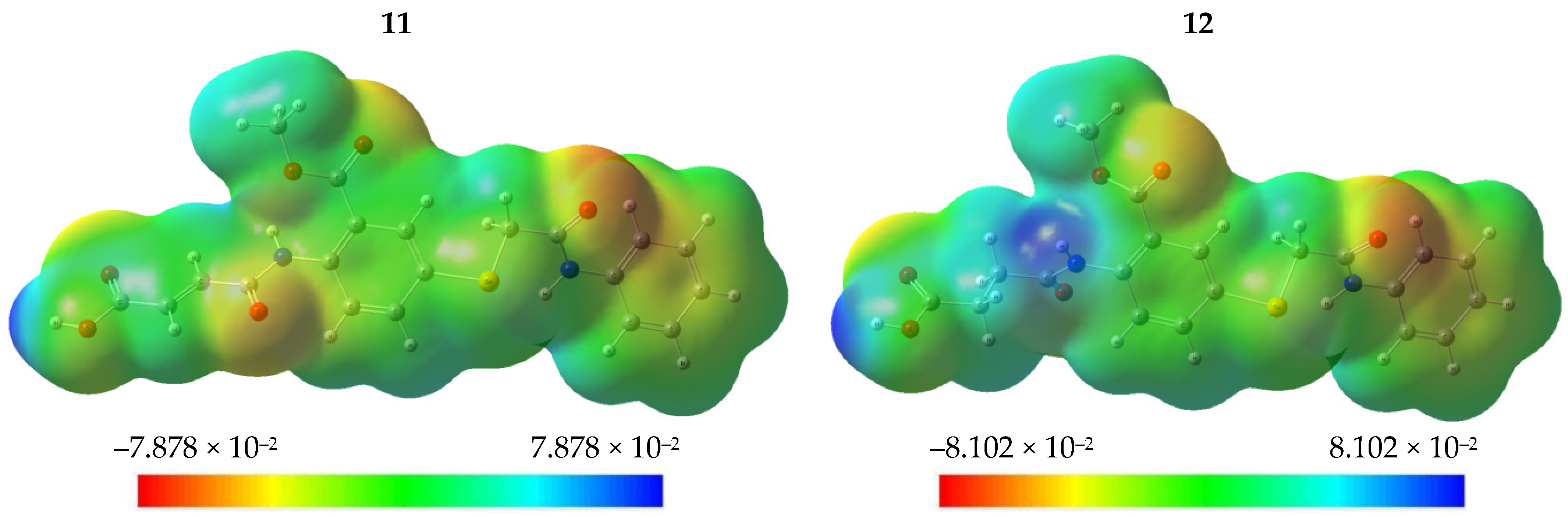
2.2.5. Molecular Electrostatic Potential (MEP)
2.2.6. Natural Charge Analysis
2.2.7. Natural Bond Orbital (NBO) Analysis
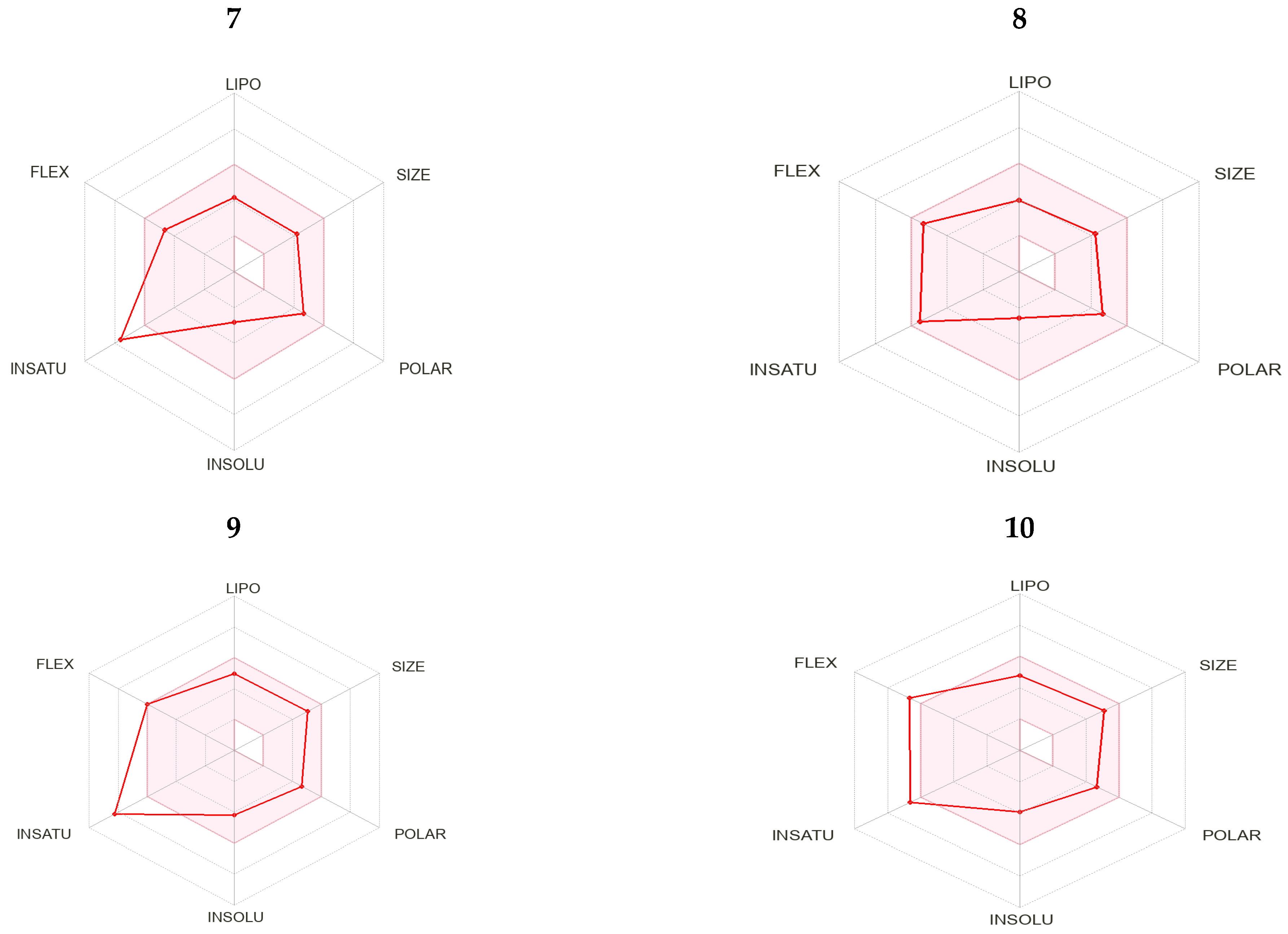
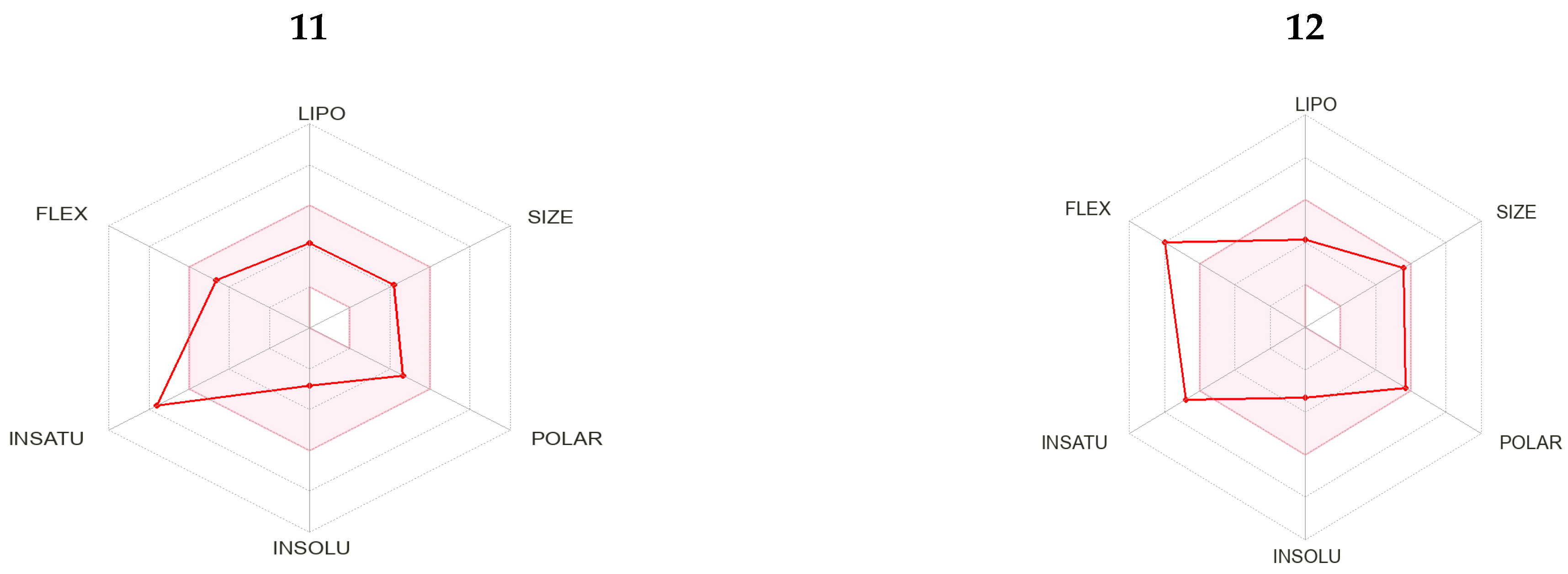
2.3. Drug Likeness Screening
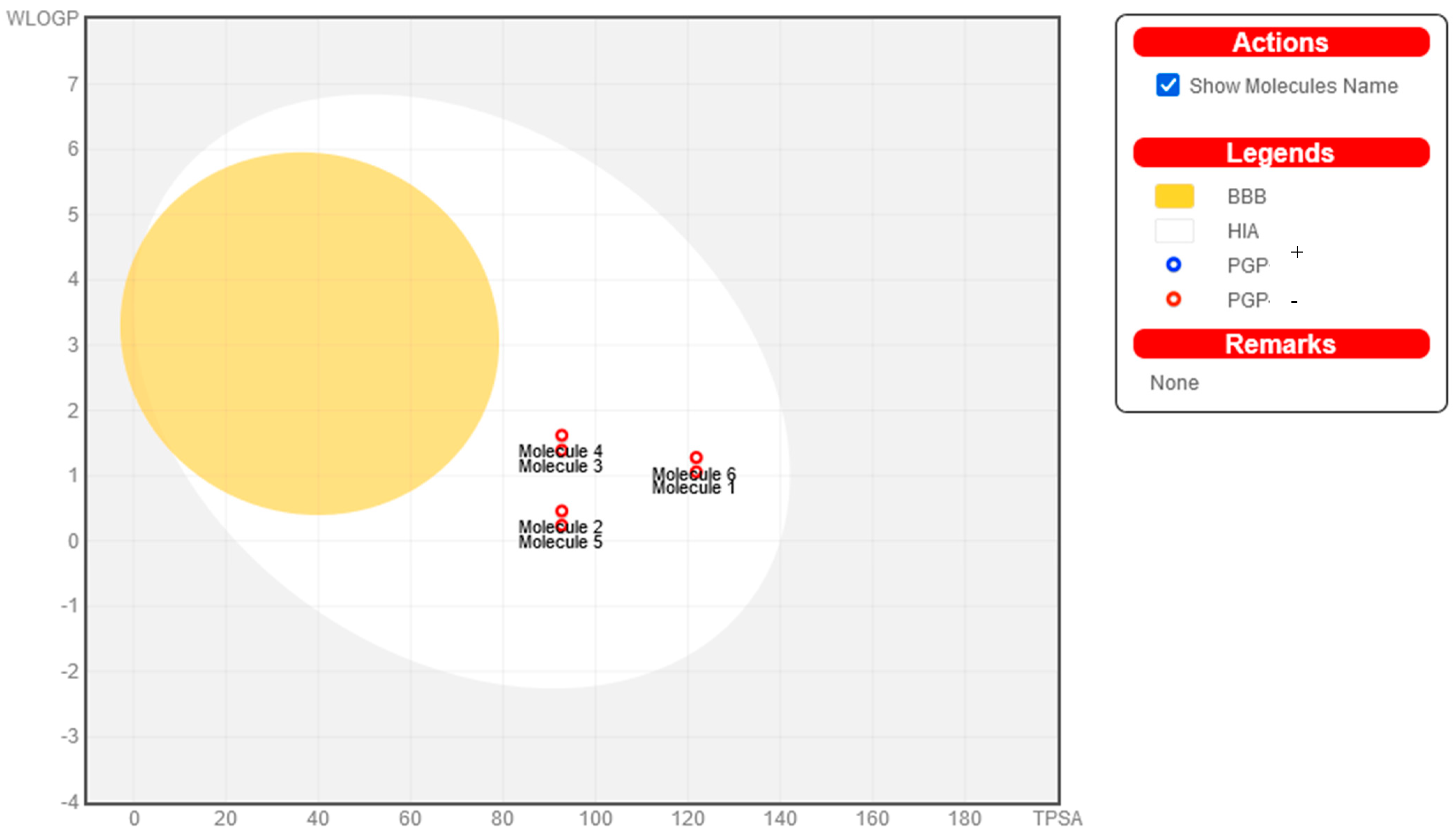
2.4. Pharmacokinetic Properties
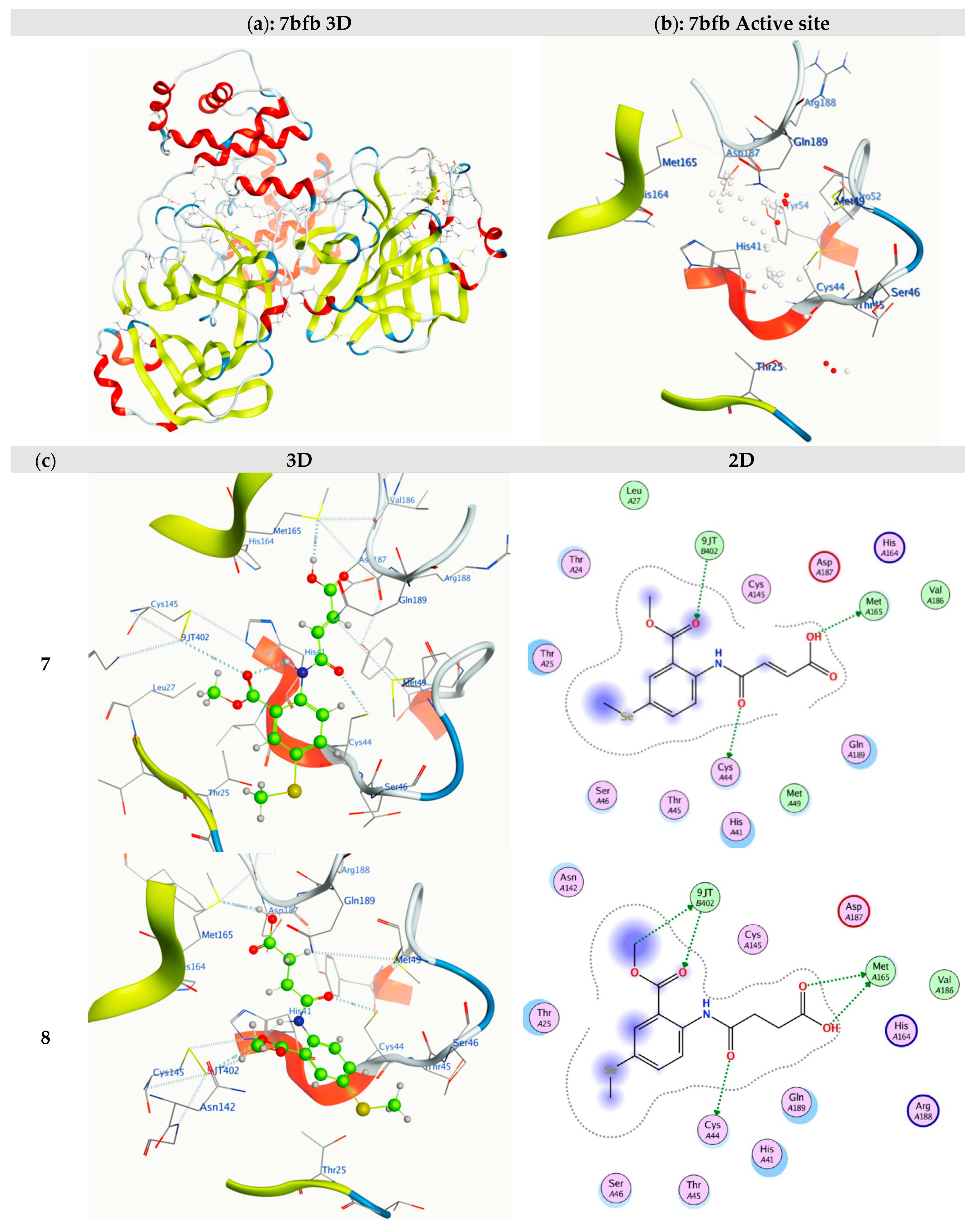
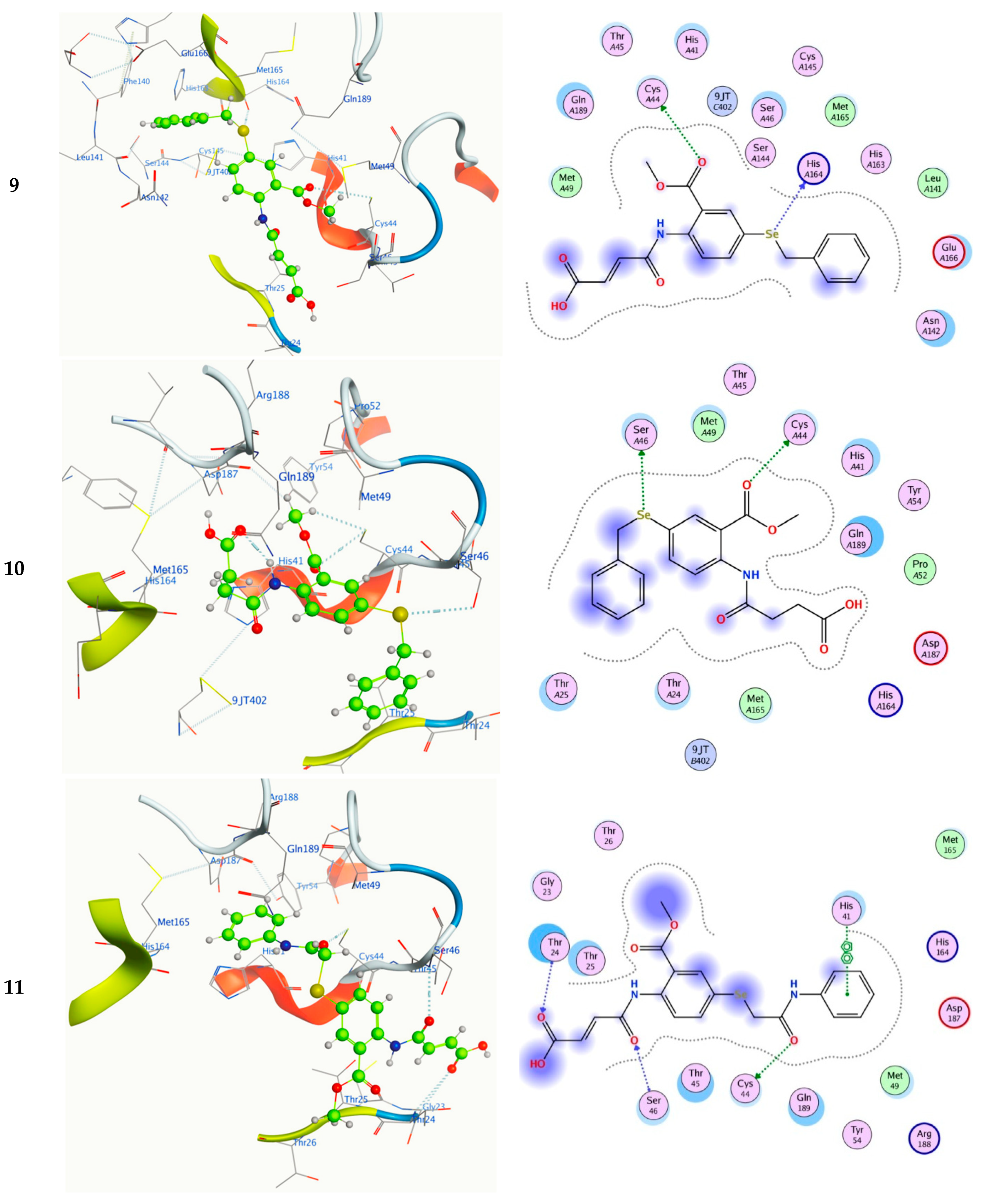
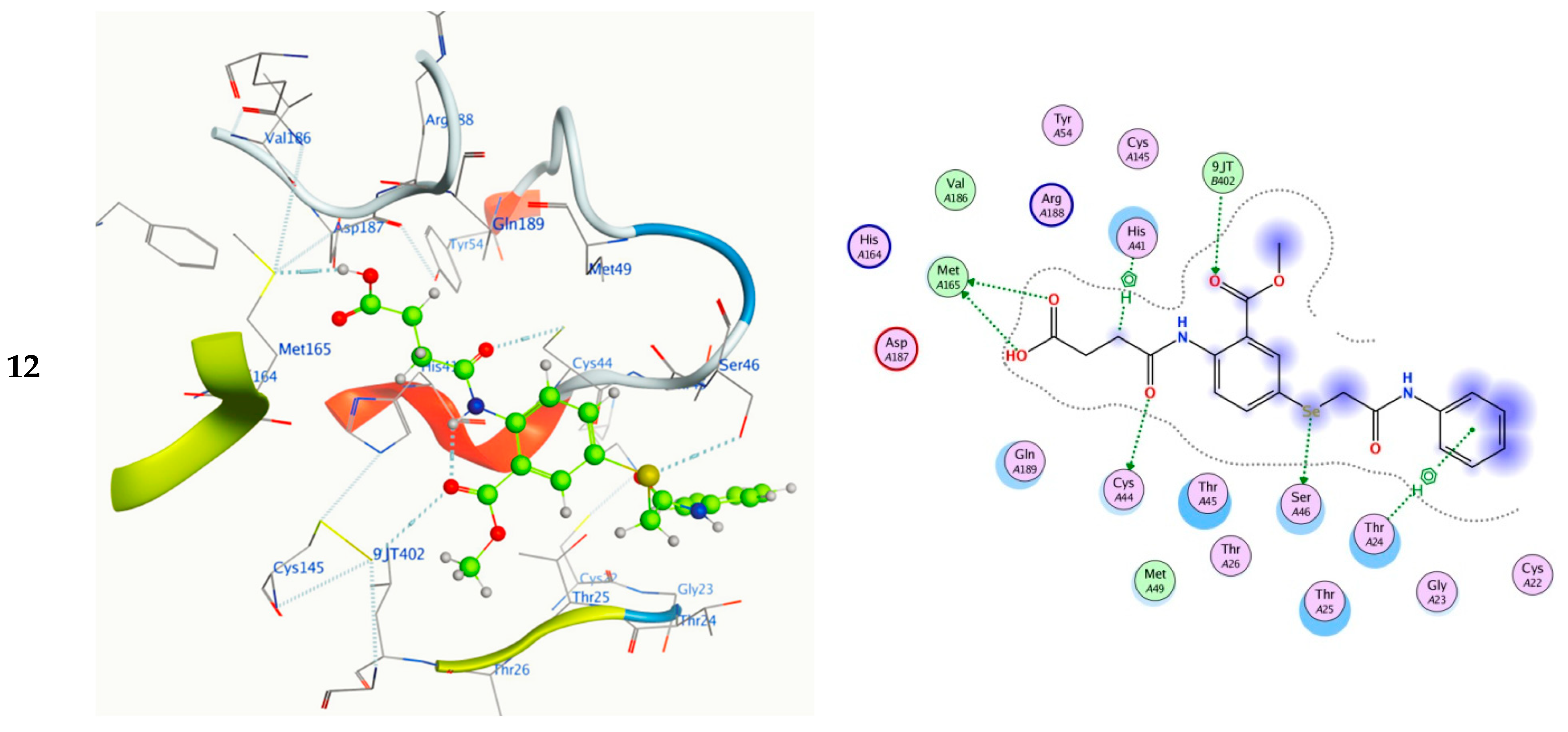
2.5. Molecular Docking
2.6. Molecular Dynamics (MD) Simulations
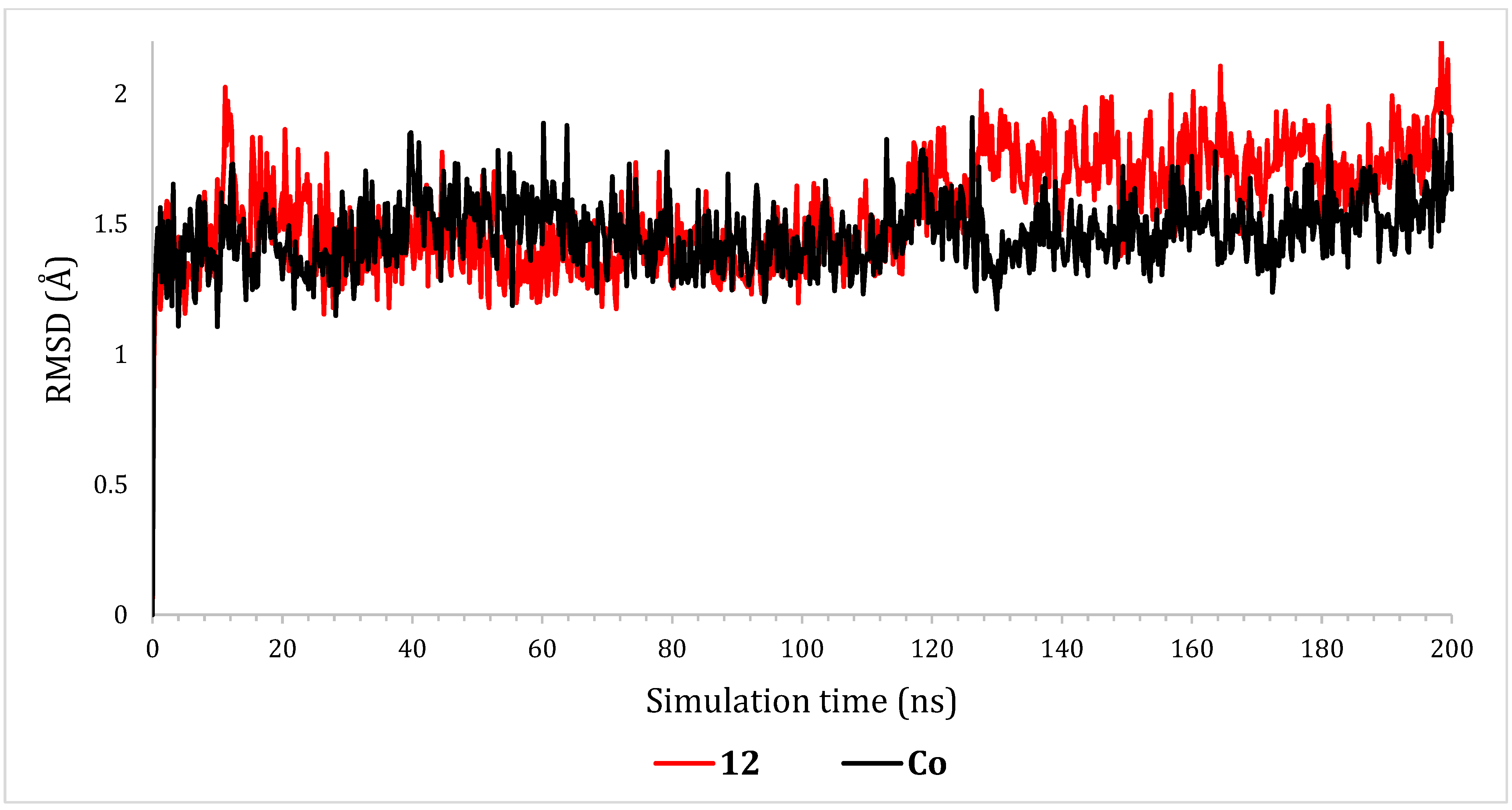
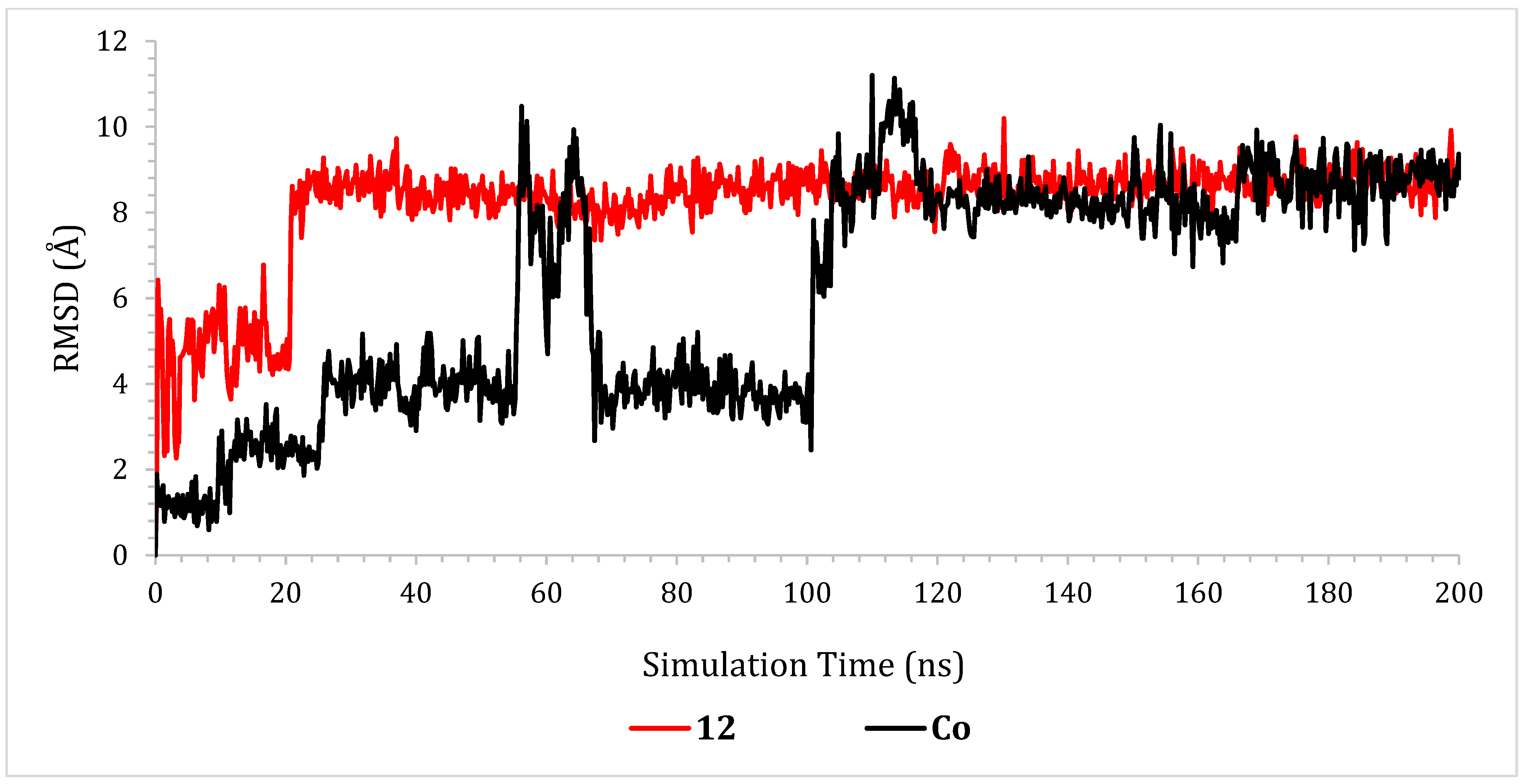
2.6.1. RMSD Analyses
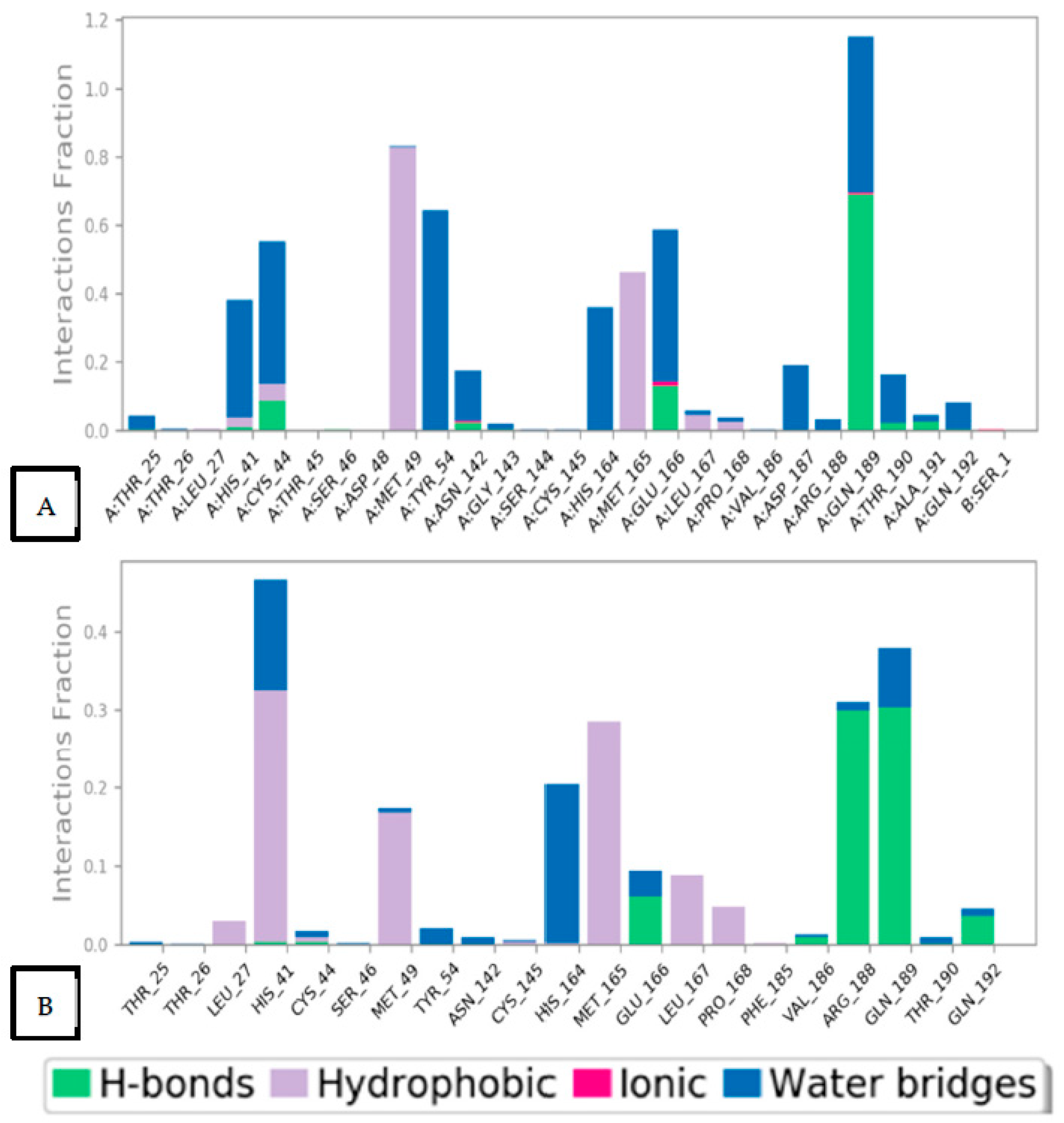
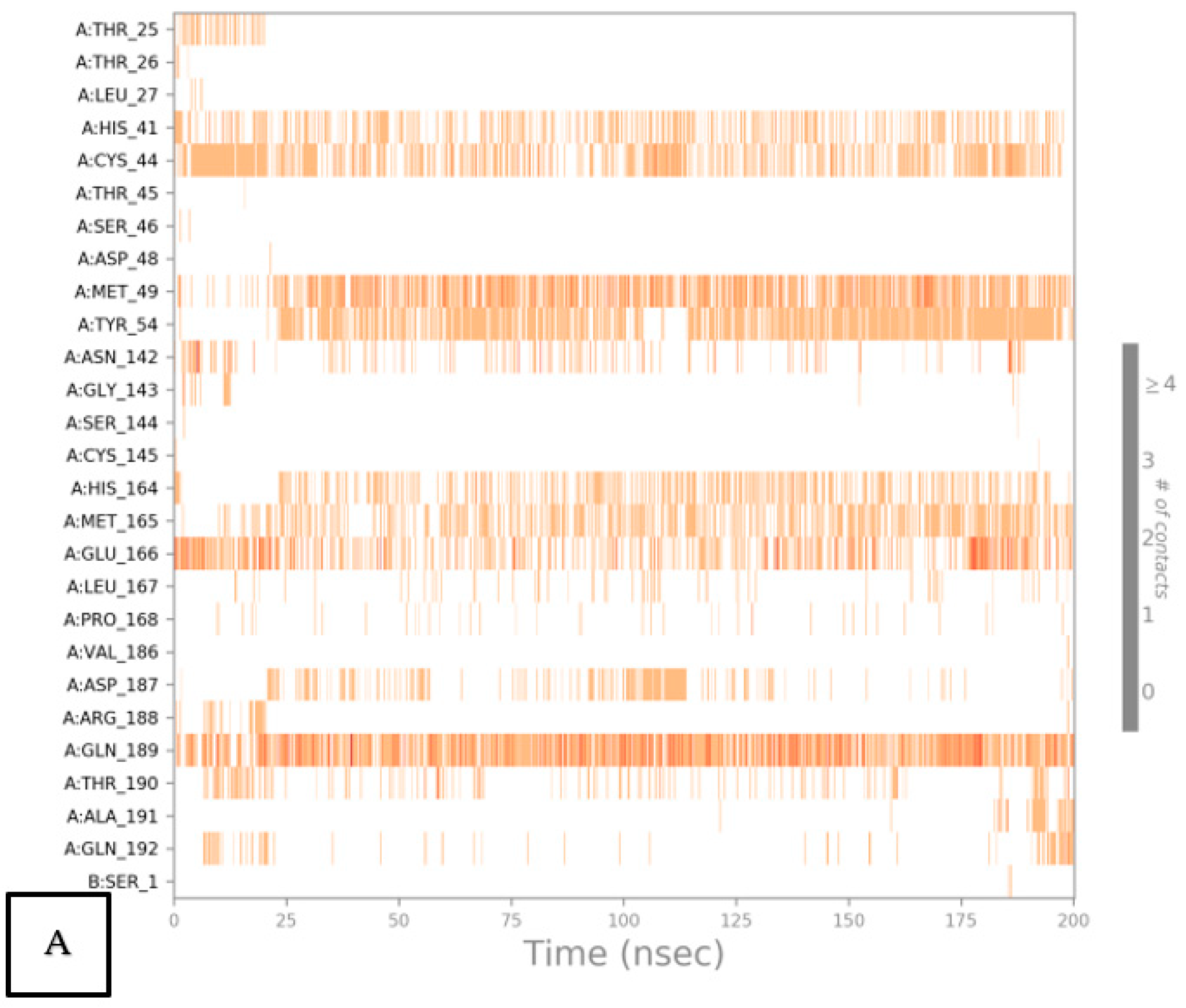
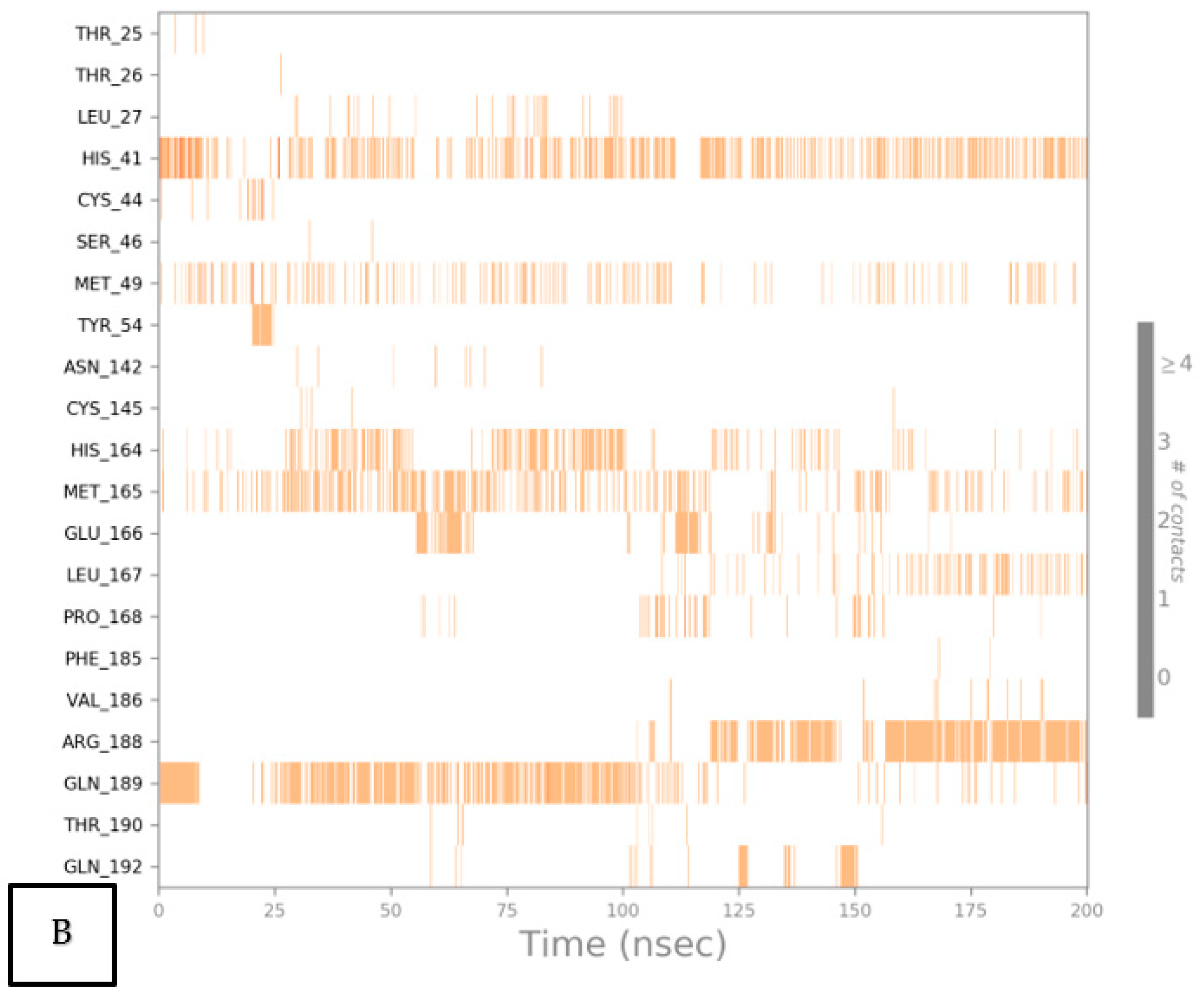
2.6.2. Histogram and Heat Map Analyses
2.7. Prime MM-GBSA Calculations and MD Trajectory Analysis
3. Experimental
3.1. Material and Methods
3.2. Synthesis and Characterization
3.2.1. Synthesis of (Z)–4–((2–(Methoxycarbonyl)–4–(Methylselanyl)Phenyl)Amino)–4–Oxobut–2–Enoic Acid (7)
3.2.2. Synthesis of 4–((2–(Methoxycarbonyl)–4–(Methylselanyl)Phenyl)Amino)–4–Oxobut–2–Enoic Acid (8)
3.2.3. Synthesis of (Z)–4–((2–(Methoxycarbonyl)–4–(Methylselanyl)Phenyl)Amino)–4–Oxobut–2–Enoic Acid (9)
3.2.4. Synthesis of 4–((2–(Methoxycarbonyl)–4–(Methylselanyl)Phenyl)Amino)–4–Oxobut–2–Enoic Acid (10)
3.2.5. Synthesis of (Z)–4–((2–(Methoxycarbonyl)–4–(Methylselanyl)Phenyl)Amino)–4–Oxobut–2–Enoic Acid (11)
3.2.6. Synthesis of4–((2–(Methoxycarbonyl)–4–(Methylselanyl)Phenyl)Amino)–4–Oxobut–2–Enoic Acid (12)
3.3. Computational Calculations
3.3.1. DFT Calculations
3.3.2. Molecular Docking Investigation
3.4. Drug Likeness and ADMET Prediction
3.5. Molecular Dynamics (MD) Simulations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Radomska, D.; Czarnomysy, R.; Radomski, D.; Bielawska, A.; Bielawski, K. Selenium as a Bioactive Micronutrient in the Human Diet and Its Cancer Chemopreventive Activity. Nutrients 2021, 13, 1649. [Google Scholar] [CrossRef] [PubMed]
- Schomburg, L. The other view: The trace element selenium as a micronutrient in thyroid disease, diabetes, and beyond. Hormones 2020, 19, 15–24. [Google Scholar] [PubMed]
- Saleem, U.; Sabir, S.; Niazi, S.G.; Naeem, M.; Ahmad, B. Role of Oxidative Stress and Antioxidant Defense Biomarkers in Neurodegenerative Diseases. Crit. Rev. Eukaryot. Gene Expr. 2020, 30, 311–322. [Google Scholar] [CrossRef] [PubMed]
- Bevinakoppamath, S.; Saleh Ahmed, A.M.; Ramachandra, S.C.; Vishwanath, P.; Prashant, A. Chemopreventive and Anticancer Property of Selenoproteins in Obese Breast Cancer. Front. Pharmacol. 2021, 12, 618172. [Google Scholar] [CrossRef]
- Shaaban, S.; El-Lateef, H.M.A.; Khalaf, M.M.; Gouda, M.; Youssef, I. One-Pot Multicomponent Polymerization, Metal-, and Non-Metal-Catalyzed Synthesis of Organoselenium Compounds. Polymers 2022, 14, 2208. [Google Scholar] [CrossRef]
- Arora, A.; Oswal, P.; Kumar Rao, G.; Kumar, S.; Kumar, A. Organoselenium ligands for heterogeneous and nanocatalytic systems: Development and applications. Dalton Trans. 2021, 50, 8628–8656. [Google Scholar] [CrossRef]
- Phadnis, P.P. Synthesis Strategies for Organoselenium Compounds and Their Potential Applications in Human Life. In Handbook on Synthesis Strategies for Advanced Materials; Springer: Berlin/Heidelberg, Germany, 2021; pp. 537–641. [Google Scholar]
- Pons, D.G.; Moran, C.; Alorda-Clara, M.; Oliver, J.; Roca, P.; Sastre-Serra, J. Micronutrients Selenomethionine and Selenocysteine Modulate the Redox Status of MCF-7 Breast Cancer Cells. Nutrients 2020, 12, 865. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Yang, Z.; Fu, J.; Yin, H.; Xiong, K.; Tan, Q.; Jin, H.; Li, J.; Wang, T.; Tang, W.; et al. Ethaselen: A potent mammalian thioredoxin reductase 1 inhibitor and novel organoselenium anticancer agent. Free Radic. Biol. Med. 2012, 52, 898–908. [Google Scholar] [CrossRef]
- Benelli, J.L.; Poester, V.R.; Munhoz, L.S.; Melo, A.M.; Trápaga, M.R.; Stevens, D.A.; Xavier, M.O. Ebselen and diphenyl diselenide against fungal pathogens: A systematic review. Med. Mycol. 2021, 59, 409–421. [Google Scholar] [CrossRef]
- Sies, H.; Parnham, M.J. Potential therapeutic use of ebselen for COVID-19 and other respiratory viral infections. Free Radic. Biol. Med. 2020, 156, 107–112. [Google Scholar] [CrossRef]
- Harrison, N.L.; Sachs, J.D. A call for an independent inquiry into the origin of the SARS-CoV-2 virus. Proc. Natl. Acad. Sci. USAS 2022, 119, e2202769119. [Google Scholar] [CrossRef] [PubMed]
- Sancineto, L.; Ostacolo, C.; Ortega-Alarcon, D.; Jimenez-Alesanco, A.; Ceballos-Laita, L.; Vega, S.; Abian, O.; Velazquez-Campoy, A.; Moretti, S.; Dabrowska, A. L-arginine improves solubility and ANTI SARS-CoV-2 Mpro activity of Rutin but not the antiviral activity in cells. Molecules 2021, 26, 6062. [Google Scholar] [PubMed]
- Xu, Z.; Yang, L.; Zhang, X.; Zhang, Q.; Yang, Z.; Liu, Y.; Wei, S.; Liu, W. Discovery of potential flavonoid inhibitors against COVID-19 3CL proteinase based on virtual screening strategy. Front. Mol. Biosci. 2020, 7, 556481. [Google Scholar]
- Amporndanai, K.; Meng, X.; Shang, W.; Jin, Z.; Rogers, M.; Zhao, Y.; Rao, Z.; Liu, Z.-J.; Yang, H.; Zhang, L. Inhibition mechanism of SARS-CoV-2 main protease by ebselen and its derivatives. Nat. Commun. 2021, 12, 3061. [Google Scholar] [CrossRef]
- La Monica, G.; Bono, A.; Lauria, A.; Martorana, A. Targeting SARS-CoV-2 main protease for treatment of COVID-19: Covalent inhibitors structure–activity relationship insights and evolution perspectives. J. Med. Chem. 2022, 65, 12500–12534. [Google Scholar]
- Shivakoti, R.; Gupte, N.; Yang, W.-T.; Mwelase, N.; Kanyama, C.; Tang, A.M.; Pillay, S.; Samaneka, W.; Riviere, C.; Berendes, S. Pre-antiretroviral therapy serum selenium concentrations predict WHO stages 3, 4 or death but not virologic failure post-antiretroviral therapy. Nutrients 2014, 6, 5061–5078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature 2020, 7811, 289–293. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.Y.; Liu, F.; Jiang, R.; Yang, X.; You, T.; Liu, X.; Xiao, C.Q.; Shi, Z.; Jiang, H.; Rao, Z. Structure of Mpro from COVID-19 virus and discovery of its inhibitors. Nature 2020, 582, 289–293. [Google Scholar]
- Li, Q.; Kang, C. Progress in developing inhibitors of SARS-CoV-2 3C-like protease. Microorganisms 2020, 8, 1250. [Google Scholar]
- Jin, Z.; Zhao, Y.; Sun, Y.; Zhang, B.; Wang, H.; Wu, Y.; Zhu, Y.; Zhu, C.; Hu, T.; Du, X. Structural basis for the inhibition of SARS-CoV-2 main protease by antineoplastic drug carmofur. Nat. Struct. Mol. Biol. 2020, 27, 529–532. [Google Scholar]
- Weglarz-Tomczak, E.; Tomczak, J.M.; Talma, M.; Burda-Grabowska, M.; Giurg, M.; Brul, S. Identification of ebselen and its analogues as potent covalent inhibitors of papain-like protease from SARS-CoV-2. Sci. Rep. 2021, 11, 3640. [Google Scholar] [CrossRef]
- Alotaibi, J.S.; Al-Faiyz, Y.S.; Shaaban, S. Design, Synthesis, and Biological Evaluation of Novel Hydroxamic Acid-Based Organoselenium Hybrids. Pharmaceuticals 2023, 16, 367–383. [Google Scholar] [CrossRef] [PubMed]
- Shaaban, S.; Abdou, A.; Alhamzani, A.G.; Abou-Krisha, M.M.; Al-Qudah, M.A.; Alaasar, M.; Youssef, I.; Yousef, T.A. Synthesis and in silico investigation of organoselenium-clubbed schiff bases as potential Mpro inhibitors for the SARS-CoV-2 replication. Life 2023, 13, 912–939. [Google Scholar] [CrossRef] [PubMed]
- Menéndez, C.A.; Byléhn, F.; Perez-Lemus, G.R.; Alvarado, W.; de Pablo, J.J. Molecular characterization of ebselen binding activity to SARS-CoV-2 main protease. Sci. Adv. 2020, 6, eabd0345. [Google Scholar] [CrossRef]
- Alaasar, M. Unexpected Kinetically Controlled Organoselenium-Based Isomaleimide: X-Ray Structure, Hirshfeld Surface Analy-sis, 3D Energy Framework Approach, and DFT Calcula-tion. Front. Chem. 2022, 10, 879. [Google Scholar]
- Haval, K.P.; Mhaske, S.B.; Argade, N.P. Cyanuric chloride: Decent dehydrating agent for an exclusive and efficient synthesis of kinetically controlled isomaleimides. Tetrahedron 2006, 62, 937–942. [Google Scholar] [CrossRef]
- Tonglairoum, P.; Brannigan, R.P.; Opanasopit, P.; Khutoryanskiy, V.V. Maleimide-bearing nanogels as novel mucoadhesive materials for drug delivery. J. Mater. Chem. B 2016, 4, 6581–6587. [Google Scholar] [CrossRef] [Green Version]
- Viveki, A.B.; Pol, M.D.; Halder, P.; Sonavane, S.R.; Mhaske, S.B. Annulation of Enals with Carbamoylpropiolates via NHC-Catalyzed Enolate Pathway: Access to Functionalized Maleimides/Iso-maleimides and Synthesis of Aspergillus FH-X-213. J. Org. Chem. 2021, 86, 9466–9477. [Google Scholar] [CrossRef] [PubMed]
- Ravasco, J.M.; Faustino, H.; Trindade, A.; Gois, P.M. Bioconjugation with maleimides: A useful tool for chemical biology. Chem. –A Eur. J. 2019, 25, 43–59. [Google Scholar] [CrossRef] [PubMed]
- Shaaban, S.; Vervandier-Fasseur, D.; Andreoletti, P.; Zarrouk, A.; Richard, P.; Negm, A.; Manolikakes, G.; Jacob, C.; Cherkaoui-Malki, M. Cytoprotective and antioxidant properties of organic selenides for the myelin-forming cells, oligodendrocytes. Bioorganic Chem. 2018, 80, 43–56. [Google Scholar]
- Al-Abdallah, B.; Al-Faiyz, Y.S.; Shaaban, S. Organoselenocyanates Tethered Methyl Anthranilate Hybrids with Promising Anticancer, Antimicrobial, and Antioxidant Activities. Inorganics 2022, 10, 246. [Google Scholar] [CrossRef]
- Zhang, L.; Hao, G.F.; Tan, Y.; Xi, Z.; Huang, M.Z.; Yang, G.F. Bioactive conformation analysis of cyclic imides as protoporphyrinogen oxidase inhibitor by combining DFT calculations, QSAR and molecular dynamic simulations. Bioorganic Med. Chem. 2009, 17, 4935–4942. [Google Scholar] [CrossRef] [PubMed]
- Hossain, M.; Khushy, K.; Latif, M.; Hossen, M.F.; Asraf, M.A.; Kudrat-E-Zahan, M.; Abdou, A. Co (II), Ni (II), and Cu (II) Complexes Containing Isatin-Based Schiff Base Ligand: Synthesis, Physicochemical Characterization, DFT Calculations, Antibacterial Activity, and Molecular Docking Analysis. Russ. J. General. Chem. 2022, 92, 2723–2733. [Google Scholar]
- Pokharia, S.; Joshi, R.; Pokharia, M.; Yadav, S.K.; Mishra, H. A density functional theory insight into the structure and reactivity of diphenyltin (IV) derivative of glycylphenylalanine. Main. Group. Metal. Chem. 2016, 39, 77–86. [Google Scholar] [CrossRef]
- Singh, M.B.; Jain, P.; Tomar, J.; Kumar, V.; Bahadur, I.; Arya, D.K.; Singh, P. An In Silico investigation for acyclovir and its derivatives to fight the COVID-19: Molecular docking, DFT calculations, ADME and td-Molecular dynamics simulations. J. Indian. Chem. Soc. 2022, 99, 100433. [Google Scholar]
- Fukui, K.; Yonezawa, T.; Shingu, H. A molecular orbital theory of reactivity in aromatic hydrocarbons. J. Chem. Phys. 1952, 20, 722–725. [Google Scholar] [CrossRef]
- Fukui, K. Role of frontier orbitals in chemical reactions. science 1982, 218, 747–754. [Google Scholar] [CrossRef] [Green Version]
- Houchi, S.; Messasma, Z. Exploring the inhibitory potential of Saussurea costus and Saussurea involucrata phytoconstituents against the Spike glycoprotein receptor binding domain of SARS-CoV-2 Delta (B. 1.617. 2) variant and the main protease (Mpro) as therapeutic candidates, using Molecular docking, DFT, and ADME/Tox studies. J. Mol. Struct. 2022, 1263, 133032. [Google Scholar] [PubMed]
- Mohapatra, R.K.; Perekhoda, L.; Azam, M.; Suleiman, M.; Sarangi, A.K.; Semenets, A.; Pintilie, L.; Al-Resayes, S.I. Computational investigations of three main drugs and their comparison with synthesized compounds as potent inhibitors of SARS-CoV-2 main protease (Mpro): DFT, QSAR, molecular docking, and in silico toxicity analysis. J. King Saud. Univ. -Sci. 2021, 33, 101315. [Google Scholar]
- Ghazy, A.; Hemeda, O.; Al-Hossainy, A.; Ghazy, R.; Henaish, A. Docking of COVID-19 Main Protease and TD-DFT/DMOl3 Simulated method, Synthesis, and Characterization with hybrid nanocomposite thin films and its applications. Surf. Interfaces 2023, 37, 102722. [Google Scholar] [CrossRef]
- Sagaama, A.; Brandan, S.A.; Issa, T.B.; Issaoui, N. Searching potential antiviral candidates for the treatment of the 2019 novel coronavirus based on DFT calculations and molecular docking. Heliyon 2020, 6, e04640. [Google Scholar] [CrossRef]
- Guidara, S.; Ahmed, A.B.; Abid, Y.; Feki, H. Molecular structure, vibrational spectra and nonlinear optical properties of 2, 5-dimethylanilinium chloride monohydrate: A density functional theory approach. Spectrochim. Acta Part. A Mol. Biomol. Spectrosc. 2014, 127, 275–285. [Google Scholar]
- Liu, C.; Luo, Y.; Lu, X.-B. DFT Studies on the Origin of Regioselective Ring-opening of Terminal Epoxides during Copolymerization with CO2. Chin. J. Polym. Sci. 2016, 34, 439–445. [Google Scholar]
- Reed, A.E.; Curtiss, L.A.; Weinhold, F. Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem. Rev. 1988, 88, 899–926. [Google Scholar]
- Al-Wabli, R.I.; Resmi, K.; Mary, Y.S.; Panicker, C.Y.; Attia, M.I.; El-Emam, A.A.; Van Alsenoy, C. Vibrational spectroscopic studies, Fukui functions, HOMO-LUMO, NLO, NBO analysis and molecular docking study of (E)-1-(1, 3-benzodioxol-5-yl)-4, 4-dimethylpent-1-en-3-one, a potential precursor to bioactive agents. J. Mol. Struct. 2016, 1123, 375–383. [Google Scholar]
- Chaudhary, A.P.; Bharti, S.K.; Kumar, S.; Ved, K.; Padam, K. Study of molecular structure, chemical reactivity and first hyperpolarizability of a newly synthesized N-(4-oxo-2-phenylquinazolin-3 (4H)-yl)-1H-indole-2-carboxamide using spectral analysis. J. Mol. Struct. 2017, 1148, 356–363. [Google Scholar]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2012, 64, 4–17. [Google Scholar] [CrossRef]
- Martin, Y.C. A bioavailability score. J. Med. Chem. 2005, 48, 3164–3170. [Google Scholar] [CrossRef]
- Al-Gaber, M.A.I.; Abd El-Lateef, H.M.; Khalaf, M.M.; Shaaban, S.; Shawky, M.; Mohamed, G.G.; Abdou, A.; Gouda, M.; Abu-Dief, A.M. Design, Synthesis, Spectroscopic Inspection, DFT and Molecular Docking Study of Metal Chelates Incorporating Azo Dye Ligand for Biological Evaluation. Materials 2023, 16, 897. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar]
- Bodnar, M.; Szczyglowska, M.; Konieczka, P.; Namiesnik, J. Methods of selenium supplementation: Bioavailability and determination of selenium compounds. Crit. Rev. Food Sci. Nutr. 2016, 56, 36–55. [Google Scholar]
- Levander, O.A.; Alfthan, G.; Arvilommi, H.; Gref, C.; Huttunen, J.; Kataja, M.; Koivistoinen, P.; Pikkarainen, J. Bioavailability of selenium to Finnish men as assessed by platelet glutathione peroxidase activity and other blood parameters. Am. J. Clin. Nutr. 1983, 37, 887–897. [Google Scholar] [CrossRef] [PubMed]
- Ali, J.; Camilleri, P.; Brown, M.B.; Hutt, A.J.; Kirton, S.B. Revisiting the general solubility equation: In silico prediction of aqueous solubility incorporating the effect of topographical polar surface area. J. Chem. Inf. Model. 2012, 52, 420–428. [Google Scholar] [PubMed]
- Shabaan, S.; Ba, L.A.; Abbas, M.; Burkholz, T.; Denkert, A.; Gohr, A.; Wessjohann, L.A.; Sasse, F.; Weber, W.; Jacob, C. Multicomponent reactions for the synthesis of multifunctional agents with activity against cancer cells. Chem. Comm. 2009, 31, 4702–4704. [Google Scholar] [CrossRef] [PubMed]
- Pires, D.E.; Blundell, T.L.; Ascher, D.B. pkCSM: Predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef] [PubMed]
- Dahlgren, D.; Lennernäs, H. Intestinal permeability and drug absorption: Predictive experimental, computational and in vivo approaches. Pharmaceutics 2019, 11, 411. [Google Scholar]
- Zanger, U.M.; Schwab, M. Pharmacology & Therapeutics Cytochrome P450 enzymes in drug metabolism: Regulation of gene expression, enzyme activities, and impact of genetic variation. Pharmacol. Ther. 2013, 138, 103–141. [Google Scholar] [PubMed]
- Horde, G.W.; Gupta, V. Drug Clearance; StatPearls Publishing: St. Petersburg, FL, USA, 2023. [Google Scholar] [PubMed]
- Han, Y.; Zhang, J.; Hu, C.; Zhang, X.; Ma, B.; Zhang, P. In silico ADME and toxicity prediction of ceftazidime and its impurities. Front Pharmacol. 2019, 10, 434. [Google Scholar] [CrossRef]
- Arafath, M.A.; Adam, F.; Ahamed, M.B.K.; Karim, M.R.; Uddin, M.N.; Yamin, B.M.; Abdou, A. Ni (II), Pd (II) and Pt (II) complexes with SNO-group thiosemicarbazone and DMSO: Synthesis, characterization, DFT, molecular docking and cytotoxicity. J. Mol. Struct. 2023, 1278, 134887. [Google Scholar]
- Nagasundaram, N.; Padmasree, K.; Santhosh, S.; Vinoth, N.; Sedhu, N.; Lalitha, A. Ultrasound promoted synthesis of new azo fused dihydropyrano [2, 3-c] pyrazole derivatives: In vitro antimicrobial, anticancer, DFT, in silico ADMET and Molecular docking studies. J. Mol. Struct. 2022, 1263, 133091. [Google Scholar]
- Elagawany, M.; Elmaaty, A.A.; Mostafa, A.; Abo Shama, N.M.; Santali, E.Y.; Elgendy, B.; Al-Karmalawy, A.A. Ligand-based design, synthesis, computational insights, and in vitro studies of novel N-(5-Nitrothiazol-2-yl)-carboxamido derivatives as potent inhibitors of SARS-CoV-2 main protease. J. Enzym. Inhib. Med. Chem. 2022, 37, 2112–2132. [Google Scholar]
- Khalid, Z.; Khan, A.T.-A.; Alnajjar, R.; Santali, E.; Shakoori, A.R. In silico mutational analysis of ACE2 to check the susceptibility of lung cancer patients towards COVID-19. Sci. Rep. 2022, 12, 7798. [Google Scholar]
- Sak, M.; Al-Faiyz, Y.S.; Elsawy, H.; Shaaban, S. Novel organoselenium redox modulators with potential anticancer, antimicrobial, and antioxidant activities. Antioxidants 2022, 11, 1231. [Google Scholar] [PubMed]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098. [Google Scholar]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785. [Google Scholar]
- Raghavachari, K. Perspective on “Density functional thermochemistry. III. The role of exact exchange” Becke AD (1993) J Chem Phys 98: 5648–52. Theor. Chem. Acc. 2000, 103, 361–363. [Google Scholar]
- Hay, P.J.; Wadt, W.R. Ab initio effective core potentials for molecular calculations. Potentials for the transition metal atoms Sc to Hg. J. Chem. Phys. 1985, 82, 270–283. [Google Scholar]
- McLean, A.; Chandler, G. Contracted Gaussian basis sets for molecular calculations. I. Second row atoms, Z= 11–18. J. Chem. Phys. 1980, 72, 5639–5648. [Google Scholar]
- Krishnan, R.; Binkley, J.S.; Seeger, R.; Pople, J.A. Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions. J. Chem. Phys. 1980, 72, 650–654. [Google Scholar]
- Tomberg, A. Gaussian 09w tutorial. Introd. Comput. Chem. Using G09W Avogadro Softw. 2013, 1–36. [Google Scholar]
- Abbas, G.; Irfan, A.; Ahmed, I.; Al-Zeidaneen, F.K.; Muthu, S.; Fuhr, O.; Thomas, R. Synthesis and investigation of anti-COVID19 ability of ferrocene Schiff base derivatives by quantum chemical and molecular docking. J. Mol. Struct. 2022, 1253, 132242. [Google Scholar] [CrossRef] [PubMed]
- Oyeneyin, O.E.; Ojo, N.D.; Ipinloju, N.; James, A.C.; Agbaffa, E.B. Investigation of corrosion inhibition potentials of some aminopyridine schiff bases using density functional theory and Monte Carlo simulation. Chem. Afr. 2022, 5, 319–332. [Google Scholar] [CrossRef]
- Qiao, Z.; Wei, N.; Jin, L.; Zhang, H.; Luo, J.; Zhang, Y.; Wang, K. The Mpro structure-based modifications of ebselen derivatives for improved antiviral activity against SARS-CoV-2 virus. Bioorganic Chem. 2021, 117, 105455. [Google Scholar] [CrossRef]
- Scholz, C.; Knorr, S.; Hamacher, K.; Schmidt, B. DOCKTITE—A Highly Versatile Step-by-Step Workflow for Covalent Docking and Virtual Screening in the Molecular Operating Environment. J. Chem. Inf. Model. 2015, 55, 398–406. [Google Scholar] [CrossRef] [PubMed]
- Vilar, S.; Cozza, G.; Moro, S. Medicinal chemistry and the molecular operating environment (MOE): Application of QSAR and molecular docking to drug discovery. Curr. Top. Med. Chem. 2008, 8, 1555–1572. [Google Scholar] [PubMed]
- Yunta, M.J. Docking and ligand binding affinity: Uses and pitfalls. Am. J. Model. Optim. 2016, 4, 74–114. [Google Scholar]
- Antonopoulou, I.; Sapountzaki, E.; Rova, U.; Christakopoulos, P. Inhibition of the main protease of SARS-CoV-2 (Mpro) by repurposing/designing drug-like substances and utilizing nature’s toolbox of bioactive compounds. Comput. Struct. Biotechnol. J. 2022, 20, 1306–1344. [Google Scholar] [CrossRef]
- Alghuwainem, Y.A.; El-Lateef, H.M.A.; Khalaf, M.M.; Amer, A.A.; Abdelhamid, A.A.; Alzharani, A.A.; Alfarsi, A.; Shaaban, S.; Gouda, M.; Abdou, A. Synthesis, DFT, biological and molecular docking analysis of novel manganese (II), Iron (III), Cobalt (II), Nickel (II), and Copper (II) Chelate Complexes Ligated by 1-(4-Nitrophenylazo)-2-naphthol. Int. J. Mol. Sci. 2022, 23, 15614. [Google Scholar]
- Azzam, K.A. SwissADME and pkCSM webservers predictors: An integrated online platform for accurate and comprehensive predictions for in silico ADME/T properties of artemisinin and its derivatives. Kompleks. Ispolz. Miner. Syra Complex. Use Miner. Resour. 2023, 325, 14–21. [Google Scholar]
- Zoete, V.; Daina, A.; Bovigny, C.; Michielin, O. SwissSimilarity: A web tool for low to ultra high throughput ligand-based virtual screening. J. Chem. Inf. Model 2016, 56, 1399–1404. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Shi, S.; Wang, Q.; Liu, S.; Qu, Z.; Li, K.; Geng, X.; Wang, T.; Gao, J. Characterization the performances of twofold resveratrol integrated compounds in binding with SIRT1 by molecular dynamics simulation and molecular mechanics/generalized born surface area (MM/GBSA) calculation. Chem. Phys. 2021, 544, 111108. [Google Scholar] [CrossRef]
- El-Masry, R.M.; Al-Karmalawy, A.A.; Alnajjar, R.; Mahmoud, S.H.; Mostafa, A.; Kadry, H.H.; Abou-Seri, S.M.; Taher, A.T. Newly synthesized series of oxoindole–oxadiazole conjugates as potential anti-SARS-CoV-2 agents: In silico and in vitro studies. New J. Chem. 2022, 46, 5078–5090. [Google Scholar]
- Release, S. 3: Desmond Molecular Dynamics System, DE Shaw Research, Maestro-Desmond Interoperability Tools, Schrödinger, New York, NY, USA. 2017. Available online: https://www.schrodinger.com/products/desmond#block-3767 (accessed on 7 July 2023).
- Hammoud, M.M.; Khattab, M.; Abdel-Motaal, M.; Van der Eycken, J.; Alnajjar, R.; Abulkhair, H.; Al-Karmalawy, A.A. Synthesis, structural characterization, DFT calculations, molecular docking, and molecular dynamics simulations of a novel ferrocene derivative to unravel its potential antitumor activity. J. Biomol. Struct. Dyn. 2023, 41, 5199–5216. [Google Scholar]
- Ezz Eldin, R.R.; Saleh, M.A.; Alotaibi, M.H.; Alsuair, R.K.; Alzahrani, Y.A.; Alshehri, F.A.; Mohamed, A.F.; Hafez, S.M.; Althoqapy, A.A.; Khirala, S.K.; et al. Ligand-based design and synthesis of N′-Benzylidene-3,4-dimethoxybenzohydrazide derivatives as potential antimicrobial agents; evaluation by in vitro, in vivo, and in silico approaches with SAR studies. J. Enzym. Inhib. Med. Chem. 2022, 37, 1098–1119. [Google Scholar] [CrossRef] [PubMed]
- Abo Elmaaty, A.; Eldehna, W.M.; Khattab, M.; Kutkat, O.; Alnajjar, R.; El-Taweel, A.N.; Al-Rashood, S.T.; Abourehab, M.A.; Binjubair, F.A.; Saleh, M.A.; et al. Anticoagulants as Potential SARS-CoV-2 Mpro Inhibitors for COVID-19 Patients: In Vitro, Molecular Docking, Molecular Dynamics, DFT, and SAR Studies. Int. J. Mol. Sci. 2022, 23, 12235. [Google Scholar] [PubMed]




















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| EHOMO (eV) | ELUMO (eV) | ∆E (eV) | IP (eV) | EA (eV) | χ (eV) | CP (eV) | η (eV) | σ (eV–1) | ω (eV) | Nu (eV–1) | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 7 | –8.94 | –0.90 | 8.04 | 8.94 | 0.90 | 4.92 | –4.92 | 4.02 | 0.12 | 3.01 | 0.33 |
| 8 | –8.70 | –0.93 | 7.77 | 8.70 | 0.93 | 4.82 | –4.82 | 3.88 | 0.13 | 2.99 | 0.33 |
| 9 | –8.58 | –1.74 | 6.85 | 8.58 | 1.74 | 5.16 | –5.16 | 3.42 | 0.15 | 3.89 | 0.26 |
| 10 | –8.67 | –1.17 | 7.50 | 8.67 | 1.17 | 4.92 | –4.92 | 3.75 | 0.13 | 3.23 | 0.31 |
| 11 | –8.56 | –1.79 | 6.77 | 8.56 | 1.79 | 5.18 | –5.18 | 3.39 | 0.15 | 3.96 | 0.25 |
| 12 | –8.23 | –1.90 | 6.33 | 8.23 | 1.90 | 5.06 | –5.06 | 3.16 | 0.16 | 4.05 | 0.25 |
| 7bfb | Ligand | Receptor | Interaction | Distance | E (kcal/mol) | S (kcal/mol) | RMSD | Ki (µM) |
|---|---|---|---|---|---|---|---|---|
| 7 | O16 | CYS 44 | H-donor | 3.16 | −1.00 | −6.97 | 1.17 | 7.85 |
| O19 | MET 165 | H-donor | 3.42 | −3.20 | ||||
| O9 | 9JT 402 | H-acceptor | 3.93 | −0.70 | ||||
| 8 | C10 | 9JT 402 | H-donor | 4.04 | −0.60 | −6.84 | 1.56 | 9.77 |
| O16 | CYS 44 | H-donor | 3.37 | −0.90 | ||||
| O19 | MET 165 | H-donor | 3.05 | −3.80 | ||||
| O20 | MET 165 | H-donor | 3.31 | −0.20 | ||||
| O9 | 9JT 402 | H-acceptor | 3.46 | −0.70 | ||||
| 9 | O9 | CYS 44 | H-donor | 3.95 | −0.90 | −7.23 | 1.43 | 5.09 |
| SE11 | HIS 164 | H-donor | 3.37 | −0.90 | ||||
| 10 | O9 | CYS 44 | H-donor | 3.17 | −0.90 | −7.62 | 1.49 | 2.65 |
| SE11 | SER 46 | H-donor | 3.44 | −1.90 | ||||
| 11 | O22 | CYS 44 | H-donor | 3.31 | −0.80 | −8.20 | 1.36 | 0.99 |
| O16 | SER 46 | H-acceptor | 2.91 | −1.10 | ||||
| O20 | THR 24 | H-acceptor | 3.25 | −0.70 | ||||
| 6-ring | HIS 41 | pi–pi | 3.99 | −0.54 | ||||
| 12 | SE11 | SER 46 | H-donor | 3.25 | −0.70 | −8.49 | 1.08 | 0.61 |
| O16 | CYS 44 | H-donor | 3.33 | −1.50 | ||||
| O19 | MET 165 | H-donor | 3.16 | −4.10 | ||||
| O20 | MET 165 | H-donor | 3.30 | −0.20 | ||||
| O9 | 9JT 402 | H-acceptor | 3.65 | −0.90 | ||||
| C15 | HIS 41 | H–pi | 3.73 | −1.00 | ||||
| 6-ring | THR 24 | pi–H | 4.10 | −0.60 |
| Complex | ΔG Binding | Coulomb | Covalent | H-bond | Lipo | Bind Packing | Solv_GB | VdW | St. Dev. |
|---|---|---|---|---|---|---|---|---|---|
| 12 | −59.07 | 13.64 | −0.60 | −1.00 | −16.09 | −1.47 | −2.55 | −50.99 | 4.91 |
| Co | −47.53 | −5.46 | 1.70 | −0.45 | −15.27 | −1.27 | 11.56 | −38.34 | 4.10 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shaaban, S.; Al-Faiyz, Y.S.; Alsulaim, G.M.; Alaasar, M.; Amri, N.; Ba-Ghazal, H.; Al-Karmalawy, A.A.; Abdou, A. Synthesis of New Organoselenium-Based Succinanilic and Maleanilic Derivatives and In Silico Studies as Possible SARS-CoV-2 Main Protease Inhibitors. Inorganics 2023, 11, 321. https://doi.org/10.3390/inorganics11080321
Shaaban S, Al-Faiyz YS, Alsulaim GM, Alaasar M, Amri N, Ba-Ghazal H, Al-Karmalawy AA, Abdou A. Synthesis of New Organoselenium-Based Succinanilic and Maleanilic Derivatives and In Silico Studies as Possible SARS-CoV-2 Main Protease Inhibitors. Inorganics. 2023; 11(8):321. https://doi.org/10.3390/inorganics11080321
Chicago/Turabian StyleShaaban, Saad, Yasair S. Al-Faiyz, Ghayah M. Alsulaim, Mohamed Alaasar, Nasser Amri, Hussein Ba-Ghazal, Ahmed A. Al-Karmalawy, and Aly Abdou. 2023. "Synthesis of New Organoselenium-Based Succinanilic and Maleanilic Derivatives and In Silico Studies as Possible SARS-CoV-2 Main Protease Inhibitors" Inorganics 11, no. 8: 321. https://doi.org/10.3390/inorganics11080321