
Structural Characteristics, Stability, and Electronic Properties of 001 Surface with Point Defects of Zinc Stannate: A First-Principle Study
Abstract
:1. Introduction
2. Results and Discussion
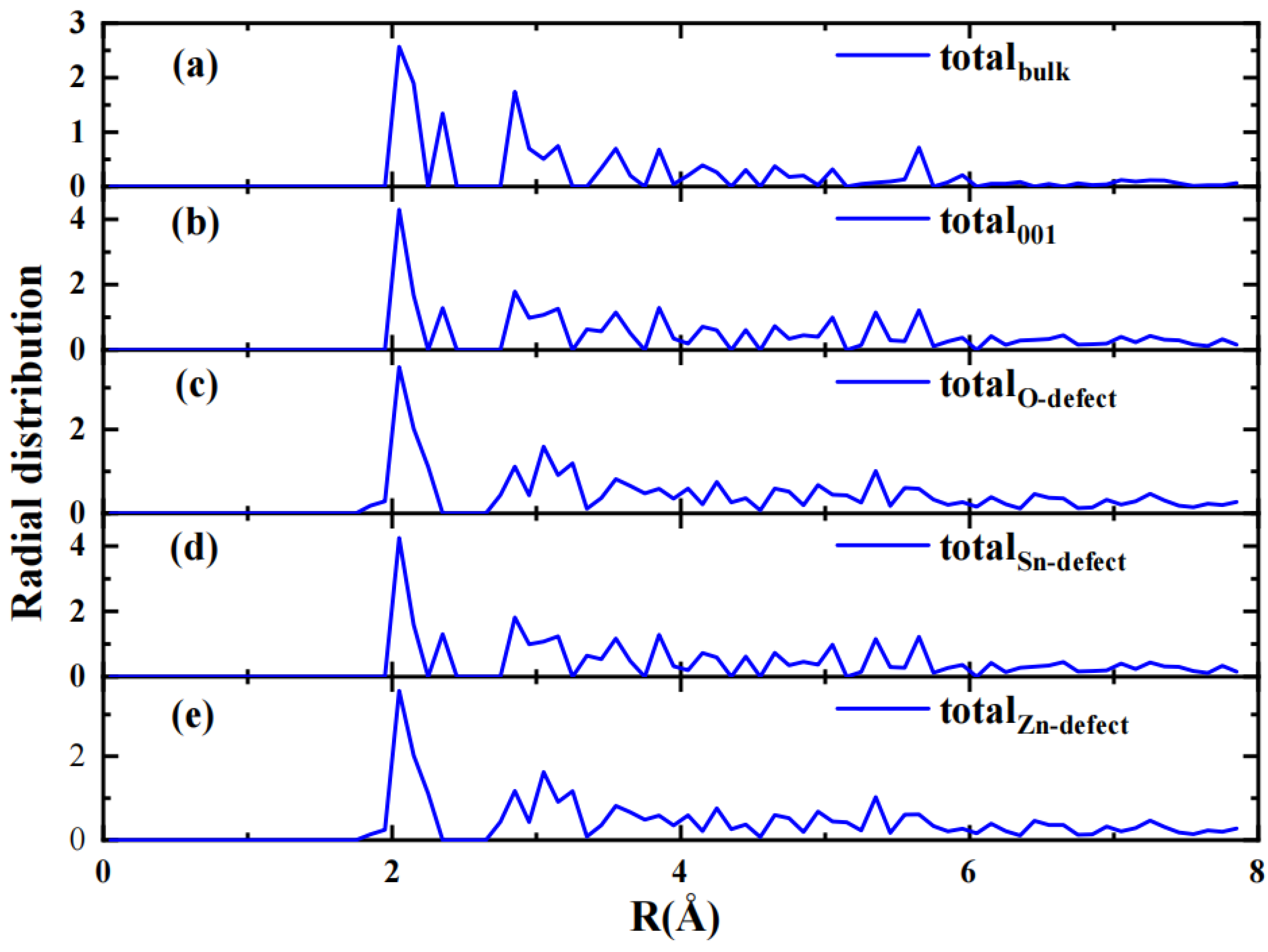
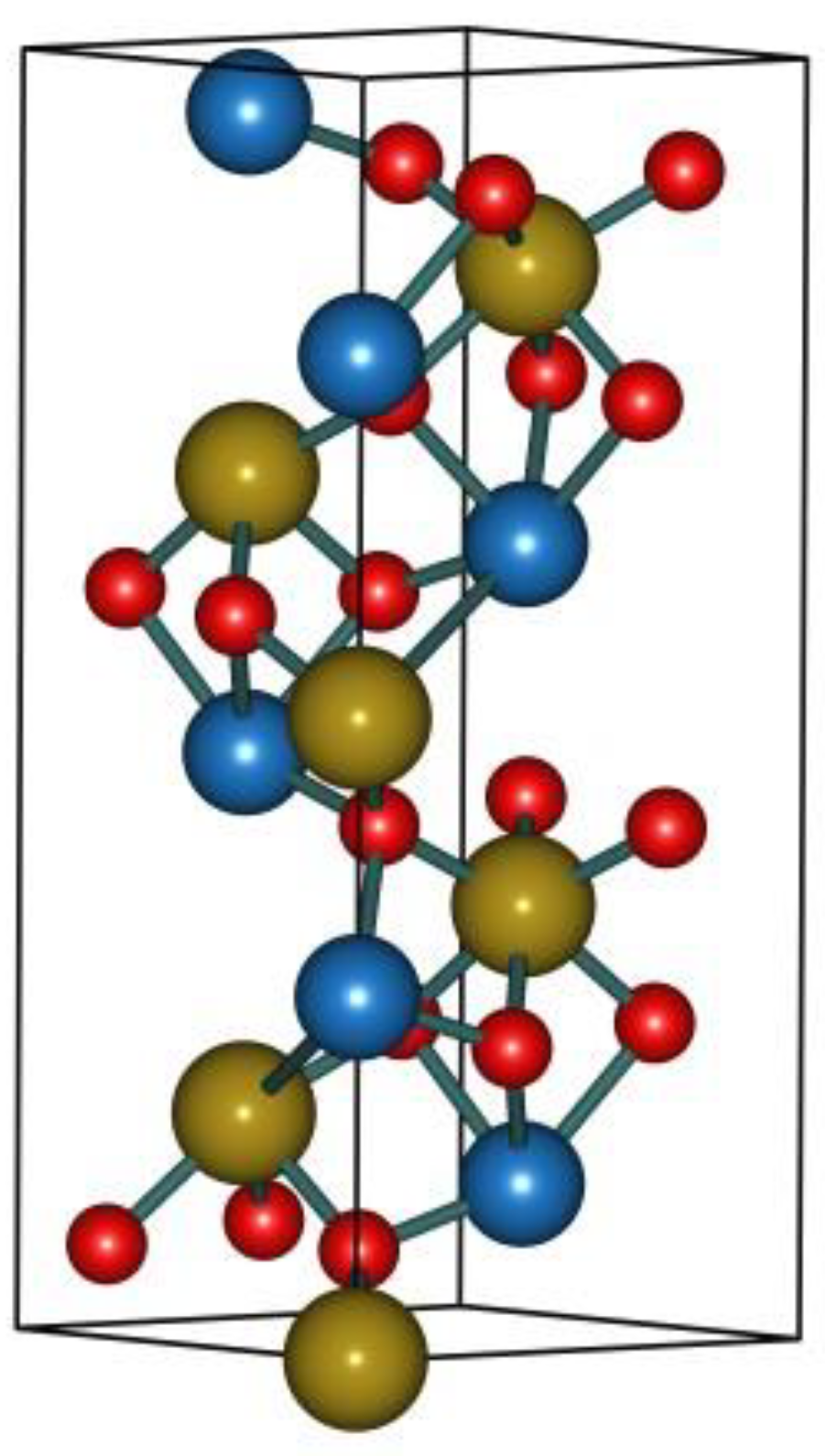
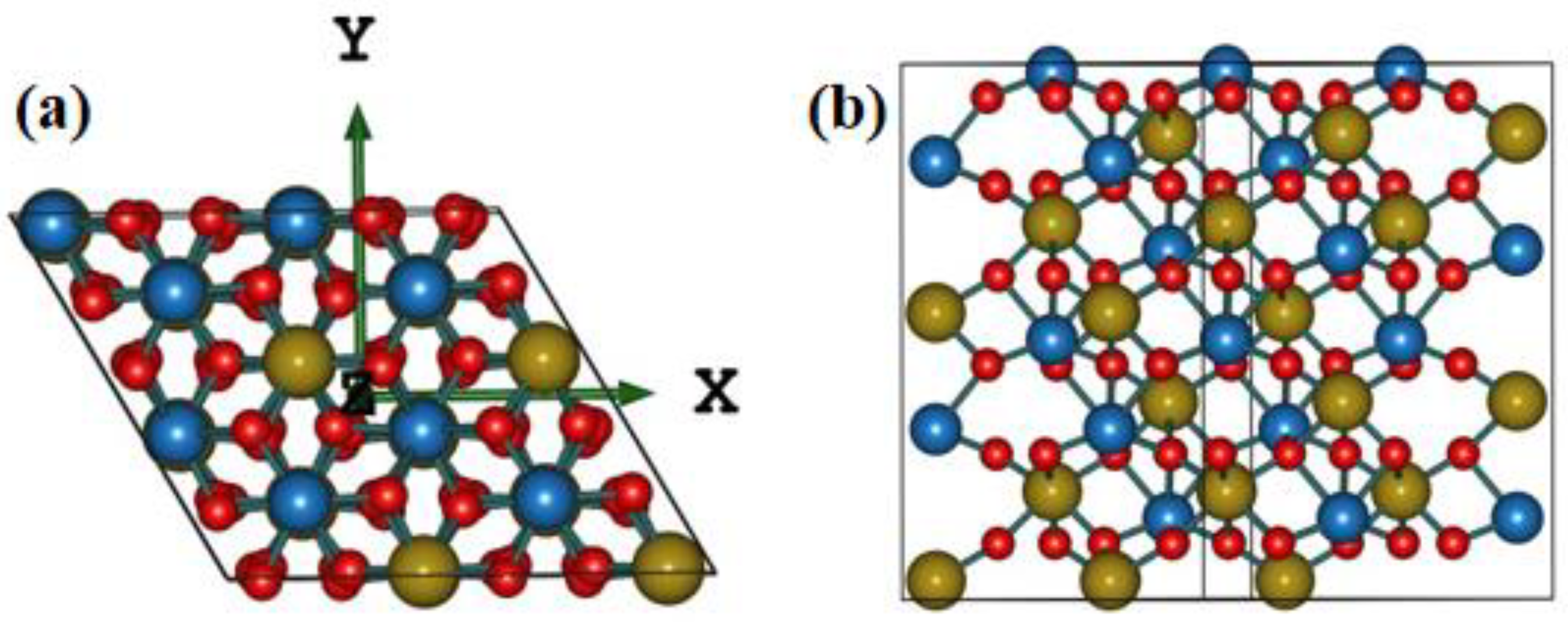
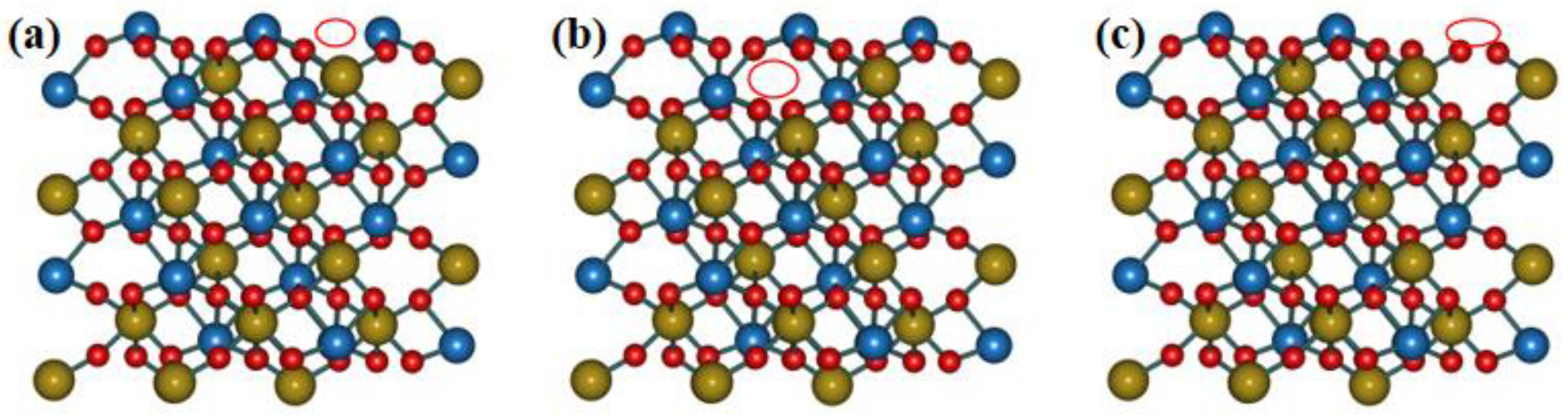
2.1. Geometry Structure
2.2. Energy and Stability
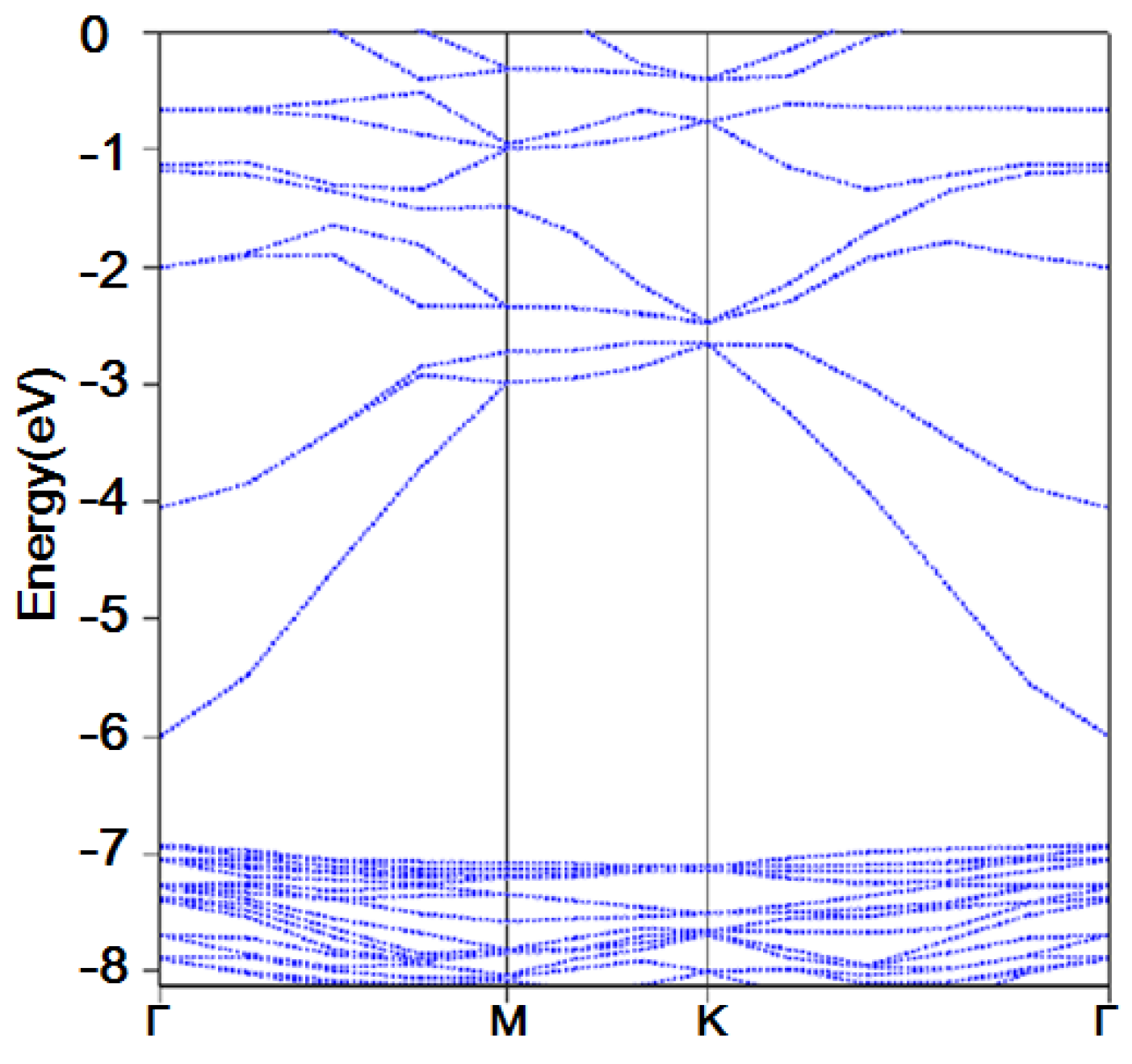
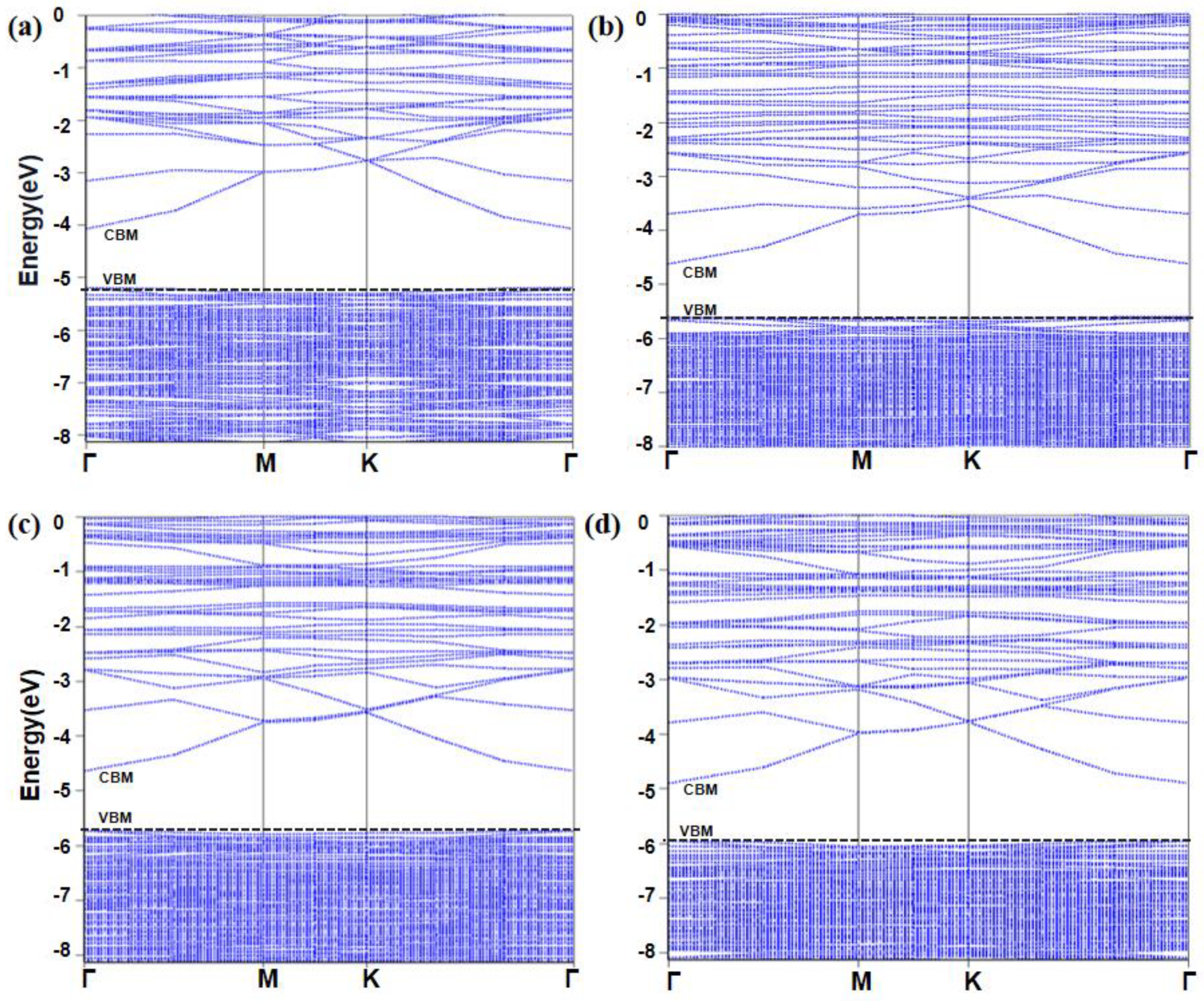
2.3. Band Structure
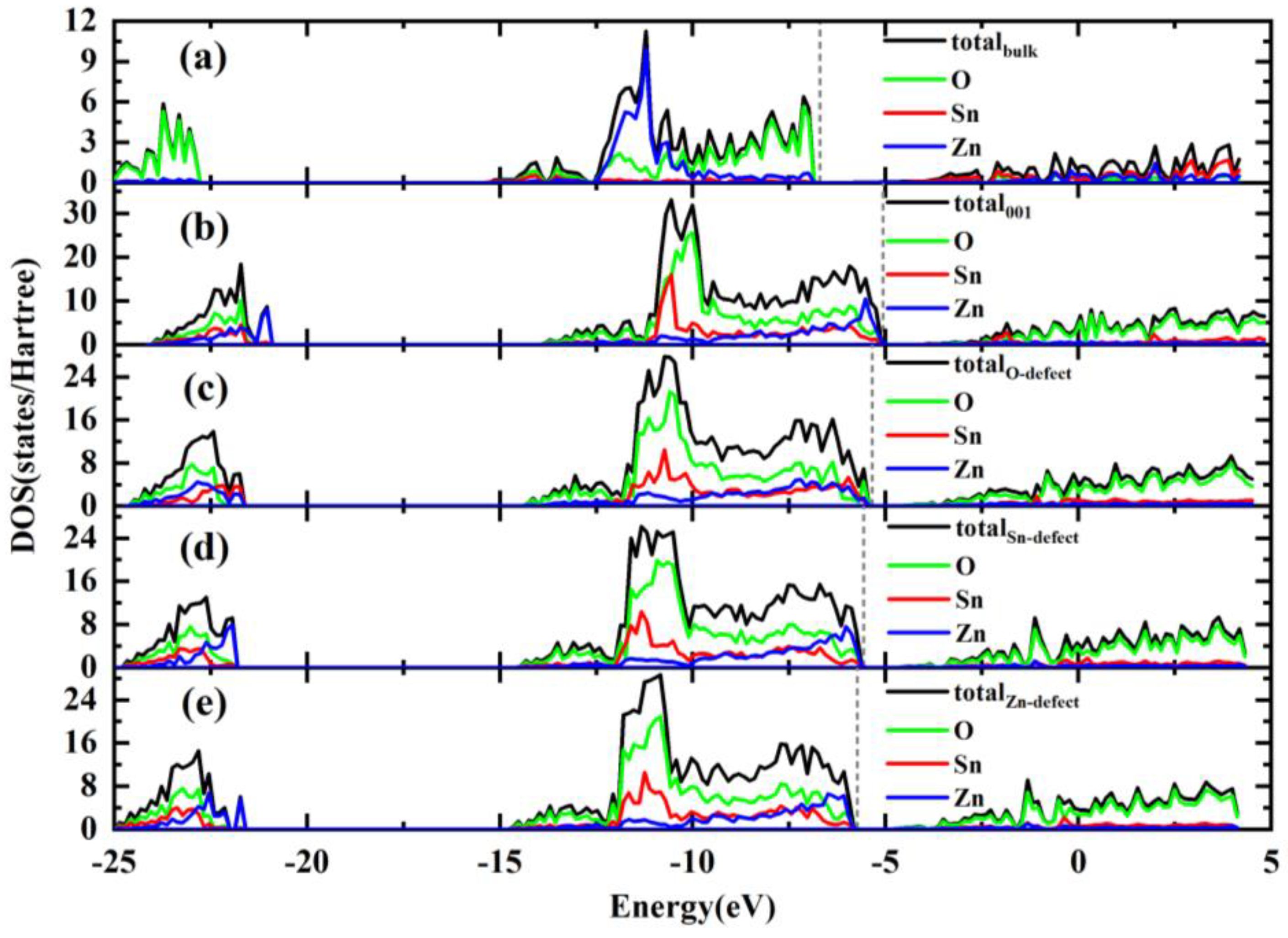
2.4. Density of States
3. Models and Computational Methods
3.1. Structure and Models
3.2. Computational Methods
4. Conclusions
- (1)
- The perfect 001 surface and the those with VO, VSn, and VZn defects lead to a decrease in the cohesive energy compared with the bulk ZS. VO vacancy defects system presents both the largest cohesive energy and the most negative defect formation energy. Therefore, from a thermodynamic point of view, VO defects on the 001 surface of ZS are easier to form than other defects.
- (2)
- The formation of the VO, VSn, and VZn vacancies on the 001 surface of ZS slightly changes the band structure and band gap compared with that of the bulk. The existence of these defects makes the relative positions of CBM and VBM move toward the valence band, while the overall band gap does not change greatly compared with that of the perfect surface.
- (3)
- According to DOS analysis, the electronic properties close to the Fermi levels of bulk ZS materials are mainly controlled by the O 2p and Sn 5s orbitals. The introduced surface and defects could lead to the rearrangement of the geometric structure. These defects and structural changes have affected the electronic properties to a certain extent and are slightly different to those without defects.
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Kanat, M.; Eren, T. Synthesis of phosphorus containing flame retardants and investigation of their flame retardant behavior in textile applications. J. Appl. Polym. Sci. 2019, 136, 47935. [Google Scholar] [CrossRef]
- Lu, S.; Hong, W.; Chen, X. Nanoreinforcements of Two-Dimensional Nanomaterials for Flame Retardant Polymeric Composites: An Overview. Adv. Polym. Technol. 2019, 2019, 4273253. [Google Scholar] [CrossRef] [Green Version]
- Oliveira, M.C.; Ribeiro, R.; Longo, E.; Bomio, M.; Lazaro, S. Quantum mechanical modeling of Zn-based spinel oxides: Assessing the structural, vibrational, and electronic properties. Int. J. Quantum Chem. 2020, 120, 26368. [Google Scholar] [CrossRef]
- Wang, H. First-principles study of structural, electronic, and optical properties of ZnSnO3. Solid State Commun. 2009, 149, 1849–1852. [Google Scholar] [CrossRef]
- He, W.; Song, P.; Yu, B.; Fang, Z.; Wang, H. Flame retardant polymeric nanocomposites through the combination of nanomaterials and conventional flame retardants. Prog. Mater. Sci. 2020, 114, 100687. [Google Scholar] [CrossRef]
- Paul, S.; Basak, S.; Ali, W. Zinc Stannate Nanostructure: Is It a New Class of Material for Multifunctional Cotton Textiles? ACS Omega 2019, 4, 2719. [Google Scholar] [CrossRef] [Green Version]
- Khan, M.; Dutta, J. Comparison of photocatalytic activity of zinc stannate particles and zinc stannate/zinc oxide composites for the removal of phenol from water, and a study on the effect of pH on photocatalytic efficiency. Mater. Sci. Semicond. Process. 2015, 36, 124–133. [Google Scholar] [CrossRef]
- Jose, M.; Nithya, G.; Robert, R.; Dhas, S. Formation and optical characterization of unique zinc hydroxy stannate nanostructures by a simple hydrothermal method. J. Mater. Sci. Mater. Electron. 2018, 29, 2628–2637. [Google Scholar] [CrossRef]
- Aziz, S.; Jung, K.; Chang, S. Stretchable strain sensor based on a nanocomposite of zinc stannate nanocubes and silver nanowires. Compos. Struct. 2019, 224, 111005. [Google Scholar] [CrossRef]
- Montenegro, J.; Ochoa-Muoz, Y.; Rodríguez-Páez, J.E. Nanoparticles of zinc stannates (ZTO): Synthesis, characterization and electrical behavior in oxygen and acetone vapors. Ceram. Int. 2019, 46, 2016–2032. [Google Scholar] [CrossRef]
- Qu, H.; Wu, W.; Xie, J.; Xu, J. Zinc hydroxystannate-coated metal hydroxides as flame retardant and smoke suppression for flexible poly vinyl chloride. Fire Mater. 2010, 33, 201–210. [Google Scholar] [CrossRef]
- Zhang, B.; Han, J. Synthesis of microencapsulated zinc stannate and its application in flame-retardant poly(vinyl chloride) membrane material. Fire Mater. 2018, 42, 109–118. [Google Scholar] [CrossRef]
- Xu, J.; Zhang, C.; Qu, H.; Tian, C. Zinc hydroxystannate and zinc stannate as flame-retardant agents for flexible poly(vinyl chloride). J. Appl. Polym. Sci. 2010, 98, 1469–1475. [Google Scholar] [CrossRef]
- Sun, S.; Liang, S. Morphological zinc stannate: Synthesis, fundamental properties and applications. J. Mater. Chem. A 2017, 5, 20534–20560. [Google Scholar] [CrossRef]
- Zeng, W.; Liu, T.M.; Lin, L.Y. Ethanol gas sensing property and mechanism of ZnSnO3 doped with Ti ions. Mater. Sci. Semicond. Process. 2012, 15, 319–325. [Google Scholar] [CrossRef]
- Xie, J.; Jiao, Y.; Xu, J.; Sun, H. Synthesis of zinc stannate and zinc stannate coated nano-CaCO3 by homogeneous precipitation. J. Chem. Res. 2011, 2, 109–111. [Google Scholar] [CrossRef]
- Eglitis, R.; Kruchinin, S. Ab initio calculations of ABO3 perovskite (001), (011) and (111) nano-surfaces, interfaces and defects. Mod. Phys. Lett. B 2020, 34, 2040057. [Google Scholar] [CrossRef]
- Sang, B.; Li, Z.; Yu, L.; Li, X.; Zhang, Z. Preparation of zinc hydroxystannate-titanate nanotube flame retardant and evaluation its smoke suppression efficiency for flexible polyvinyl chloride matrix. Mater. Lett. 2017, 204, 133–137. [Google Scholar] [CrossRef]
- Pramchu, S.; Laosiritaworn, Y.; Jaroenjittichai, A.P. Electronic properties of surface/bulk iodine defects of CsSnBr3 perovskite. Surf. Coat. Technol. 2016, 306, 159–163. [Google Scholar] [CrossRef]
- Zhukovskii, Y.; Kotomin, E.; Piskunov, S.; Ellis, D. A comparative ab initio study of bulk and surface oxygen vacancies in PbTiO3, PbZrO3 and SrTiO3 perovskites. Solid State Commun. 2016, 149, 1359–1362. [Google Scholar] [CrossRef]
- Long, L.; Cao, D.; Fei, J.; Wang, J.; Zhou, Y.; Jiang, Z.; Jiao, Z.; Shu, H. Effect of surface intrinsic defects on the structural stability and electronic properties of the all-inorganic halide perovskite CsPbI3(001) film. Chem. Phys. Lett. 2019, 734, 136719. [Google Scholar] [CrossRef]
- Xiao, H.; Zhang, Y.; Weber, W. Impact of point defects on electronic structure in Y2Ti2O7. RSC Adv. 2012, 2, 7235–7240. [Google Scholar] [CrossRef]
- Ahangari, M.; Mashhadzadeh, A.; Fathalian, M.; Dadrasi, A.; Rostamiyan, Y.; Mallahi, A. Effect of various defects on mechanical and electronic properties of zinc-oxide graphene-like structure: A DFT study. Vacuum 2019, 165, 26–34. [Google Scholar] [CrossRef]
- Zheng, M.; Li, X.; Wang, M.; Guo, L. Dynamic profiles of tar products during Naomaohu coal pyrolysis revealed by large-scale reactive molecular dynamic simulation. Fuel 2019, 253, 910–920. [Google Scholar] [CrossRef]
- Boonchun, A.; Dabsamut, K.; Lambrecht, W. First-principles study of point defects in LiGaO2. J. Appl. Phys. 2019, 126, 155703. [Google Scholar] [CrossRef] [Green Version]
- Yuan, Y.; Chen, G.; Ye, H.; Jin, W.; Zhu, Y.; Wu, Y. First-principles study on native point defects of cubic cuprite Ag2O. J. Appl. Phys. 2016, 120, 215707. [Google Scholar] [CrossRef]
- Gou, H.; Gao, F.; Zhang, J. Structural identification, electronic and optical properties of ZnSnO3: First principle calculations. Comput. Mater. Sci. 2010, 49, 552–555. [Google Scholar] [CrossRef]
- Wang, Y.; Yang, C.; Zhang, Y.; Guo, L.; Wang, Y.; Gao, G.; Fu, F.; Xu, B.; Wang, D. Nanoarchitectonics of CdS/ZnSnO3 heterostructures for Z-Scheme mediated directional transfer of photo-generated charges with enhanced photocatalytic performance. Int. J. Hydrogen Energy 2022, 16, 47. [Google Scholar] [CrossRef]
- Liu, Q.J.; Qin, H.; Jiao, Z.; Liu, F.; Liu, Z. First-principles calculations of structural, elastic, and electronic properties of trigonal ZnSnO3 under pressure. Mater. Chem. Phys. 2016, 180, 75–81. [Google Scholar] [CrossRef]
- Ni, J.; Liu, N.; Yang, G.; Zhang, X. First-principle study on electronic structure of BaTiO3 (001) surfaces. J. Phys. 2008, 57, 4434–4440. [Google Scholar]
- Ong, K.; Fan, X.; Subedi, A.; Sullivan, M.; Singh, D. Transparent conducting properties of SrSnO3 and ZnSnO3. APL Mater. 2015, 3, 062505. [Google Scholar] [CrossRef]
- Deml, A.M.; Stevanović, V.; Muhich, C.L.; Musgrave, C.B.; O’Hayre, R. Oxide enthalpy of formation and band gap energy as accurate descriptors of oxygen vacancy formation energetics. Energy Environ. Sci. 2014, 7, 1996–2004. [Google Scholar] [CrossRef]
- Zhu, Y.; Bai, H.; Huang, Y. Crystal orbital studies on the 1D silic-diyne nanoribbons and nanotubes. J. Phys. Condens. Matter Inst. Phys. J. 2016, 28, 045303. [Google Scholar] [CrossRef] [PubMed]
- Liao, H.; Li, J.; Wei, T.; Wen, P.; Li, M.; Hu, X. First-principles study of C-N point defects on sidewall surface of [0001]-oriented GaN nanowires. Appl. Surf. Sci. 2019, 467, 293–297. [Google Scholar] [CrossRef]
- Alay-E-Abbas, S.; Nazir, S.; Ali, S. Formation energies and electronic structure of intrinsic vacancy defects and oxygen vacancy clustering in BaZrO3. Phys. Chem. Chem. Phys. Pccp 2016, 18, 23737. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.; Peng, W.; Liu, Z.; Deng, X. First-principles calculations for formation energy and magnetism of defect structures in Heusler alloys Mg-V-Z (Z=Al, Ga, In). Phys. B 2020, 600, 412388. [Google Scholar] [CrossRef]
- He, R.; Wan, Y.; Guo, P.; Jiang, Z.; Zheng, J. First-principles investigation of native point defects in two-dimensional Ti3C2. Comput. Theor. Chem. 2019, 1150, 26–39. [Google Scholar] [CrossRef]
- Yuan, H.; Li, J. Effect of annealing temperature on the growth of Zn-Sn-O nanocomposite thin films. J. Alloys Compd. 2017, 714, 114–119. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, H.; Yuan, N.; Meng, L.; Geng, C.; Bai, H. Insights into reactive behaviors and mechanisms of nickel-based oxygen carriers doped by Fe/Co during chemical looping combustion from multiple-scale molecular modeling combined with experiments. Fuel Process. Technol. 2022, 229, 107181. [Google Scholar] [CrossRef]
- Xie, H.; Su, W.; Lu, H.; Mo, Z.; Wang, D.; Sun, H.; Tian, L.; Gao, X.; Li, Z.; Shen, J. Enhanced low-field magnetocaloric effect in Nb and Al co-substituted EuTiO3 compounds. Mater. Sci. Technol. 2022, 118, 128. [Google Scholar] [CrossRef]
- Goesten, M.; Xia, Y.; Aschauer, U.; Amsler, M. Conformational Gap Control in CsTaS3. J. Am. Chem. Soc. 2022, 144. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Chen, X.; Zhao, Y.; Lai, T. Theoretical study of stability and electronic structure of the new type of ferroelectric materials XSnO3(X = Mn, Zn, Fe, Mg). Int. J. Mod. Phys. B 2014, 28, 1450224. [Google Scholar] [CrossRef]
- Sun, X.; Liu, Y.; Song, Z.; Li, Y.; Wang, W.; Lin, H.; Wang, L.; Li, Y. Structures, mobility and electronic properties of point defects in arsenene, antimonene and an antimony arsenide alloy. J. Mater. Chem. C 2017, 5, 4159–4166. [Google Scholar] [CrossRef]
- Mishra, N.; Makov, G. Point defects in lead sulfide: A first-principles study. Comput. Mater. Sci. 2021, 190, 110285. [Google Scholar] [CrossRef]
- Liu, L.; Diao, Y.; Xia, S. Intrinsic point defects in pristine and Zn-doped GaAs nanowire surfaces: A first-principles investigation. Appl. Surf. Sci. 2020, 514, 145906. [Google Scholar] [CrossRef]
- Jiang, L.; Chen, Z.; Cui, Q.; Xu, S.; Tang, F. Experimental and DFT-D3 study of sensitivity and sensing mechanism of ZnSnO3 nanosheets to C3H6O gas. J. Mater. Sci. 2022, 57, 1–21. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Lattice Constants | a = b | c | References |
|---|---|---|---|
| Exp. | 5.344 | 14.221 | [4] |
| Cal. | 5.562 | 14.003 | [31] |
| This work | 5.351 | 14.224 | / |
| Models | Ecoh/eV/Atom | Ef/eV |
|---|---|---|
| Bulk ZS | 5.324 | / |
| 001 surface | 5.099 | / |
| VZn surface | 5.122 | 1.175 |
| VO surface | 5.143 | −4.318 |
| VSn surface | 5.069 | 4.928 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, J.; Zhu, M.; Feng, R.; Yuan, Y.; Fu, Z.; Meng, L.; Wang, Y.; Zhou, Y.; Zhang, H.; Bai, H. Structural Characteristics, Stability, and Electronic Properties of 001 Surface with Point Defects of Zinc Stannate: A First-Principle Study. Inorganics 2022, 10, 258. https://doi.org/10.3390/inorganics10120258
Li J, Zhu M, Feng R, Yuan Y, Fu Z, Meng L, Wang Y, Zhou Y, Zhang H, Bai H. Structural Characteristics, Stability, and Electronic Properties of 001 Surface with Point Defects of Zinc Stannate: A First-Principle Study. Inorganics. 2022; 10(12):258. https://doi.org/10.3390/inorganics10120258
Chicago/Turabian StyleLi, Jun, Meilin Zhu, Rou Feng, Yingjie Yuan, Zewei Fu, Liangliang Meng, Yingwu Wang, Ying Zhou, Hui Zhang, and Hongcun Bai. 2022. "Structural Characteristics, Stability, and Electronic Properties of 001 Surface with Point Defects of Zinc Stannate: A First-Principle Study" Inorganics 10, no. 12: 258. https://doi.org/10.3390/inorganics10120258