

Hydrogen Evolution Reaction, Electrochemical CO2 Reduction, and Oxidative Photodegradation of Organic Dyes Catalyzed by Co(II) Trimethoxy-Meso-Arylporphyrin
 , and
, and
Abstract
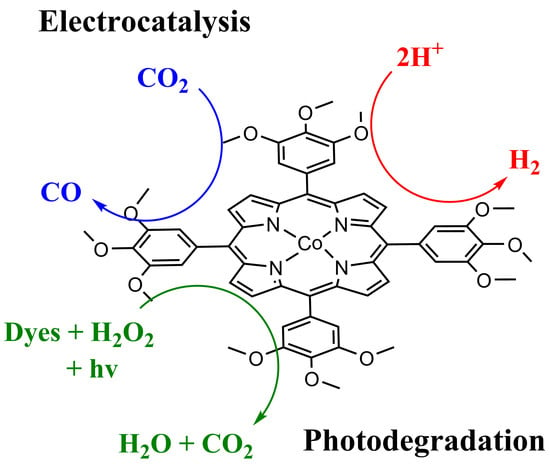
:
1. Introduction
2. Results and Discussion
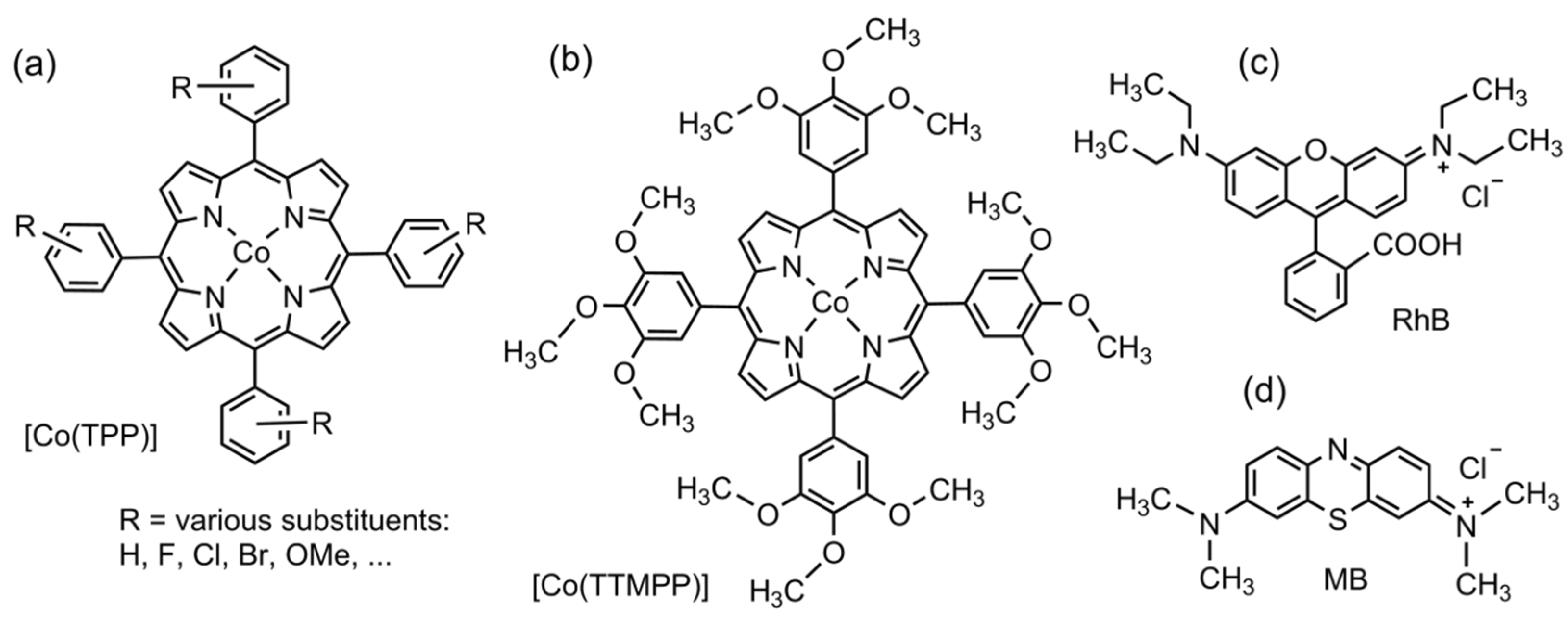
2.1. Synthesis and Analysis
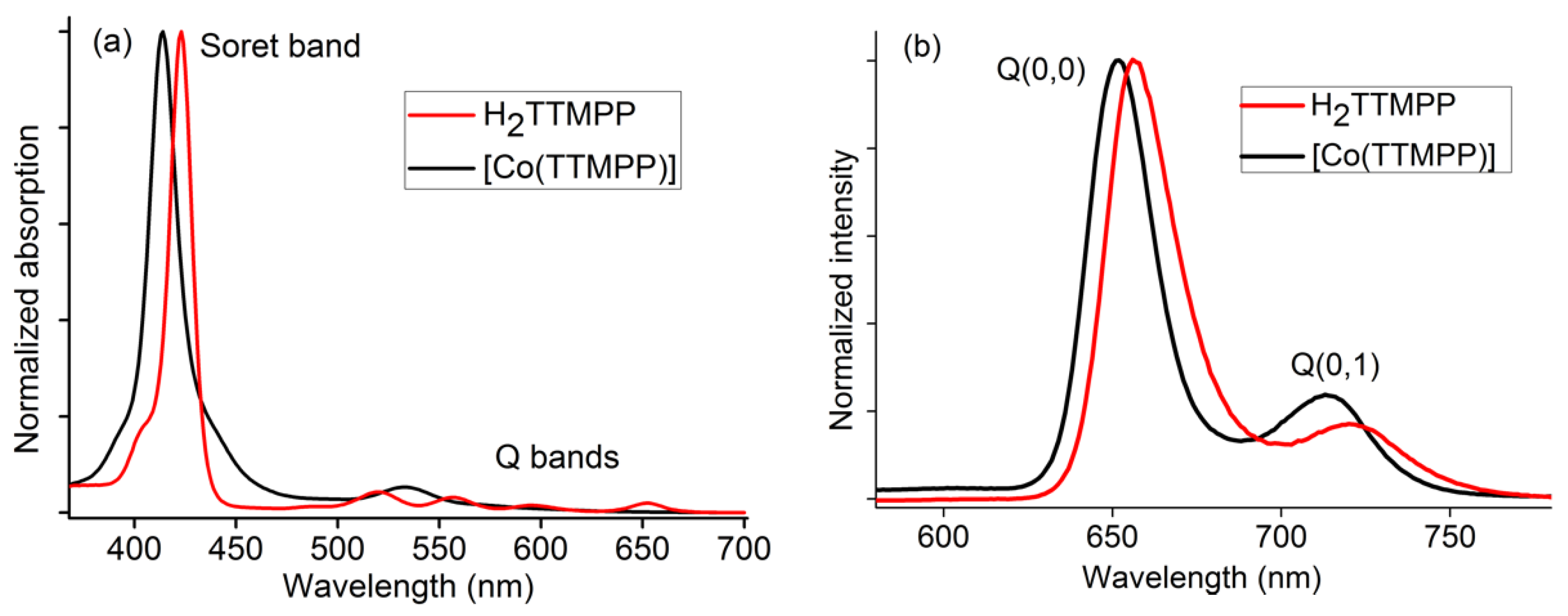
2.2. Photophysical Properties
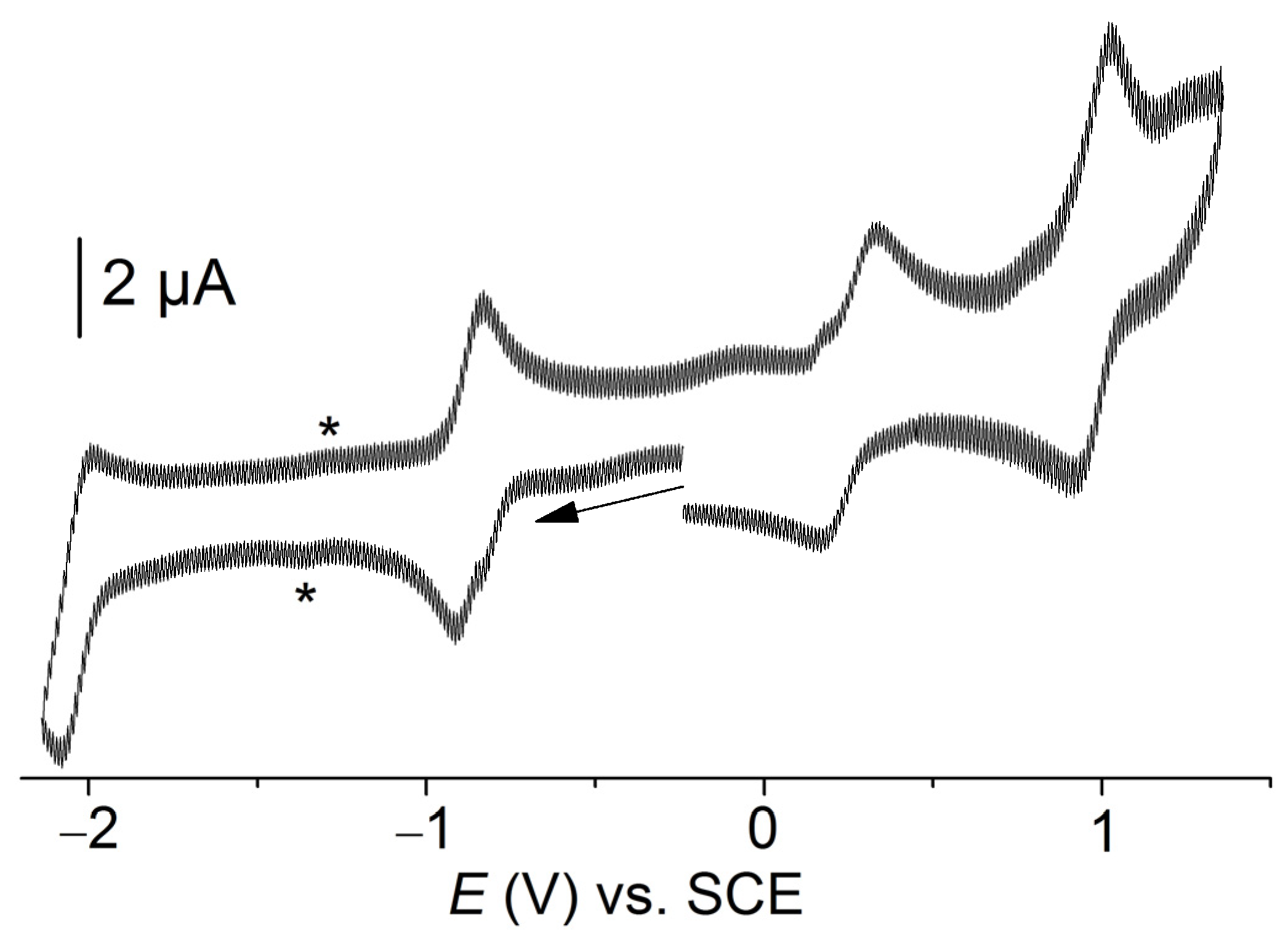
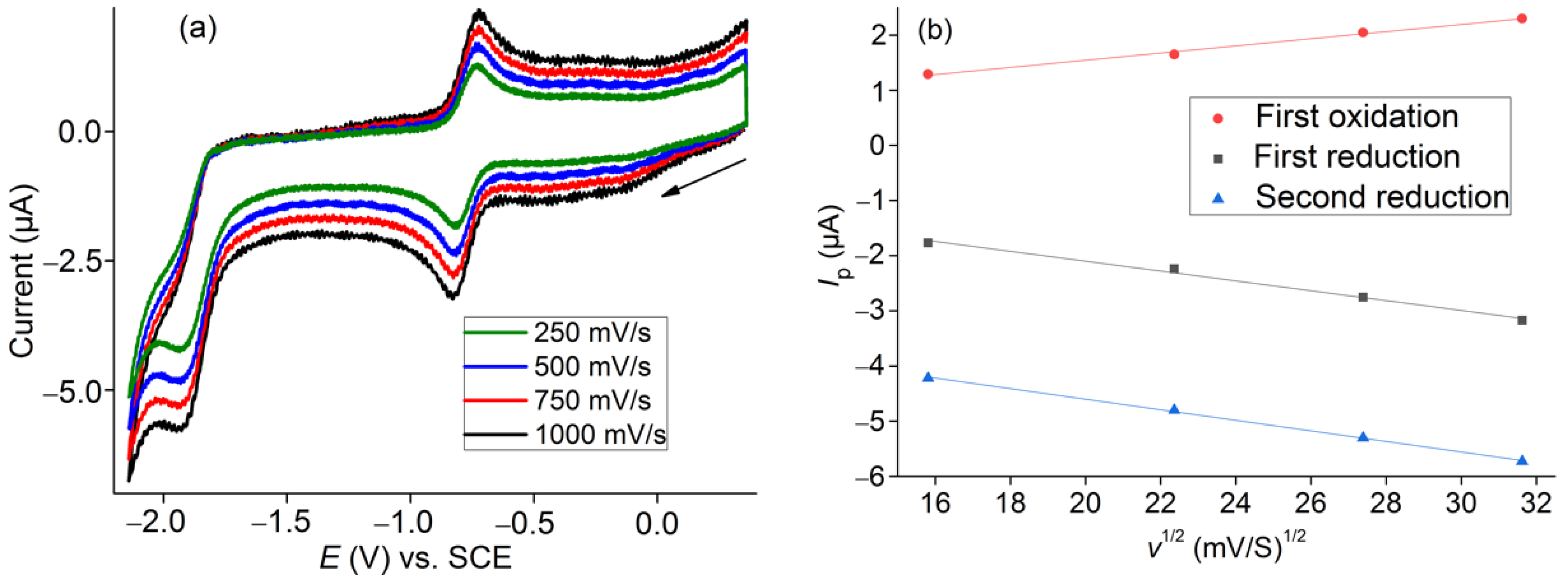
2.3. Electrochemical Characterization of [Co(TTMPP)]
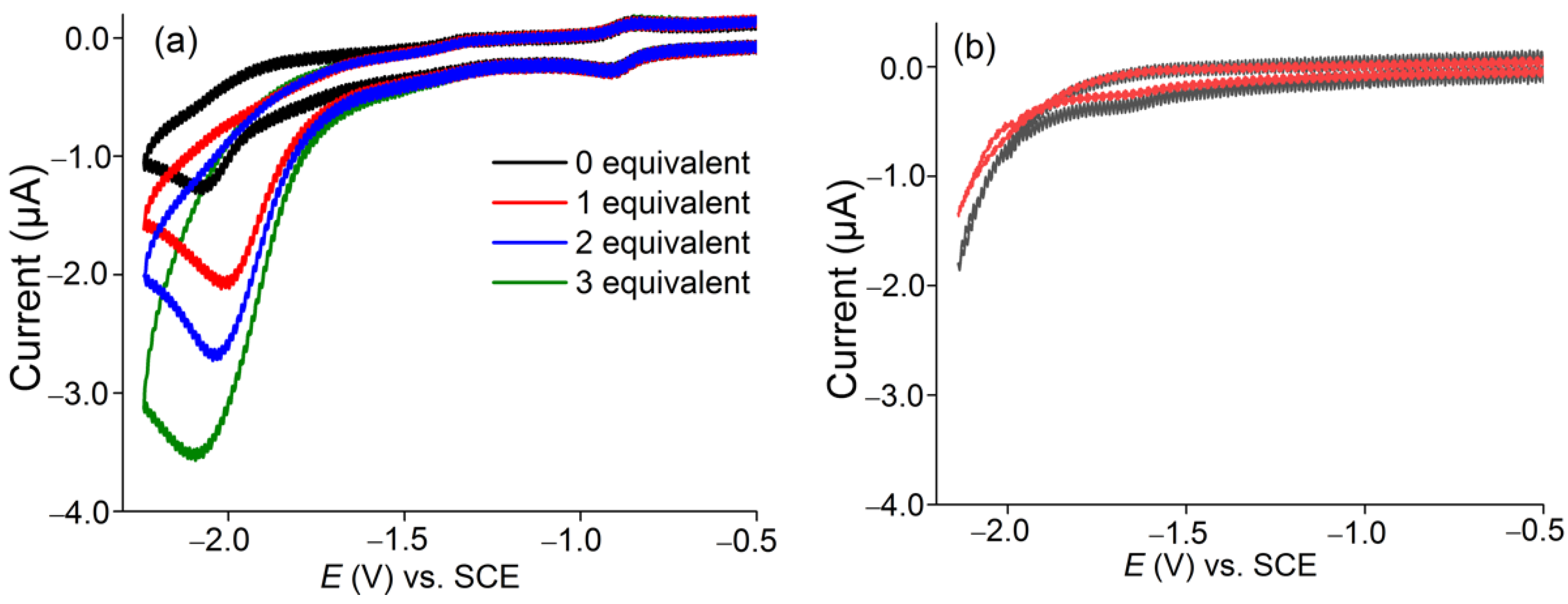
2.4. Electrocatalytic H2 Production
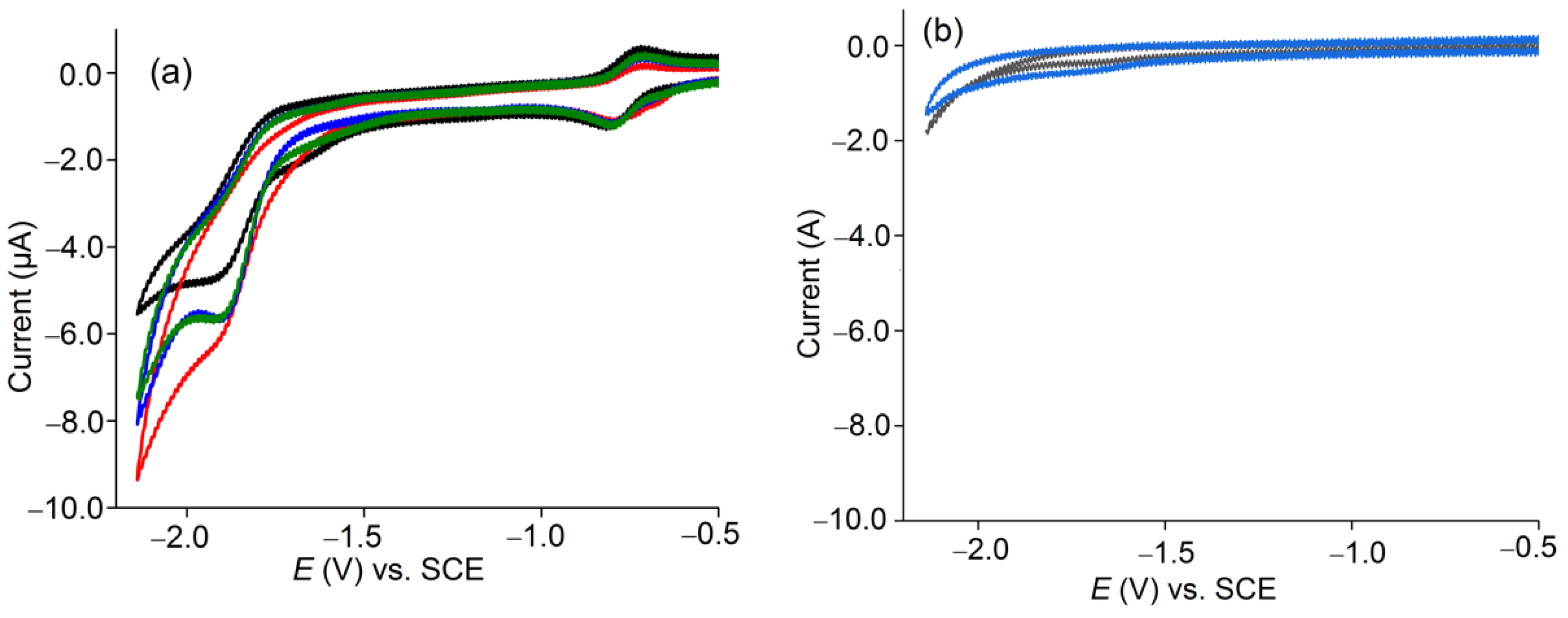
2.5. Electroreduction of CO2 to CO
2.5.1. Catalytic Behavior under CO2
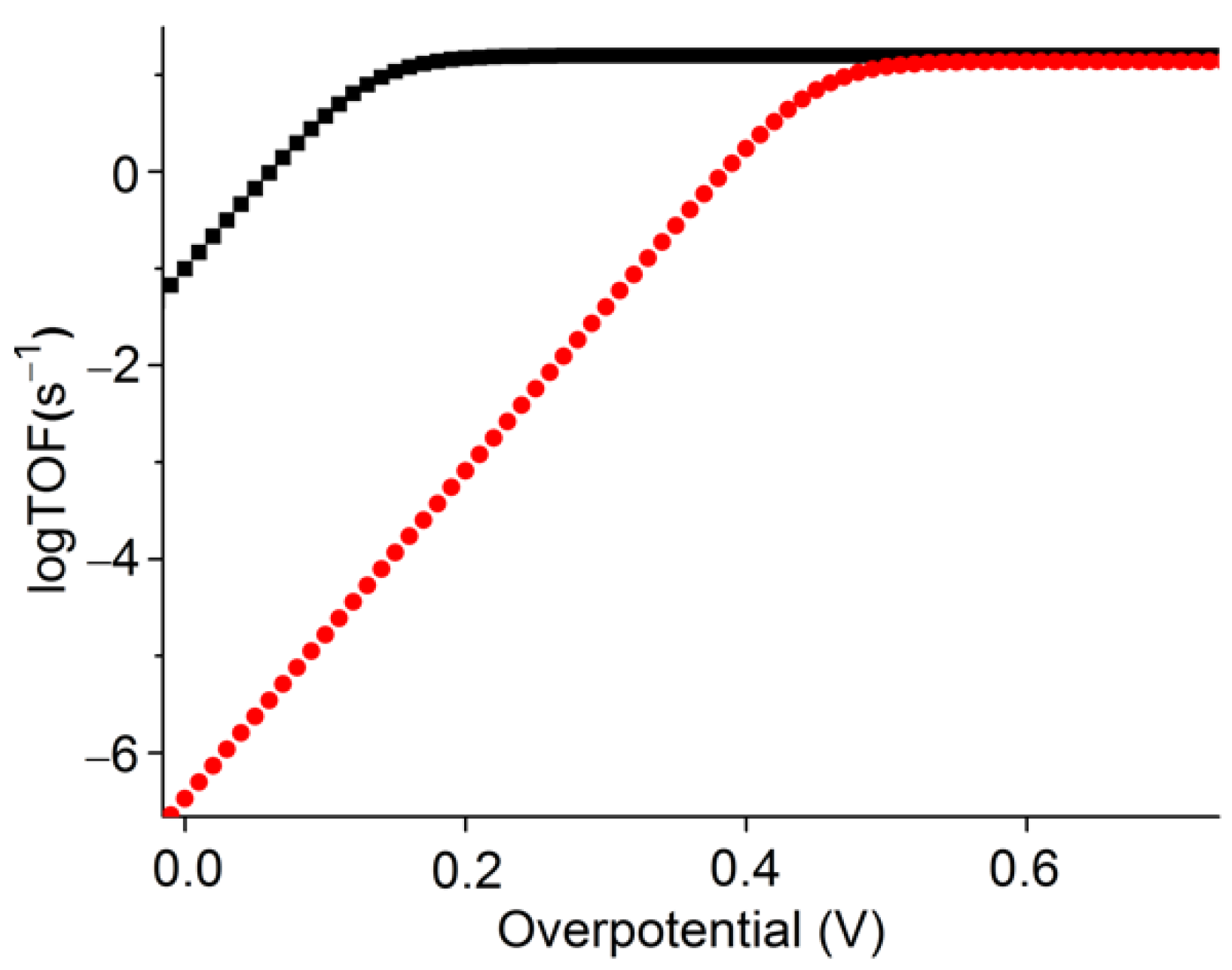
2.5.2. Benchmarking of the Catalyst
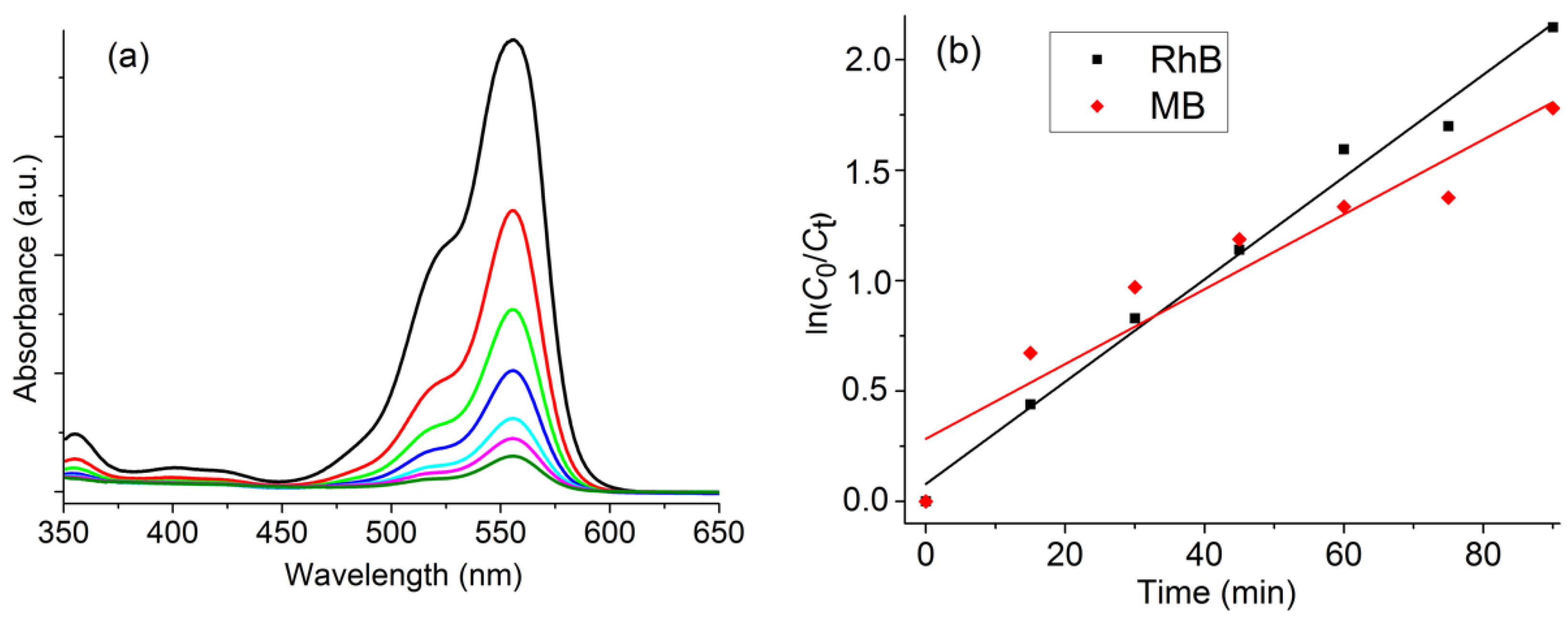
2.6. Photocatalytic Degradation of Methylene Blue and Rhodamine B Using H2O2
3. Experimental Section
3.1. Materials
3.2. Syntheses
3.2.1. Meso-Tetrakis(3,4,5-Trimethoxyphenyl)Porphyrin (H2TTMPP)
3.2.2. Meso-Tetrakis(3,4,5-Trimethoxyphenyl)Porphyrinato Cobalt(II) [Co(TTMPP)]
3.3. Methods and Instrumentation
3.4. Electrochemistry
3.5. Electrocatalytic CO2 Reduction
3.6. Gas Chromatography (GC)
3.7. Faradaic Efficiency, Turnover Number, Turnover Frequency Calculation
3.8. Gas Phase Analyses
3.9. Photo-Decomposition of RhB and MB with H2O2
3.10. Oxidative Photodegradation Mechanism–Trapping Experiments
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sun, T.; Zhang, Z.; Xu, J.; Liang, L.; Mai, C.-L.; Ren, L.; Zhou, Q.; Yu, Y.; Zhang, B.; Gao, P. Structural, photophysical, electrochemical and spintronic study of first-row metal Tetrakis(meso-triphenylamine)-porphyrin complexes: A combined experimental and theoretical study. Dye. Pigm. 2021, 193, 109469. [Google Scholar] [CrossRef]
- Zhang, R.; Warren, J.J. Recent Developments in Metalloporphyrin Electrocatalysts for Reduction of Small Molecules: Strategies for Managing Electron and Proton Transfer Reactions. ChemSusChem 2021, 14, 293–302. [Google Scholar] [CrossRef] [PubMed]
- Ke, X.; Kumar, R.; Sankar, M.; Kadish, K.M. Electrochemistry and Spectroelectrochemistry of Cobalt Porphyrins with π-Extending and/or Highly Electron-Withdrawing Pyrrole Substituents. In Situ Electrogeneration of σ-Bonded Complexes. Inorg. Chem. 2018, 57, 1490–1503. [Google Scholar] [CrossRef] [PubMed]
- Ye, L.; Fang, Y.; Ou, Z.; Xue, S.; Kadish, K.M. Cobalt Tetrabutano- and Tetrabenzotetraarylporphyrin Complexes: Effect of Substituents on the Electrochemical Properties and Catalytic Activity of Oxygen Reduction Reactions. Inorg. Chem. 2017, 56, 13613–13626. [Google Scholar] [CrossRef] [PubMed]
- Ke, X.; Yadav, P.; Cong, L.; Kumar, R.; Sankar, M.; Kadish, K.M. Facile and Reversible Electrogeneration of Porphyrin Trianions and Tetraanions in Nonaqueous Media. Inorg. Chem. 2017, 56, 8527–8537. [Google Scholar] [CrossRef]
- Klein, A. Spectroelectrochemistry of Metalloporphyrins. In Spectroelectrochemistry; Kaim, W., Klein, A., Eds.; RSC Publishing: Cambridge, UK, 2008; pp. 91–122. ISBN 978-1-84755-840-4. [Google Scholar]
- Sun, H.; Smirnov, V.V.; DiMagno, S.G. Slow Electron Transfer Rates for Fluorinated Cobalt Porphyrins: Electronic and Conformational Factors Modulating Metalloporphyrin ET. Inorg. Chem. 2003, 42, 6032–6040. [Google Scholar] [CrossRef]
- Chaudhri, N.; Cong, L.; Bulbul, A.S.; Grover, N.; Osterloh, W.R.; Fang, Y.; Sankar, M.; Kadish, K.M. Structural, Photophysical, and Electrochemical Properties of Doubly Fused Porphyrins and Related Fused Chlorins. Inorg. Chem. 2020, 59, 1481–1495. [Google Scholar] [CrossRef]
- Guergueb, M.; Loiseau, F.; Molton, F.; Nasri, H.; Klein, A. CO2 to CO Electroreduction, electrocatalytic H2 evolution, and catalytic degradation of organic dyes using a Co(II) meso-tetraarylporphyrin. Molecules 2022, 27, 1705. [Google Scholar] [CrossRef]
- Liu, Y.; Fu, L.-Z.; Yang, L.-M.; Liu, X.-P.; Zhan, S.-Z.; Ni, C.-L. The impact of modifying the ligands on hydrogen production electro-catalyzed by meso-tetra-p-X-phenylporphin cobalt complexes, CoT(X)PP. J. Mol. Catal. A Chem. 2016, 417, 101–106. [Google Scholar] [CrossRef] [Green Version]
- Nasri, S.; Hajji, M.; Guergueb, M.; Dhifaoui, S.; Marvaud, V.; Loiseau, F.; Molton, F.; Roisnel, T.; Guerfel, T.; Nasri, H. Spectroscopic, Electrochemical, Magnetic and Structural Characterization of an hexamethylenetetramine Co(II) Porphyrin Complex–Application in the Catalytic Degradation of Vat Yellow 1 dye. J. Mol. Struct. 2021, 1231, 129676. [Google Scholar] [CrossRef]
- Guergueb, M.; Nasri, S.; Brahmi, J.; Al-Ghamdi, Y.O.; Loiseau, F.; Molton, F.; Roisnel, T.; Guerineau, V.; Nasri, H. Spectroscopic characterization, X-ray molecular structures and cyclic voltammetry study of two (piperazine) cobalt(II) meso-arylporphyin complexes. Application as a catalyst for the degradation of 4-nitrophenol. Polyhedron 2021, 209, 115468. [Google Scholar] [CrossRef]
- Guergueb, M.; Nasri, S.; Brahmi, J.; Loiseau, F.; Molton, F.; Roisnel, T.; Guerineau, V.; Turowska-Tyrk, I.; Aouadi, K.; Nasri, H. Effect of the coordination of π-acceptor 4-cyanopyridine ligand on the structural and electronic properties of meso-tetra(para-methoxy) and meso-tetra(para-chlorophenyl) porphyrin cobalt(II) coordination compounds. Application in the catalytic degradation of methylene blue dye. RSC Adv. 2020, 10, 6900–6918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pamin, K.; Tabor, E.; Gjrecka, S.; Kubiak, W.W.; Rutkowska-Zbik, D.; Połtowicz, J. Three Generations of Cobalt Porphyrins as Catalysts in the Oxidation of Cycloalkanes. ChemSusChem 2019, 12, 684–691. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.-M.; Rønne, M.H.; Pedersen, S.U.; Skrydstrup, T.; Daasbjerg, K. Enhanced Catalytic Activity of Cobalt Porphyrin in CO2 Electroreduction upon Immobilization on Carbon Materials. Angew. Chem. Int. Ed. 2017, 56, 6468–6472. [Google Scholar] [CrossRef]
- Lin, S.; Diercks, C.S.; Zhang, Y.-B.; Kornienko, N.; Nichols, E.M.; Zhao, Y.; Paris, A.R.; Kim, D.; Yang, P.; Yaghi, O.M.; et al. Covalent organic frameworks comprising cobalt porphyrins for catalytic CO2 reduction in water. Science 2015, 349, 1208–1221. [Google Scholar] [CrossRef] [Green Version]
- Harvey, P.D. Porphyrin-based MOFs as heterogeneous photocatalysts for the eradication of organic pollutants and toxins. J. Porphyr. Phthalocyanines 2021, 25, 583–604. [Google Scholar] [CrossRef]
- Amiri, N.; Guergueb, M.; Al-Fakeh, M.S.; Bourguiba, M.; Nasri, H. A new cobalt(II) meso-porphyrin: Synthesis, characterization, electric properties and application in catalytic degradation of dyes. RSC Adv. 2020, 10, 44920–44932. [Google Scholar] [CrossRef]
- Nishiori, D.; Wadsworth, B.L.; Reyes Cruz, E.A.; Nguyen, N.P.; Hensleigh, L.K.; Karcher, T.; Moore, G.F. Photoelectrochemistry of metalloporphyrin-modified GaP semiconductors. Photosynth. Res. 2022, 151, 195–204. [Google Scholar] [CrossRef]
- Hong, Y.H.; Han, J.W.; Jung, J.; Nakagawa, T.; Lee, Y.-M.; Nam, W.; Fukuzumi, S. Photocatalytic Oxygenation Reactions with a Cobalt Porphyrin Complex Using Water as an Oxygen Source and Dioxygen as an Oxidant. J. Am. Chem. Soc. 2019, 141, 9155–9159. [Google Scholar] [CrossRef]
- Li, W.; He, X.; Ge, R.; Zhu, M.; Feng, L.; Li, Y. Cobalt porphyrin (CoTCPP) advanced visible light response of g-C3N4 nanosheets. Sust. Mater. Technol. 2019, 22, e00114. [Google Scholar] [CrossRef]
- Francis, S.; Rajith, L. Selective Fluorescent Sensing of Adenine Via the Emissive Enhancement of a Simple Cobalt Porphyrin. J. Fluoresc. 2021, 31, 577–586. [Google Scholar] [CrossRef] [PubMed]
- Delmarre, D.; Bied-Charreton, C. Grafting of cobalt porphyrins in sol–gel matrices: Application to the detection of amines. Sens. Actuators B Chem. 2000, 62, 136–142. [Google Scholar] [CrossRef]
- Lawrence, M.A.W.; Celestine, M.J.; Artis, E.T.; Joseph, L.S.; Esquivel, D.L.; Ledbetter, A.J.; Cropek, D.M.; Jarrett, W.L.; Bayse, C.A.; Brewer, M.I.; et al. Computational, electrochemical, and spectroscopic studies of two mononuclear cobaloximes: The influence of an axial pyridine and solvent on the redox behaviour and evidence for pyridine coordination to cobalt(I) and cobalt(II) metal centres. Dalton Trans. 2016, 45, 10326–10342. [Google Scholar] [CrossRef]
- Kaim, W.; Schwederski, B.; Klein, A. Bioinorganic Chemistry: Inorganic Elements in the Chemistry of Life–An Introduction and Guide, 2nd ed.; John Wiley & Sons: Chichester, UK, 2013; ISBN 978-0-470-97523-7. [Google Scholar]
- Simonova, O.R.; Zdanovich, S.A.; Zaitseva, S.V.; Koifman, O.I. Kinetic Study of the Redox Properties of [5,10,15,20-Tetrakis(2,5-dimethoxyphenyl)porphyrinato]cobalt(II) in the Reaction with Hydrogen Peroxide. Russ. J. Gen. Chem. 2020, 90, 863–869. [Google Scholar] [CrossRef]
- Pu, G.; Yang, Z.; Wu, Y.; Wang, Z.; Deng, Y.; Gao, Y.J.; Zhang, Z.; Lu, X. Investigation into the Oxygen-Involved Electrochemiluminescence of Porphyrins and Its Regulation by Peripheral Substituents/Central Metals. Anal. Chem. 2019, 91, 2319–2328. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.Q.; Boisselier-Cocolios, B.; Kadish, K.M. Electrochemistry, Spectroelectrochemistry, and Ligand Addition Reactions of an Easily Reducible Cobalt Porphyrin. Reactions of Tetracyanotetraphenylporphinato)cobalt(II) ((CN)4TPP)CoII) in Pyridine and in Pyridine/Methylene Chloride Mixtures. Inorg. Chem. 1986, 25, 3242–3248. [Google Scholar] [CrossRef]
- Mansour, A.; Belghith, Y.; Belkhiria, M.S.; Bujacz, A.; Guérineau, V.; Nasri, H. Synthesis, crystal structures and spectroscopic characterization of Co(II) bis(4,4′-bipyridine) with mesoporphyrins α,β,α,β-tetrakis(o-pivalamidophenyl) porphyrin (α,β,α,β-TpivPP) and tetraphenylporphyrin (TPP). J. Porphyr. Phthalocyanines 2013, 17, 1094–1103. [Google Scholar] [CrossRef]
- Puerres, H.; Díaz, M.; Hurtado, J.; Ortiz, P.; Cortés, M.T. Photoelectrochemical Stability under Anodic and Cathodic Conditions of Meso-Tetra-(4-Sulfonatophenyl)-Porphyrinato Cobalt (II) Immobilized in Polypyrrole Thin Films. Polymers 2021, 13, 657. [Google Scholar] [CrossRef]
- Smith, P.T.; Benke, B.P.; An, L.; Kim, Y.; Kim, K.; Chang, C.J. A Supramolecular Porous Organic Cage Platform Promotes Electrochemical Hydrogen Evolution from Water Catalyzed by Cobalt Porphyrins. ChemElectroChem 2021, 8, 1653–1657. [Google Scholar] [CrossRef]
- Lv, X.; Chen, Y.; Wu, Y.; Wang, H.; Wang, X.; Wei, C.; Xiao, Z.; Yang, G.; Jiang, J. A Br-regulated transition metal active-site anchoring and exposure strategy in biomass derived carbon nanosheets for obtaining robust ORR/HER electrocatalysts at all pH values. J. Mater. Chem. A 2019, 7, 27089–27098. [Google Scholar] [CrossRef]
- Wu, Y.; Veleta, J.M.; Tang, D.; Price, A.D.; Botez, C.E.; Villagrán, D. Efficient electrocatalytic hydrogen gas evolution by a cobalt–porphyrin-based crystalline polymer. Dalton Trans. 2018, 47, 8801–8806. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.-J.; Yu, Z.-T.; Chen, D.-Q.; Zou, Z.-G. Metal-complex chromophores for solar hydrogen generation. Chem. Soc. Rev. 2017, 46, 603–631. [Google Scholar] [CrossRef] [PubMed]
- Pal, R.; Laureanti, J.A.; Groy, T.L.; Jones, A.K.; Trovitch, R.J. Hydrogen production from water using a bis(imino)pyridine molybdenum electrocatalyst. Chem. Commun. 2016, 52, 11555–11558. [Google Scholar] [CrossRef] [PubMed]
- Zee, D.Z.; Chantarojsiri, T.; Long, J.R.; Chang, C.J. Metal polypyridyl catalysts for electro- and photochemical reduction of water to hydrogen. Acc. Chem. Res. 2015, 48, 2027–2036. [Google Scholar] [CrossRef] [Green Version]
- McKone, J.R.; Marinescu, S.C.; Brunschwig, B.S.; Winkler, J.J.; Gray, H.B. Earth-abundant hydrogen evolution electrocatalysts. Chem. Sci. 2014, 5, 865–878. [Google Scholar] [CrossRef] [Green Version]
- Roubelakis, M.M.; Bediako, D.K.; Dogutan, D.K.; Nocera, G. Proton-coupled electron transfer kinetics for the hydrogen evolution reaction of hangman porphyrins. Energy Environ. Sci. 2012, 5, 7737–7740. [Google Scholar] [CrossRef]
- Du, P.; Eisenberg, R. Catalysts made of earth-abundant elements (Co, Ni, Fe) for water splitting: Recent progress and future challenges. Energy Environ. Sci. 2012, 5, 6012–6021. [Google Scholar] [CrossRef]
- Artero, V.; Chavarot-Kerlidou, M.; Fontecave, M. Splitting water with cobalt. Angew. Chem. Int. Ed. 2011, 50, 7238–7266. [Google Scholar] [CrossRef]
- Kellett, R.M.; Spiro, T.G. Cobalt(I) porphyrin catalysts of hydrogen production from water. Inorg. Chem. 1985, 24, 2373–2377. [Google Scholar] [CrossRef]
- Attatsi, I.K.; Weihua Zhu, W.; Liang, X. Noncovalent immobilization of Co(II)porphyrin through axial coordination as an enhanced electrocatalyst on carbon electrodes for oxygen reduction and evolution. New J. Chem. 2020, 44, 4340–4345. [Google Scholar] [CrossRef]
- Wang, Y.-H.; Schneider, P.E.; Goldsmith, Z.K.; Mondal, B.; Hammes-Schiffer, S.; Stahl, S.S. Brønsted Acid Scaling Relationships Enable Control Over Product Selectivity from O2 Reduction with a Mononuclear Cobalt Porphyrin Catalyst. ACS Cent. Sci. 2019, 5, 1024–1034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Z.-S.; Chen, C.; Liu, J.; Parvez, K.; Liang, H.; Shu, J.; Sachdev, H.; Graf, R.; Feng, X.; Müllen, K. High-Performance Electrocatalysts for Oxygen Reduction Derived from Cobalt Porphyrin-Based Conjugated Mesoporous Polymers. Adv. Mater. 2014, 26, 1450–1455. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, S.; Basu, O.; Das, S.K. ZIF-8 MOF Encapsulated Co-porphyrin, an Efficient Electrocatalyst for Water Oxidation in a Wide pH Range: Works Better at Neutral pH. ChemCatChem. 2020, 12, 5430–5438. [Google Scholar] [CrossRef]
- Long, C.; Wan, K.; Qiu, X.; Zhang, X.; Han, J.; An, P.; Yang, Z.; Li, X.; Guo, J.; Shi, X.; et al. Single site catalyst with enzyme-mimic micro-environment for electroreduction of CO2. Nano Res. 2022, 15, 1817–1823. [Google Scholar] [CrossRef]
- Usman, M.; Humayun, M.; Garba, M.D.; Ullah, L.; Zeb, Z.; Helal, A.; Suliman, M.H.; Alfaifi, B.Y.; Iqbal, N.; Abdinejad, M.; et al. Electrochemical Reduction of CO2: A Review of Cobalt Based Catalysts for Carbon Dioxide Conversion to Fuels. Nanomaterials 2021, 11, 2029. [Google Scholar] [CrossRef]
- Marianov, A.N.; Kochubei, A.S.; Roman, T.; Conquest, O.J.; Stampfl, C.; Jiang, Y. Modeling and Experimental Study of the Electron Transfer Kinetics for Non-ideal Electrodes Using Variable-Frequency Square Wave Voltammetry. Anal. Chem. 2021, 93, 10175–10186. [Google Scholar] [CrossRef]
- Dou, S.; Sun, L.; Xi, S.; Li, X.; Su, T.; Fan, H.J.; Wang, X. Enlarging the π-Conjugation of Cobalt Porphyrin for Highly Active and Selective CO2 Electroreduction. ChemSusChem 2021, 14, 2126–2132. [Google Scholar] [CrossRef]
- Marianov, A.N.; Kochubei, A.S.; Roman, T.; Conquest, O.J.; Stampfl, C.; Jiang, Y. Resolving Deactivation Pathways of Co Porphyrin-Based Electrocatalysts for CO2 Reduction in Aqueous Medium. ACS Catal. 2021, 11, 3715–3729. [Google Scholar] [CrossRef]
- Chen, X.; Hu, X.-M.; Daasbjerg, K.; Ahlquist, M.S.G. Understanding the Enhanced Catalytic CO2 Reduction upon Adhering Cobalt Porphyrin to Carbon Nanotubes and the Inverse Loading Effect. Organometallics 2020, 39, 1634–1641. [Google Scholar] [CrossRef]
- Jack, J.; Park, E.; Maness, P.-C.; Huang, S.; Zhang, W.; Ren, Z.J. Selective ligand modification of cobalt porphyrins for carbon dioxide electrolysis: Generation of a renewable H2/CO feedstock for downstream catalytic hydrogenation. Inorg. Chim. Acta 2020, 507, 119594. [Google Scholar] [CrossRef]
- Wang, Z.-j.; Song, H.; Liu, H.; Ye, J. Coupling of Solar Energy and Thermal Energy for Carbon Dioxide Reduction: Status and Prospects. Angew. Chem. Int. Ed. 2020, 59, 8016–8035. [Google Scholar] [CrossRef] [PubMed]
- Sinha, S.; Zhang, R.; Warren, J.J. Low Overpotential CO2 Activation by a Graphite-Adsorbed Cobalt Porphyrin. ACS Catal. 2020, 10, 12284–12291. [Google Scholar] [CrossRef]
- Wu, Q.-J.; Mao, M.-J.; Chen, J.-X.; Huang, Y.-B.; Cao, R. Integration of metalloporphyrin into cationic covalent triazine frameworks for the synergistically enhanced chemical fixation of CO2. Catal. Sci. Technol. 2020, 10, 8026–8033. [Google Scholar] [CrossRef]
- Abdinejad, M.; Seifitokaldani, A.; Dao, C.; Sargent, E.H.; Zhang, X.-a.; Kraatz, H.B. Enhanced Electrochemical Reduction of CO2 Catalyzed by Cobalt and Iron Amino Porphyrin Complexes. ACS Appl. Energy Mater. 2019, 2, 1330–1335. [Google Scholar] [CrossRef]
- Hu, B.; Xie, W.; Li, R.; Pan, Z.; Song, S.; Wang, Y. How does the ligands structure surrounding metal-N4 of Co-based macrocyclic compounds affect electrochemical reduction of CO2 performance? Electrochim. Acta 2019, 331, 135283. [Google Scholar] [CrossRef]
- Miyamoto, K.; Asahi, R. Water Facilitated Electrochemical Reduction of CO2 on Cobalt-Porphyrin Catalysts. J. Phys. Chem. C 2019, 123, 9944–9948. [Google Scholar] [CrossRef] [Green Version]
- Behar, D.; Dhanasekaran, T.; Neta, P.; Hosten, C.M.; Ejeh, D.; Hambright, P.; Fujita, E. Cobalt Porphyrin Catalyzed Reduction of CO2, Radiation Chemical, Photochemical, and Electrochemical Studies. J. Phys. Chem. A 1998, 102, 2870–2877. [Google Scholar] [CrossRef]
- Piccirillo, G.; Aroso, R.T.; Rodrigues, F.M.S.; Carrilho, R.M.B.; Pinto, S.M.A.; Calvete, M.J.F.; Pereira, M.M. Oxidative Degradation of Pharmaceuticals: The Role of Tetrapyrrole-Based Catalysts. Catalysts 2021, 11, 11335. [Google Scholar] [CrossRef]
- Xie, J.; Xu, P.; Zhu, Y.; Wang, J.; Lee, W.-C.C.; Zhang, X.P. New Catalytic Radical Process Involving 1,4-Hydrogen Atom Abstraction: Asymmetric Construction of Cyclobutanones. J. Am. Chem. Soc. 2021, 143, 11670–11678. [Google Scholar] [CrossRef]
- Li, C.; Lang, K.; Lu, H.; Hu, Y.; Cui, X.; Wojtas, L.; Zhang, X.P. Catalytic Radical Process for Enantioselective Amination of C(sp3)–H Bonds. Angew. Chem. Int. Ed. 2018, 57, 16837–16841. [Google Scholar] [CrossRef]
- Chan, T.L.; To, C.T.; Liao, B.-S.; Liu, S.-T.; Chan, K.S. Electronic Effects of Ligands on the Cobalt(II)–Porphyrin-Catalyzed Direct C–H Arylation of Benzene. Eur. J. Inorg. Chem. 2012, 2012, 485–489. [Google Scholar] [CrossRef]
- Puchovskaya, S.G.; Ivanova, Y.B.; Chizhova, N.Z.; Syrbu, S.A. Synthesis, Spectral, Acid-Basic, and Coordination Properties of Bromine- and Methoxy-Substituted Tetraphenylporphyrins. Russ. J. Gen. Chem. 2021, 91, 1050–1056. [Google Scholar] [CrossRef]
- Mishra, E.; Worlinsky, J.L.; Gilbert, T.M.; Brückner, C.; Ryzhov, V. Axial Imidazole Binding Strengths in Porphyrinoid Cobalt(III) Complexes as Studied by Tandem Mass Spectrometry. J. Am. Soc. Mass Spectrom. 2012, 23, 1135–1146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subbaiyan, N.K.; Wijesinghe, C.A.; D’Souza, F. Supramolecular Solar Cells: Surface Modification of Nanocrystalline TiO2 with Coordinating Ligands To Immobilize Sensitizers and Dyads via Metal-Ligand Coordination for Enhanced Photocurrent Generation. J. Am. Chem. Soc. 2009, 131, 14646–14647. [Google Scholar] [CrossRef] [PubMed]
- Sugamoto, K.; Matsushita, Y.-i.; Matsui, T. Direct hydroperoxygenation of conjugated olefins catalyzed by cobalt(II) porphyrin. J. Chem. Soc., Perkin Trans. 1 1998, 1998, 3989–3998. [Google Scholar] [CrossRef]
- Lindsey, J.S.; Hsu, H.C.; Schreiman, I.C. Synthesis of tetraphenylporphyrins under very mild conditions. Tetrahedron Lett. 1986, 27, 4969–4970. [Google Scholar] [CrossRef]
- Soury, R.; Jabli, M.; Saleh, T.A.; Abdul-Hassan, W.S.; Saint-Aman, E.; Loiseau, F.; Philouze, C.; Nasri, H. Tetrakis(ethyl-4(4-butyryl)oxyphenyl)porphyrinato zinc complexes with 4,4′-bpyridine: Synthesis, characterization, and its catalytic degradation of Calmagite. RSC Adv. 2018, 8, 20143–20156. [Google Scholar] [CrossRef] [Green Version]
- Chen, P.; Finikova, O.S.; Ou, Z.; Vinogradov, S.A.; Kadish, K.M. Electrochemistry of Platinum(II) Porphyrins: Effect of Substituents and π-Extension on Redox Potentials and Site of Electron Transfer. Inorg. Chem. 2012, 51, 6200–6210. [Google Scholar] [CrossRef] [PubMed]
- Queyriaux, N.; Sun, D.; Fize, J.; Pecaut, J.; Field, M.J.; Chavarot-Kerlidou, M.; Artero, V. Electrocatalytic Hydrogen Evolution with a Cobalt Complex Bearing Pendant Proton Relays: Acid Strength and Applied Potential Govern Mechanism and Stability. J. Am. Chem. Soc. 2020, 142, 274–282. [Google Scholar] [CrossRef]
- Gu, S.; Marianov, A.N.; Jiang, Y. Covalent grafting of cobalt aminoporphyrin-based electrocatalyst onto carbon nanotubes for excellent activity in CO2 reduction. Appl. Catal. B Environm. 2022, 300, 120750. [Google Scholar] [CrossRef]
- Rountree, E.S.; McCarthy, B.D.; Eisenhart, T.T.; Dempsey, J.L. Evaluation of Homogeneous Electrocatalysts by Cyclic Voltammetry. Inorg. Chem. 2014, 53, 9983–10002. [Google Scholar] [CrossRef] [PubMed]
- Costentin, C.; Drouet, S.; Robert, M.; Saveant, J.-M. Turnover numbers, turnover frequencies, and overpotential in molecular catalysis of electrochemical reactions. Cyclic voltammetry and preparative-scale electrolysis. J. Am. Chem. Soc. 2012, 134, 11235–11242. [Google Scholar] [CrossRef] [PubMed]
- Warren, J.J.; Tronic, T.A.; Mayer, J.M. Thermochemistry of Proton-Coupled Electron Transfer Reagents and Its Implications. Chem. Rev. 2010, 110, 6961–7001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ait Ahsaine, H.; El Jaouhari, A.; Slassi, A.; Ezahri, M.; Benlhachemi, A.; Bakiz, B.; Guinneton, F.; Gavarri, J.-R. Electronic band structure and visible-light photocatalytic activity of Bi2WO6: Elucidating the effect of lutetium doping. RSC Adv. 2016, 6, 101105–101114. [Google Scholar] [CrossRef]
- Guergueb, M.; Brahmi, J.; Nasri, S.; Loiseau, F.; Aouadi, K.; Guerineau, V.; Najmudin, S.; Nasri, H. Zinc(II) triazole meso-arylsubstituted porphyrins for UV-visible chloride and bromide detection. Adsorption and catalytic degradation of malachite green dye. RSC Adv. 2020, 10, 22712–22725. [Google Scholar] [CrossRef]
- Connelly, N.G.; Geiger, W.E. Chemical Redox Agents for Organometallic Chemistry. Chem. Rev. 1996, 96, 877–910. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| H+ Source | Time CPE | ECPE | Icat/Ip | FEH2 | TON | TOF (h−1) |
|---|---|---|---|---|---|---|
| 3 eq. TFA | 2 h | −2.00 | 10.86 | 76 | 11.04 | 5.52 |
| 3 eq. HNEt3+ | 2 h | −1.89 | 13.65 | 88 | 14.60 | 7.30 |
| H+ Source | Time CPE | ECPE | FECO2 | FEH2 |
|---|---|---|---|---|
| 1 eq. TFE | 2 h | −1.94 | 95 | Not detected |
| 1 eq. PhOH | 2 h | −1.93 | 88 |
| H+ Source | ECPE | Icat/Ip | TOF(s−1) | TON b | |
|---|---|---|---|---|---|
| CPE | CVs | ||||
| 1eq. TFE | −2 | 5.6 | 9.33 | 15.80 | 113,760 |
| 1eq. PhOH | −2 | 4.9 | 8.31 | 13.85 | 99,720 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guergueb, M.; Kechiche, A.; Loiseau, F.; Molton, F.; Nasri, H.; Hohnsen, J.; Klein, A. Hydrogen Evolution Reaction, Electrochemical CO2 Reduction, and Oxidative Photodegradation of Organic Dyes Catalyzed by Co(II) Trimethoxy-Meso-Arylporphyrin. Inorganics 2023, 11, 6. https://doi.org/10.3390/inorganics11010006
Guergueb M, Kechiche A, Loiseau F, Molton F, Nasri H, Hohnsen J, Klein A. Hydrogen Evolution Reaction, Electrochemical CO2 Reduction, and Oxidative Photodegradation of Organic Dyes Catalyzed by Co(II) Trimethoxy-Meso-Arylporphyrin. Inorganics. 2023; 11(1):6. https://doi.org/10.3390/inorganics11010006
Chicago/Turabian StyleGuergueb, Mouhieddinne, Azhar Kechiche, Frédérique Loiseau, Florian Molton, Habib Nasri, Johannes Hohnsen, and Axel Klein. 2023. "Hydrogen Evolution Reaction, Electrochemical CO2 Reduction, and Oxidative Photodegradation of Organic Dyes Catalyzed by Co(II) Trimethoxy-Meso-Arylporphyrin" Inorganics 11, no. 1: 6. https://doi.org/10.3390/inorganics11010006