
Destabilization of the LiBH4–NaBH4 Eutectic Mixture through Pore Confinement for Hydrogen Storage
 , , and
, , and
Abstract
:1. Introduction
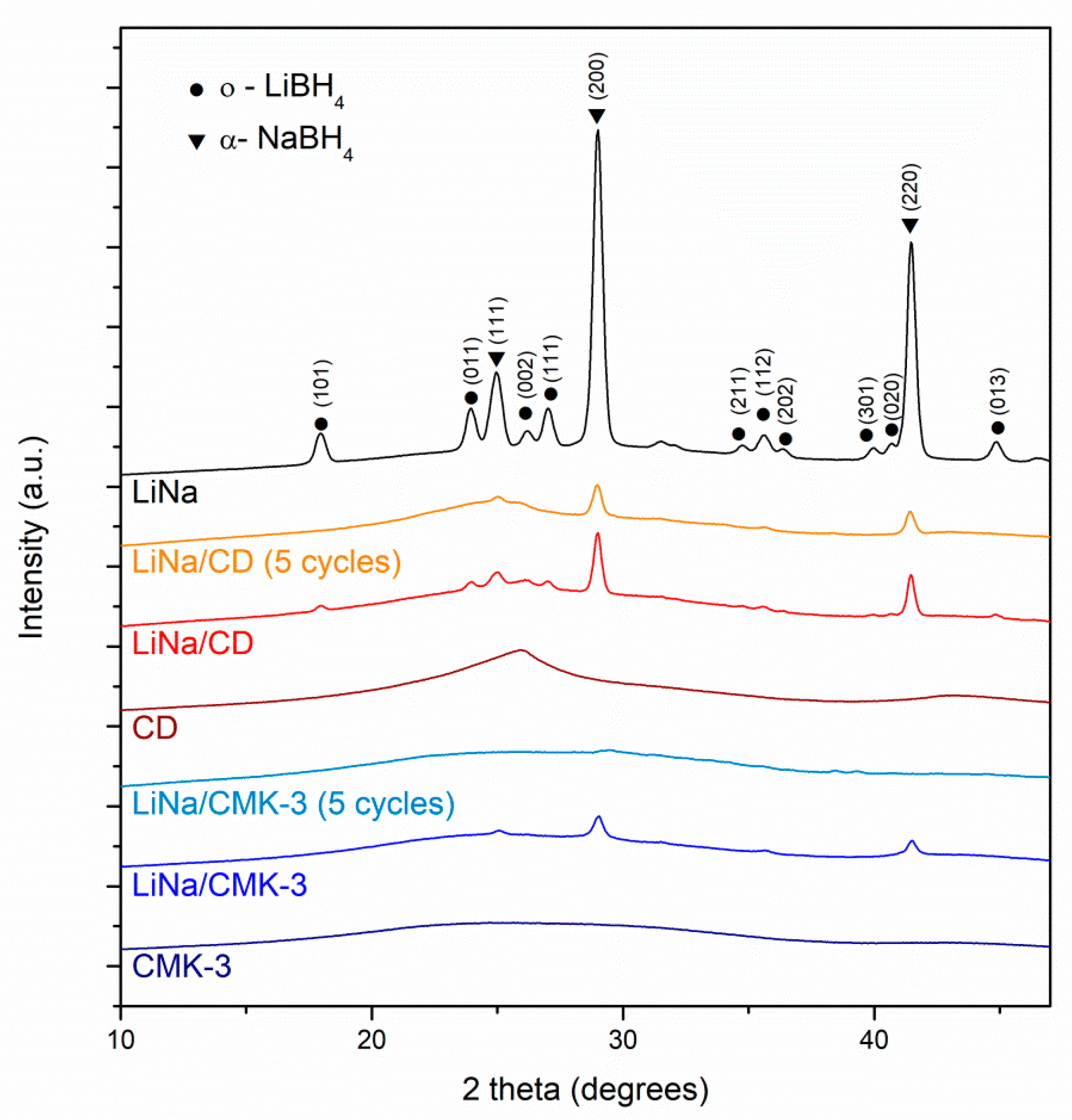
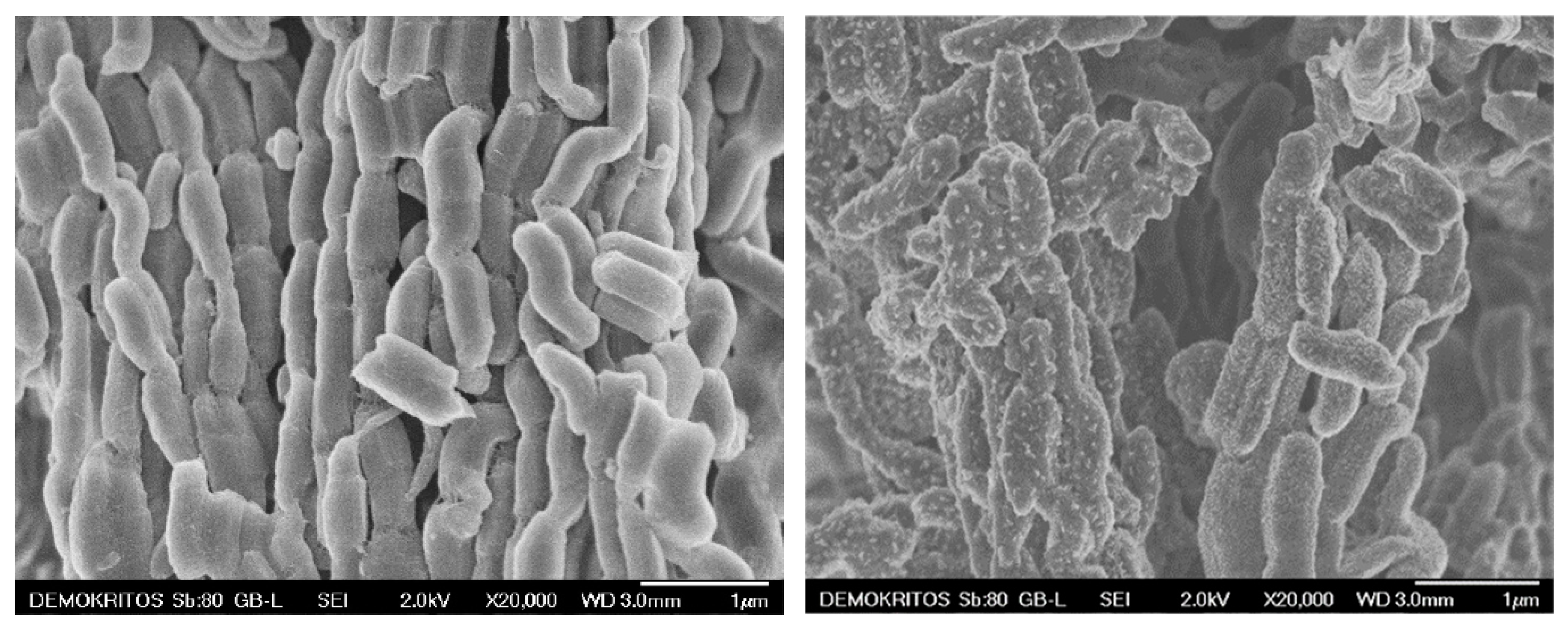
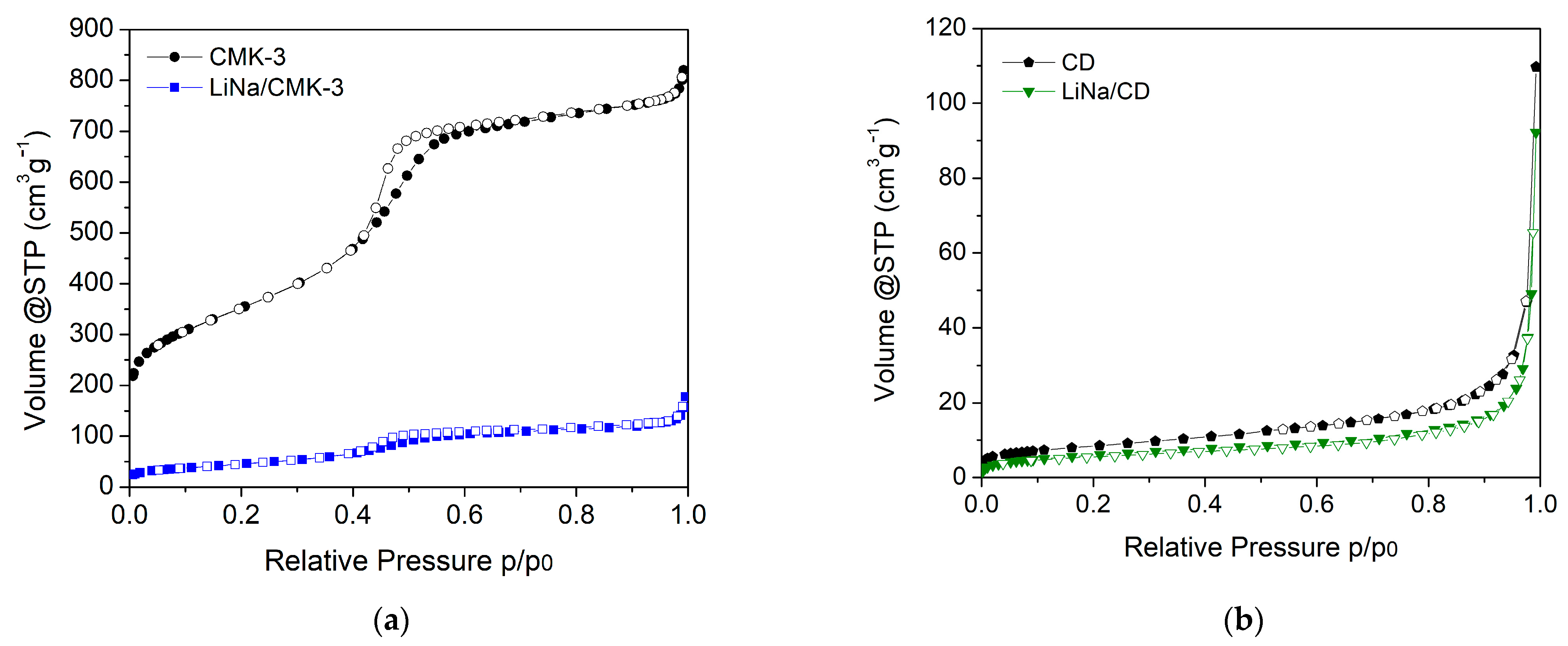
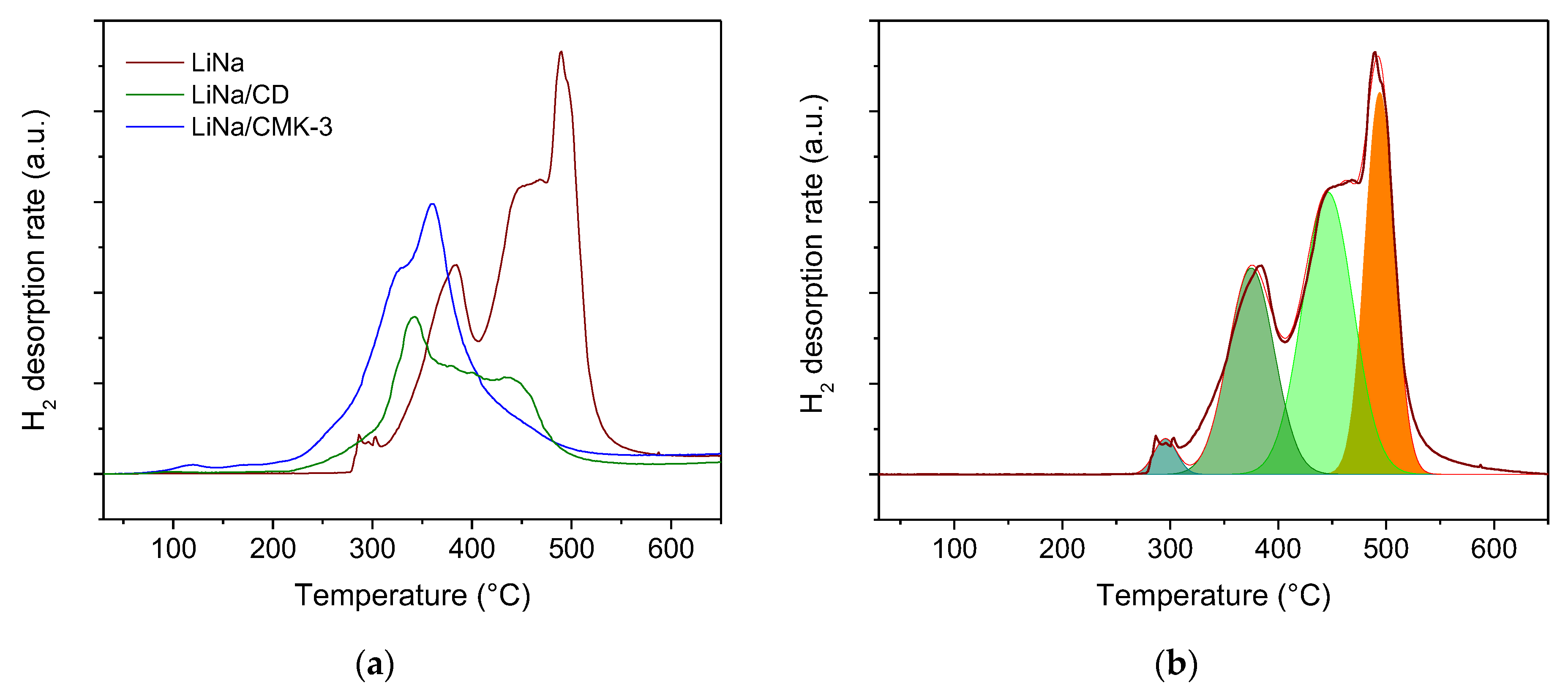
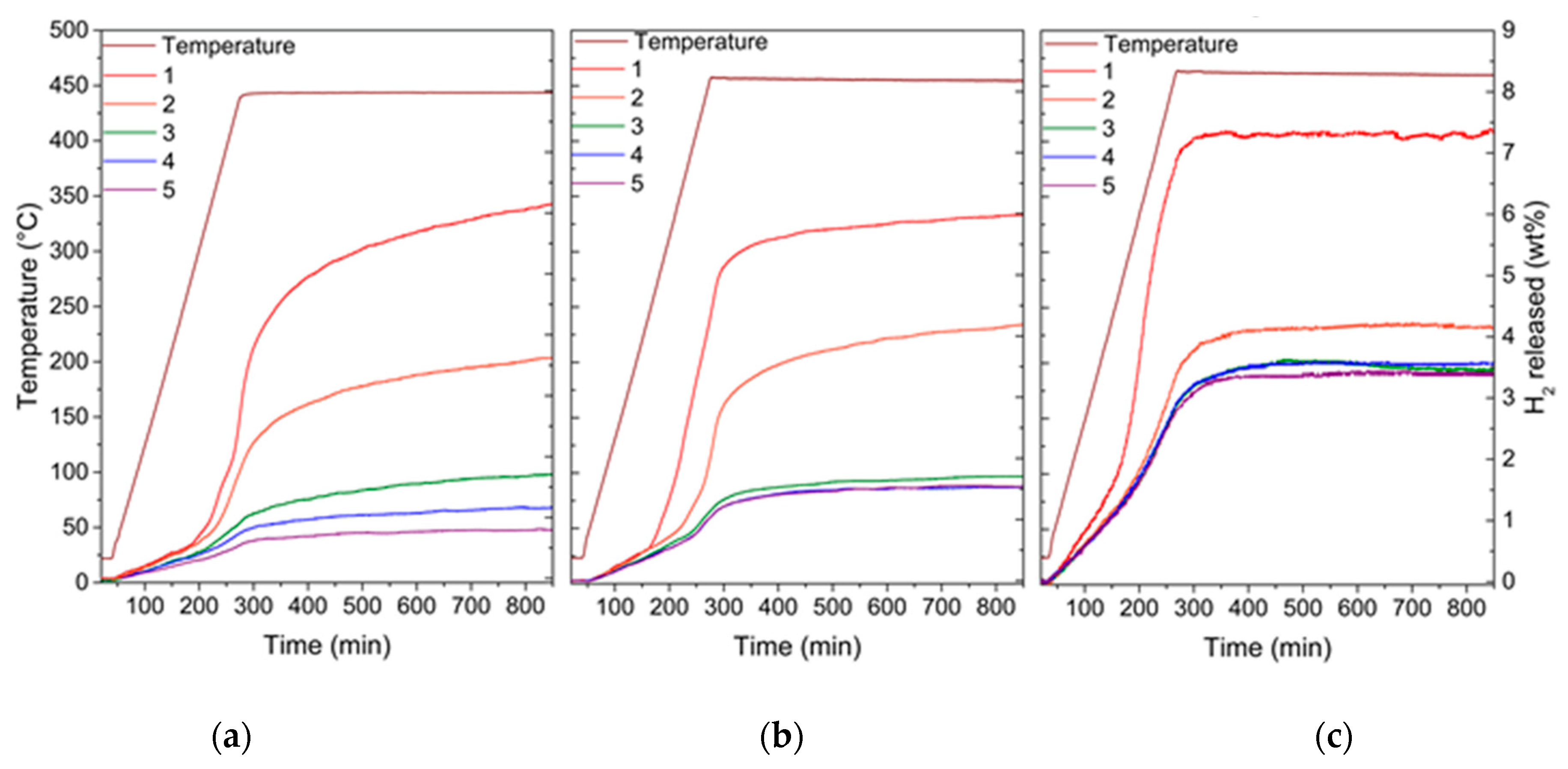
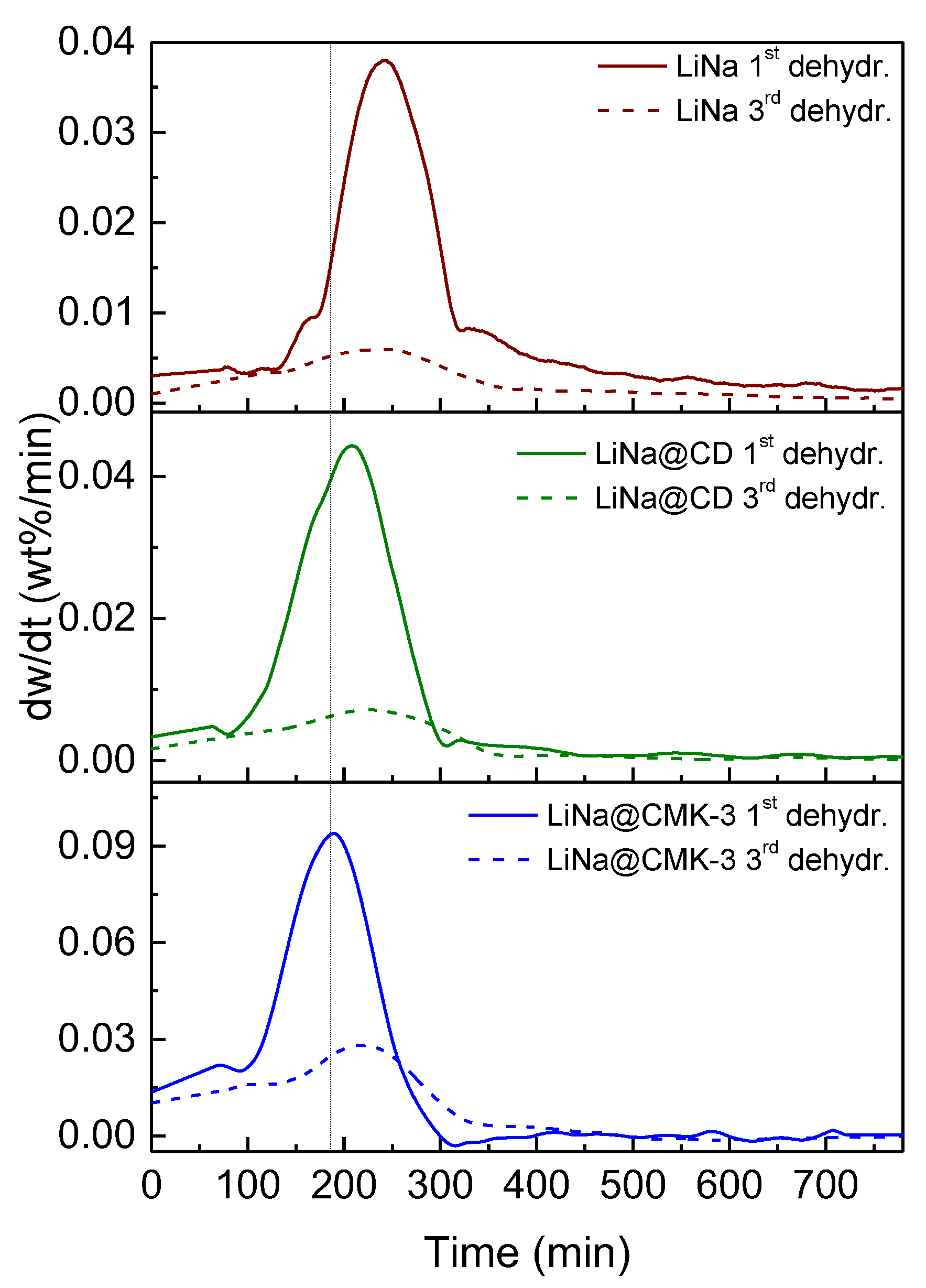
2. Results
3. Discussion
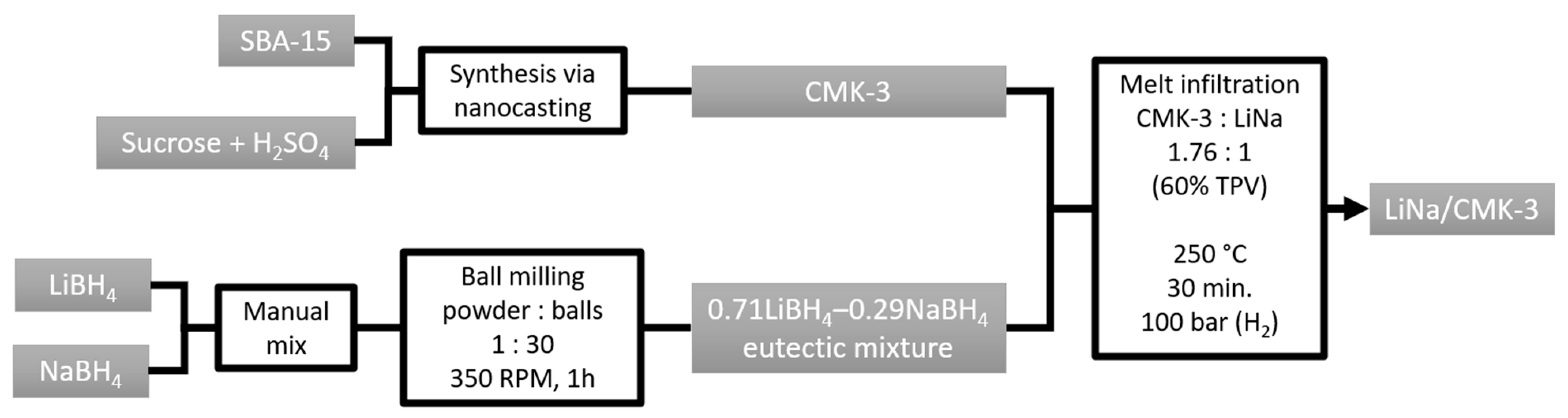
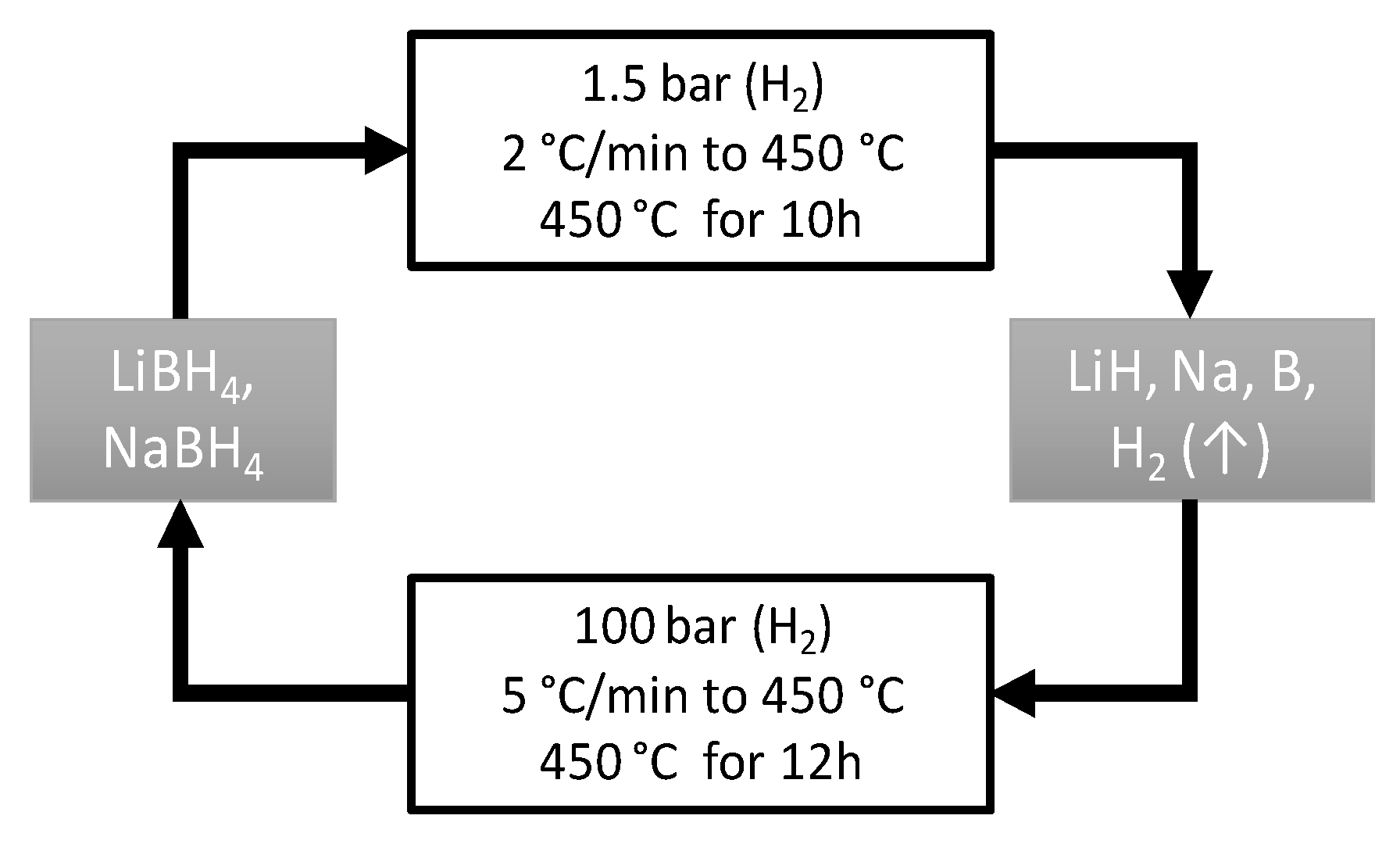
4. Materials and Methods
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Asghar, U.; Rafiq, S.; Anwar, A.; Iqbal, T.; Ahmed, A.; Jamil, F.; Khurram, M.S.; Akbar, M.M.; Farooq, A.; Shah, N.S.; et al. Review on the Progress in Emission Control Technologies for the Abatement of CO2, SOx and NOx from Fuel Combustion. J. Environ. Chem. Eng. 2021, 9, 106064. [Google Scholar] [CrossRef]
- Beringer, S.L. Energy, Climate Change and EU Development Policy. In EU Development Policies; Palgrave Macmillan: Cham, Switzerland, 2019; pp. 17–34. ISBN 9783030013073. [Google Scholar]
- Zhou, Y.; Li, R.; Lv, Z.; Liu, J.; Zhou, H.; Xu, C. Green Hydrogen: A Promising Way to the Carbon-Free Society. Chin. J. Chem. Eng. 2022, 43, 2–13. [Google Scholar] [CrossRef]
- Bongartz, D.; Dore’, L.; Eichler, K.; Grube, T.; Heuser, B.; Hombach, L.E.; Robinius, M.; Pischinger, S.; Stolten, D.; Walther, G. Comparison of Light-Duty Transportation Fuels Produced from Renewable Hydrogen and Green Carbon Dioxide. Appl. Energy 2018, 231, 757–767. [Google Scholar] [CrossRef]
- Abe, J.O.; Popoola, A.P.I.; Ajenifuja, E.; Popoola, O.M. Hydrogen Energy, Economy and Storage: Review and Recommendation. Int. J. Hydrogen Energy 2019, 44, 15072–15086. [Google Scholar] [CrossRef]
- Ball, M.; Weeda, M. The Hydrogen Economy - Vision or Reality? Int. J. Hydrogen Energy 2015, 40, 7903–7919. [Google Scholar] [CrossRef]
- Lai, Q.; Paskevicius, M.; Sheppard, D.A.; Buckley, C.E.; Thornton, A.W.; Hill, M.R.; Gu, Q.; Mao, J.; Huang, Z.; Liu, H.K.; et al. Hydrogen Storage Materials for Mobile and Stationary Applications: Current State of the Art. ChemSusChem 2015, 8, 2789–2825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hua, T.Q.; Roh, H.S.; Ahluwalia, R.K. Performance Assessment of 700-Bar Compressed Hydrogen Storage for Light Duty Fuel Cell Vehicles. Int. J. Hydrogen Energy 2017, 42, 25121–25129. [Google Scholar] [CrossRef]
- Zhang, J.; Yan, Y.; Zhang, C.; Xu, Z.; Li, X.; Zhao, G.; Ni, Z. Properties Improvement of Composite Layer of Cryo-Compressed Hydrogen Storage Vessel by Polyethylene Glycol Modified Epoxy Resin. Int. J. Hydrogen Energy 2022, 48, 5576–5594. [Google Scholar] [CrossRef]
- Zhao, X.; Yan, Y.; Zhang, J.; Zhang, F.; Wang, Z.; Ni, Z. Analysis of Multilayered Carbon Fiber Winding of Cryo-Compressed Hydrogen Storage Vessel. Int. J. Hydrogen Energy 2022, 47, 10934–10946. [Google Scholar] [CrossRef]
- Rivard, E.; Trudeau, M.; Zaghib, K. Hydrogen Storage for Mobility: A Review. Materials 2019, 12, 1973. [Google Scholar] [CrossRef] [Green Version]
- Balasooriya, W.; Clute, C.; Schrittesser, B.; Pinter, G. A Review on Applicability, Limitations, and Improvements of Polymeric Materials in High-Pressure Hydrogen Gas Atmospheres. Polym. Rev. 2022, 62, 175–209. [Google Scholar] [CrossRef]
- Grinderslev, J.B.; Amdisen, M.B.; Skov, L.N.; Møller, K.T.; Kristensen, L.G.; Polanski, M.; Heere, M.; Jensen, T.R. New Perspectives of Functional Metal Borohydrides. J. Alloys Compd. 2022, 896, 163014. [Google Scholar] [CrossRef]
- Schneemann, A.; White, J.L.; Kang, S.; Jeong, S.; Wan, L.F.; Cho, E.S.; Heo, T.W.; Prendergast, D.; Urban, J.; Wood, B.C.; et al. Nanostructured Metal Hydrides for Hydrogen Storage. Chem. Rev. 2018, 118, 10775–10839. [Google Scholar] [CrossRef]
- Hadjixenophontos, E.; Dematteis, E.M.; Berti, N.; Wołczyk, A.R.; Huen, P.; Brighi, M.; Le, T.T.; Santoru, A.; Payandeh, S.H.; Peru, F.; et al. A Review of the MSCA ITN ECOSTORE-Novel Complex Metal Hydrides for Efficient and Compact Storage of Renewable Energy as Hydrogen and Electricity. Inorganics 2020, 8, 17. [Google Scholar] [CrossRef] [Green Version]
- Hirscher, M.; Yartys, V.A.; Baricco, M.; Bellosta von Colbe, J.; Blanchard, D.; Bowman, R.C.J.; Broom, D.P.; Buckley, C.E.; Chang, F.; Chen, P.; et al. Materials for Hydrogen-Based Energy Storage—Past, Recent Progress and Future Outlook. J. Alloys Compd. 2020, 827, 153548. [Google Scholar] [CrossRef]
- Dematteis, E.M.; Amdisen, M.B.; Autrey, T.; Barale, J.; Bowden, M.E.; Buckley, C.E.; Cho, Y.W.; Deledda, S.; Dornheim, M.; De Jongh, P.; et al. Hydrogen Storage in Complex Hydrides: Past Activities and New Trends. Prog. Energy 2022, 4, 032009. [Google Scholar] [CrossRef]
- Eghbali, P.; Nişancl, B.; Metin, Ö. Graphene Hydrogel Supported Palladium Nanoparticles as an Efficient and Reusable Heterogeneous Catalysts in the Transfer Hydrogenation of Nitroarenes Using Ammonia Borane as a Hydrogen Source. Pure Appl. Chem. 2018, 90, 327–335. [Google Scholar] [CrossRef]
- Eghbali, P.; Gürbüz, M.U.; Ertürk, A.S.; Metin, Ö. In Situ Synthesis of Dendrimer-Encapsulated palladium(0) Nanoparticles as Catalysts for Hydrogen Production from the Methanolysis of Ammonia Borane. Int. J. Hydrogen Energy 2020, 45, 26274–26285. [Google Scholar] [CrossRef]
- Bellosta von Colbe, J.; Ares, J.R.; Barale, J.; Baricco, M.; Buckley, C.; Capurso, G.; Gallandat, N.; Grant, D.M.; Guzik, M.N.; Jacob, I.; et al. Application of Hydrides in Hydrogen Storage and Compression: Achievements, Outlook and Perspectives. Int. J. Hydrogen Energy 2019, 44, 7780–7808. [Google Scholar] [CrossRef]
- Manickam, K.; Mistry, P.; Walker, G.; Grant, D.; Buckley, C.E.; Humphries, T.D.; Paskevicius, M.; Jensen, T.; Albert, R.; Peinecke, K.; et al. Future Perspectives of Thermal Energy Storage with Metal Hydrides. Int. J. Hydrogen Energy 2019, 44, 7738–7745. [Google Scholar] [CrossRef]
- Mazzucco, A.; Dornheim, M.; Sloth, M.; Jensen, T.R.; Oluf, J.; Rokni, M. Bed Geometries, Fueling Strategies and Optimization of Heat Exchanger Designs in Metal Hydride Storage Systems for Automotive Applications: A Review. Int. J. Hydrogen Energy 2014, 39, 17054–17074. [Google Scholar] [CrossRef]
- Yartys, V.A.; Lototskyy, M.V.; Akiba, E.; Albert, R.; Antonov, V.E.; Ares, J.R.; Baricco, M.; Bourgeois, N.; Buckley, C.E.; Bellaosta von Colbe, J.M.; et al. Magnesium Based Materials for Hydrogen Based Energy Storage: Past, Present and Future. Int. J. Hydrogen Energy 2019, 44, 7809–7859. [Google Scholar] [CrossRef]
- Callini, E.; Aguey-Zinsou, K.F.; Ahuja, R.; Ares, J.R.; Bals, S.; Biliškov, N.; Chakraborty, S.; Charalambopoulou, G.; Chaudhary, A.L.; Cuevas, F.; et al. Nanostructured Materials for Solid-State Hydrogen Storage: A Review of the Achievement of COST Action MP1103. Int. J. Hydrogen Energy 2016, 41, 14404–14428. [Google Scholar] [CrossRef]
- Møller, K.T.; Jensen, T.R.; Akiba, E.; Li, H. wen Hydrogen - A Sustainable Energy Carrier. Prog. Nat. Sci. Mater. Int. 2017, 27, 34–40. [Google Scholar] [CrossRef]
- Puszkiel, J.; Gasnier, A.; Amica, G.; Gennari, F. Tuning LiBH4 for Hydrogen Storage: Destabilization, Additive, and Nanoconfinement Approaches. Molecules 2020, 25, 163. [Google Scholar] [CrossRef] [Green Version]
- Demirci, U.B.; Miele, P. Sodium Borohydride versus Ammonia Borane, in Hydrogen Storage and Direct Fuel Cell Applications. Energy Environ. Sci. 2009, 2, 627–637. [Google Scholar] [CrossRef]
- Møller, K.T.; Sheppard, D.; Ravnsbæk, D.B.; Buckley, C.E.; Akiba, E.; Li, H.; Jensen, T.R. Complex Metal Hydrides for Hydrogen, Thermal and Electrochemical Energy Storage. Energies 2017, 10, 1645. [Google Scholar] [CrossRef] [Green Version]
- Lai, Q.; Aguey-Zinsou, K.-F.; Lai, Q.; Aguey-Zinsou, K.-F. Borohydrides as Solid-State Hydrogen Storage Materials: Past, Current Approaches and Future Perspectives. Gen. Chem. 2018, 4, 180017. [Google Scholar] [CrossRef] [Green Version]
- Milanese, C.; Jensen, T.R.; Hauback, B.C.; Pistidda, C.; Dornheim, M.; Yang, H.; Lombardo, L.; Zuettel, A.; Filinchuk, Y.; Ngene, P.; et al. Complex Hydrides for Energy Storage. Int. J. Hydrogen Energy 2019, 44, 7860–7874. [Google Scholar] [CrossRef] [Green Version]
- Paskevicius, M.; Jepsen, L.H.; Schouwink, P.; Černý, R.; Ravnsbæk, D.B.; Filinchuk, Y.; Dornheim, M.; Besenbacher, F.; Jensen, T.R. Metal Borohydrides and Derivatives-Synthesis, Structure and Properties. Chem. Soc. Rev. 2017, 46, 1565–1634. [Google Scholar] [CrossRef]
- Callini, E.; Özlem Kocabas Atakli, Z.; Hauback, B.C.; Orimo, S.-I.; Jensen, C.; Dornheim, M.; Grant, D.; Cho, Y.W.; Chen, P.; Hjörvarsson, B.; et al. Complex and Liquid Hydrides for Energy Storage. Appl. Phys. A 2016, 122, 353. [Google Scholar] [CrossRef]
- Javadian, P.; Payandeh, S.G.; Sheppard, D.A.; Buckley, C.E.; Jensen, T.R. Reversibility of LiBH4 Facilitated by the LiBH4–Ca(BH4)2 Eutectic. J. Phys. Chem. C 2017, 121, 18439–18449. [Google Scholar] [CrossRef]
- Černý, R.; Murgia, F.; Brighi, M. Metal Hydroborates: From Hydrogen Stores to Solid Electrolytes. J. Alloys Compd. 2022, 895, 162659. [Google Scholar] [CrossRef]
- Paskevicius, M.; Ley, M.B.; Sheppard, D.A.; Jensen, T.R.; Buckley, C.E. Eutectic Melting in Metal Borohydrides. Phys. Chem. Chem. Phys. 2013, 15, 19774–19789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamori, Y.; Orimo, S.I. Destabilization of Li-Based Complex Hydrides. J. Alloys Compd. 2004, 370, 271–275. [Google Scholar] [CrossRef]
- Nickels, E.A.; Jones, M.O.; David, W.I.F.; Johnson, S.R.; Lowton, R.L.; Sommariva, M.; Edwards, P.P. Tuning the Decomposition Temperature in Complex Hydrides: Synthesis of a Mixed Alkali Metal Borohydride. Angew. Chemie Int. Ed. 2008, 47, 2817–2819. [Google Scholar] [CrossRef] [PubMed]
- Shao, J.; Xiao, X.; Fan, X.; Huang, X.; Zhai, B.; Li, S.; Ge, H.; Wang, Q.; Chen, L. Enhanced Hydrogen Storage Capacity and Reversibility of LiBH4 Nanoconfined in the Densified Zeolite-Templated Carbon with High Mechanical Stability. Nano Energy 2015, 15, 244–255. [Google Scholar] [CrossRef]
- Zhang, L.; Zheng, J.; Xiao, X.; Fan, X.; Huang, X.; Yang, X.; Chen, L. Enhanced Hydrogen Storage Properties of a Dual-Cation (Li+, Mg2+) Borohydride and Its Dehydrogenation Mechanism. RSC Adv. 2017, 7, 36852–36859. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Zheng, J.; Chen, L.; Xiao, X.; Qin, T.; Jiang, Y.; Li, S.; Ge, H.; Wang, Q. Remarkable Enhancement in Dehydrogenation Properties of Mg(BH4)2 Modified by the Synergetic Effect of Fluorographite and LiBH4. Int. J. Hydrogen Energy 2015, 40, 14163–14172. [Google Scholar] [CrossRef]
- El Kharbachi, A.; Pinatel, E.; Nuta, I.; Baricco, M. A Thermodynamic Assessment of LiBH4. Calphad Comput. Coupling Phase Diagrams Thermochem. 2012, 39, 80–90. [Google Scholar] [CrossRef] [Green Version]
- Urgnani, J.; Torres, F.J.; Palumbo, M.; Baricco, M. Hydrogen Release from Solid State NaBH4. Int. J. Hydrogen Energy 2008, 33, 3111–3115. [Google Scholar] [CrossRef]
- Dematteis, E.M.; Roedern, E.; Pinatel, E.R.; Corno, M.; Jensen, T.R.; Baricco, M. A Thermodynamic Investigation of the LiBH4-NaBH4 System. RSC Adv. 2016, 6, 60101–60108. [Google Scholar] [CrossRef] [Green Version]
- Plerdsranoy, P.; Kaewsuwan, D. Effects of Specific Surface Area and Pore Volume of Activated Carbon Nanofibers on Nanoconfinement and Dehydrogenation of LiBH4. Int. J. Hydrogen Energy 2017, 42, 6189–6201. [Google Scholar] [CrossRef]
- Sofianos, M.V.; Chaudhary, A.; Paskevicius, M.; Sheppard, D.A.; Humphries, T.D.; Dornheim, M.; Buckley, C.E. Hydrogen Storage Properties of Eutectic Metal Borohydrides Melt- Infiltrated into Porous Al Scaffolds. J. Alloys Compd. 2019, 775, 474–480. [Google Scholar] [CrossRef]
- Suryanarayana, C. Mechanical Alloying and Milling. Prog. Mater. Sci. 2001, 46, 1–184. [Google Scholar] [CrossRef]
- Le Caër, G.; Delcroix, P.; Bégin-Colin, S.; Ziller, T. High-Energy Ball-Milling of Alloys and Compounds. Hyperfine Interact. 2002, 141–142, 63–72. [Google Scholar] [CrossRef]
- Wu, C.; Cheng, H.M. Effects of Carbon on Hydrogen Storage Performances of Hydrides. J. Mater. Chem. 2010, 20, 5390–5400. [Google Scholar] [CrossRef]
- Javadian, P.; Sheppard, D.A.; Buckley, C.E.; Jensen, T.R. Hydrogen Storage Properties of Nanoconfined LiBH4 -NaBH4. Int. J. Hydrogen Energy 2015, 40, 14916–14924. [Google Scholar] [CrossRef] [Green Version]
- Adams, R.M. Preparation of Diborane. Adv. Chem. 1961, 32, 60–68. [Google Scholar] [CrossRef]
- Jun, S.; Joo, S.H.; Ryoo, R.; Kruk, M. Synthesis of New, Nanoporous Carbon with Hexagonally Ordered Mesostructure. J. Am. Chem. Soc. 2000, 122, 10712–10713. [Google Scholar] [CrossRef]
- Thommes, M.; Kaneko, K.; Neimark, A.V.; Olivier, J.P.; Rodriguez-Reinoso, F.; Rouquerol, J.; Sing, K.S.W. Physisorption of Gases, with Special Reference to the Evaluation of Surface Area and Pore Size Distribution (IUPAC Technical Report). Pure Appl. Chem. 2015, 87, 1051–1069. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Reed, D.; Paterakis, C.; Contreras, L.; Baricco, M.; Book, D. Study of the Decomposition of a 0.62LiBH4–0.38NaBH4 Mixture. Int. J. Hydrogen Energy 2017, 42, 22480–22488. [Google Scholar] [CrossRef]
- Orimo, S.; Nakamori, Y.; Eliseo, J.R.; Züttel, A.; Jensen, C.M. Complex Hydrides for Hydrogen Storage. Chem. Rev. 2007, 107, 4111–4132. [Google Scholar] [CrossRef] [PubMed]
- Zuttel, A.; Wenger, P.; Rentsch, S.; Sudan, P.; Mauron, P.; Emmenegger, C. LiBH4 a New Hydrogen Storage Material. J. Power Sources 2003, 118, 1–7. [Google Scholar] [CrossRef]
- Zuttel, A.; Rentsch, S.; Fischer, P.; Wenger, P.; Sudan, P.; Mauron, P.; Emmenegger, C. Hydrogen Storage Properties of LiBH4. J. Alloys Compd. 2003, 356-357, 515–520. [Google Scholar] [CrossRef]
- Humphries, T.D.; Kalantzopoulos, G.N.; Llamas-Jansa, I.; Olsen, J.E.; Hauback, B.C. Reversible Hydrogenation Studies of NaBH4 Milled with Ni-Containing Additives. J. Phys. Chem. C 2013, 117, 6060–6065. [Google Scholar] [CrossRef] [Green Version]
- Mao, J.; Gregory, D.H. Recent Advances in the Use of Sodium Borohydride as a Solid State Hydrogen Store. Energies 2015, 8, 430–453. [Google Scholar] [CrossRef]
- Orłowski, P.A.; Grochala, W. Effect of Vanadium Catalysts on Hydrogen Evolution from NaBH4. Solids 2022, 3, 21. [Google Scholar] [CrossRef]
- Mao, J.; Guo, Z.; Nevirkovets, I.P.; Liu, H.K.; Dou, S.X. Hydrogen De-/absorption Improvement of NaBH4 Catalyzed by Titanium-Based Additives. J. Phys. Chem. C 2012, 116, 1596–1604. [Google Scholar] [CrossRef] [Green Version]
- Orimo, S.I.; Nakamori, Y.; Ohba, N.; Miwa, K.; Aoki, M.; Towata, S.I.; Züttel, A. Experimental Studies on Intermediate Compound of LiBH4. Appl. Phys. Lett. 2006, 89, 87–90. [Google Scholar] [CrossRef]
- Reed, D.; Book, D. In-Situ Raman Study of the Thermal Decomposition of LiBH4. Mater. Res. Soc. Symp. Proc. 2010, 1216, 2–8. [Google Scholar] [CrossRef]
- Jensen, S.R.H.; Paskevicius, M.; Hansen, B.R.S.; Jakobsen, A.S.; Møller, K.T.; White, J.L.; Allendorf, M.D.; Stavila, V.; Skibsted, J.; Jensen, T.R. Hydrogenation Properties of Lithium and Sodium Hydride - Closo-Borate, [B10H10]2- and [B12H12]2-, Composites. Phys. Chem. Chem. Phys. 2018, 20, 16266–16275. [Google Scholar] [CrossRef] [Green Version]
- Ampoumogli, A.; Steriotis, T.; Trikalitis, P.; Giasafaki, D.; Bardaji, E.G.; Fichtner, M.; Charalambopoulou, G. Nanostructured Composites of Mesoporous Carbons and Boranates as Hydrogen Storage Materials. J. Alloys Compd. 2011, 509, S705–S708. [Google Scholar] [CrossRef]
- Nielsen, T.K.; Javadian, P.; Polanski, M.; Besenbacher, F.; Bystrzycki, J.; Jensen, T.R. Nanoconfined NaAlH4: Determination of Distinct Prolific Effects from Pore Size, Crystallite Size, and Surface Interactions. J. Phys. Chem. C 2012, 116, 21046–21051. [Google Scholar] [CrossRef]
- Peru, F.; Payandeh, S.; Charalambopoulou, G.; Jensen, T.R.; Steriotis, T. Hydrogen Sorption and Reversibility of the LiBH4—KBH4 Eutectic System Confined in a CMK-3 Type Carbon via Melt Infiltration. J. Carbon Res. 2020, 2, 19. [Google Scholar] [CrossRef] [Green Version]
- Ampoumogli, A.; Charalambopoulou, G.; Javadian, P.; Richter, B.; Jensen, T.R.; Steriotis, T. Hydrogen Desorption and Cycling Properties of Composites Based on Mesoporous Carbons and a LiBH4–Ca(BH4)2 Eutectic Mixture. J. Alloys Compd. 2015, 645, S480–S484. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SBET (m2 g−1) | TPV (cm3 g−1) | <d> (nm) | |
|---|---|---|---|
| CMK-3 | 1250 | 1.2 | 4.6 |
| LiNa/CMK-3 | 170 | 0.21 | 4.6 |
| CD | ~30 | - | - |
| LiNa/CD | ~25 | - | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peru, F.; Payandeh, S.; Jensen, T.R.; Charalambopoulou, G.; Steriotis, T. Destabilization of the LiBH4–NaBH4 Eutectic Mixture through Pore Confinement for Hydrogen Storage. Inorganics 2023, 11, 128. https://doi.org/10.3390/inorganics11030128
Peru F, Payandeh S, Jensen TR, Charalambopoulou G, Steriotis T. Destabilization of the LiBH4–NaBH4 Eutectic Mixture through Pore Confinement for Hydrogen Storage. Inorganics. 2023; 11(3):128. https://doi.org/10.3390/inorganics11030128
Chicago/Turabian StylePeru, Filippo, Seyedhosein Payandeh, Torben R. Jensen, Georgia Charalambopoulou, and Theodore Steriotis. 2023. "Destabilization of the LiBH4–NaBH4 Eutectic Mixture through Pore Confinement for Hydrogen Storage" Inorganics 11, no. 3: 128. https://doi.org/10.3390/inorganics11030128