

Spontaneous Release of Metalloradicals and Coordinatively Unsaturated Species in Asymmetric Iridium Dimers to Promote C-N Bond Formation

Abstract
:1. Introduction
2. Results and Discussion
2.1. Synthesis and Structural Characterization
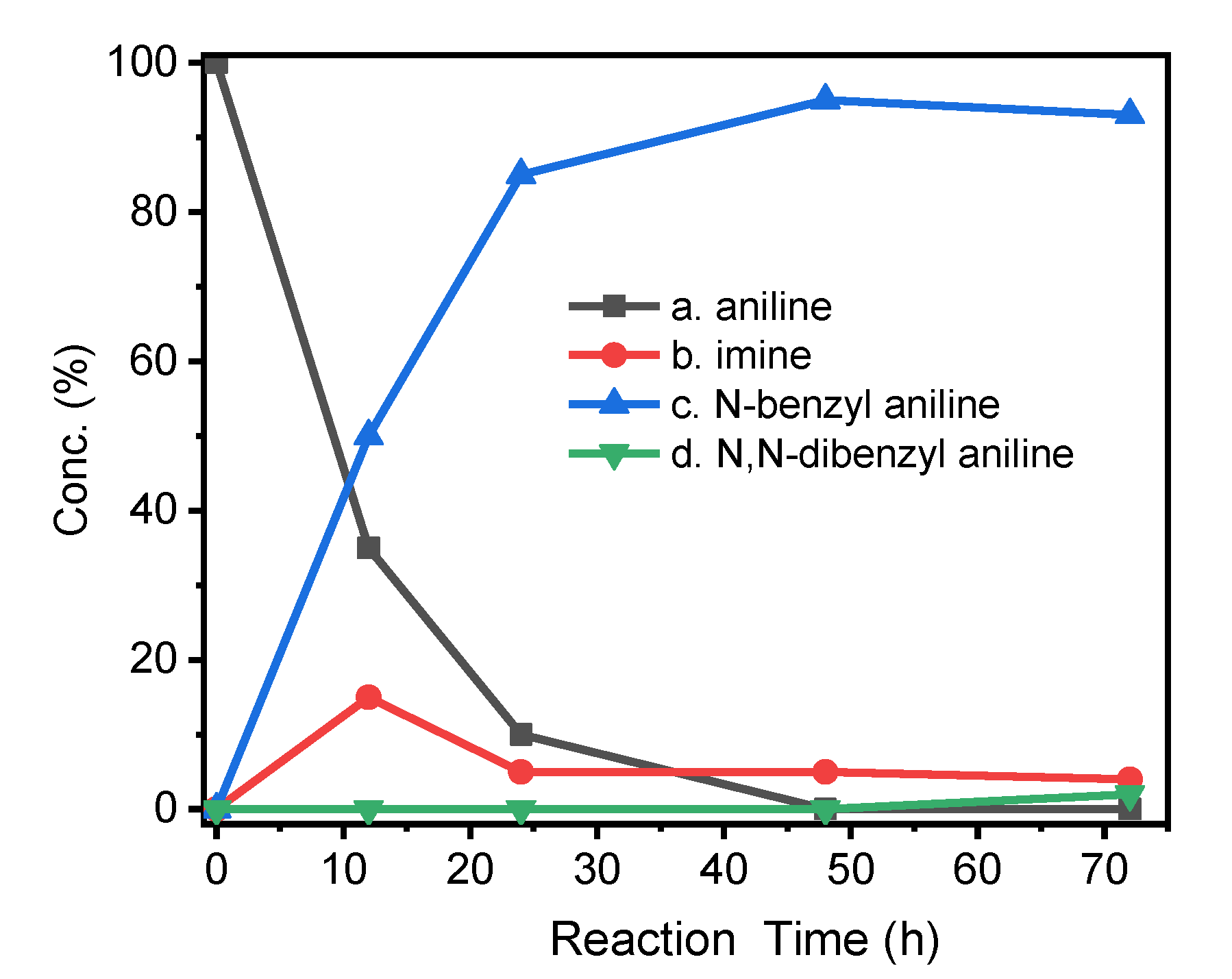
2.2. Initial Catalytic Studies
2.3. Substrate Scope
2.4. Synthetic Application
2.5. Conclusions
3. Experimental Section
3.1. Materials and Methods
3.2. Synthetic Procedures
3.2.1. Synthesis of 4-Chlorophenylbenzoxazole, (4-cpboH)
3.2.2. Synthesis of [(4-cpbo)2Ir(μ-Cl)]2
3.2.3. Synthesis of Compound 1, [(4-cpbo)Ir(μ-Cl) (μ-O)Ir(4-cpbo)]
3.2.4. General Procedure for N-alkylation Reaction
3.2.5. Intermolecular Cyclyzation to Synthesize Cyclizine
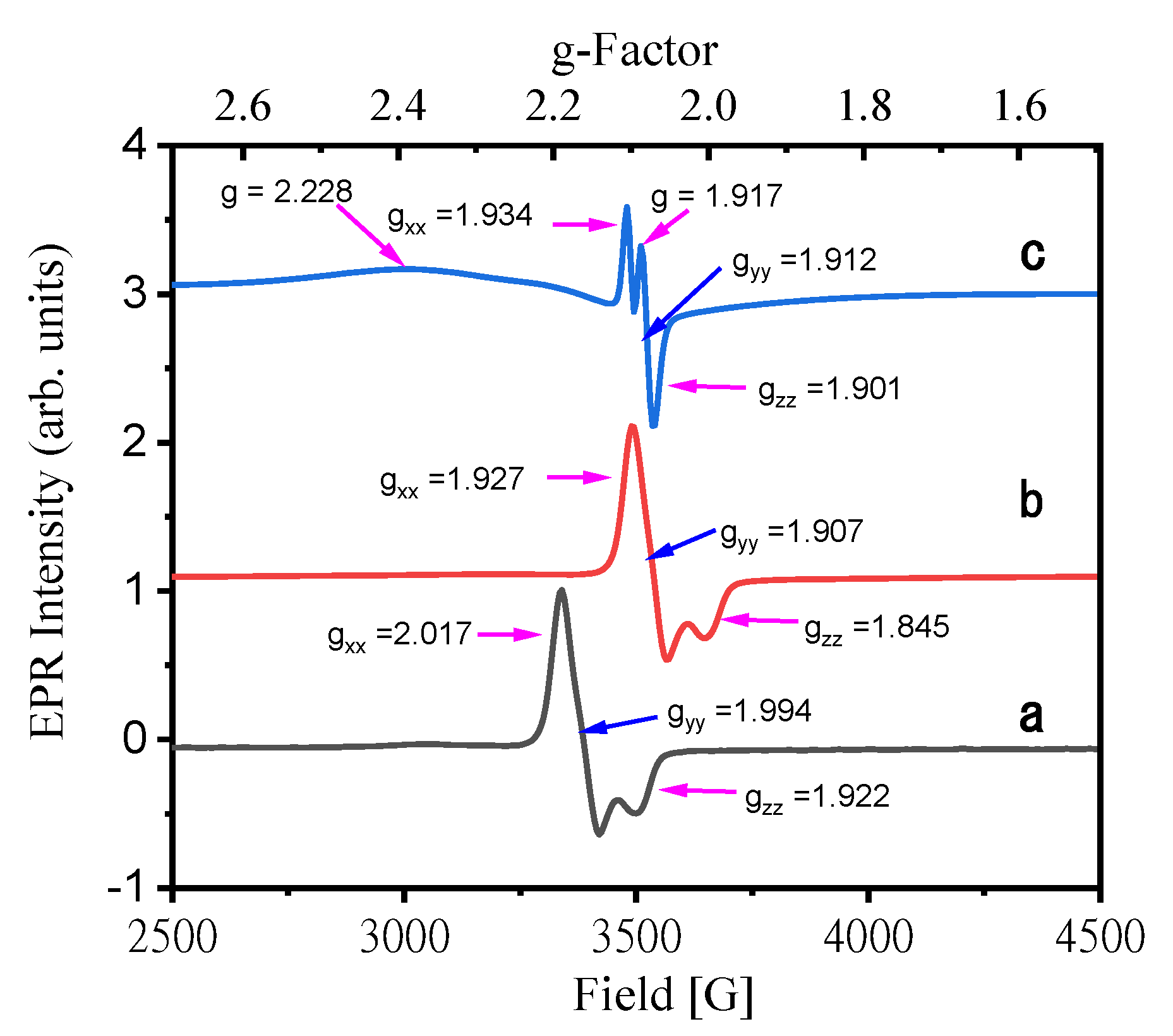
3.3. Electron Paramagnetic Resonance (EPR)
3.4. Single-Crystal X-ray Diffraction
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Roy, S.; Das, S.K.; Khatua, H.; Das, S.; Chattopadhyay, B. Road Map for the Construction of High-Valued N-Heterocycles via Denitrogenative Annulation. Acc. Chem. Res. 2021, 54, 4395–4409. [Google Scholar] [CrossRef] [PubMed]
- Mondal, S.; Dumur, F.; Gigmes, D.; Sibi, M.P.; Bertr, M.P.; Nechab, M. Enantioselective Radical Reactions Using Chiral Catalysts. Chem. Rev. 2022, 122, 5842–5967. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wen, X.; Cui, X.; Wojtas, L.; Zhang, X.P. Asymmetric Radical Cyclopropanation of Alkenes with In Situ-Generated Donor-Substituted Diazo Reagents via Co(II)-Based Metalloradical Catalysis. J. Am. Chem. Soc. 2017, 139, 1049–1052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; Wang, D.-S.; Lee, W.-C.C.; McKillop, A.M.; Zhang, X.P. Controlling Enantioselectivity and Diastereoselectivity in Radical Cascade Cyclization for Construction of Bicyclic Structures. J. Am. Chem. Soc. 2021, 143, 11130–11140. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Xu, P.; Zhu, Y.; Wang, J.; Lee, W.-C.C.; Zhang, X.P. New Catalytic Radical Process Involving 1,4-Hydrogen Atom Abstraction: Asymmetric Construction of Cyclobutanones. J. Am. Chem. Soc. 2021, 143, 11670–11678. [Google Scholar] [CrossRef]
- Zhou, M.; Wolzak, L.A.; Li, Z.; de Zwart, F.J.; Mathew, S.; de Bruin, B.J. Catalytic Synthesis of 1H-2-Benzoxocins: Cobalt(III)-Carbene Radical Approach to 8-Membered Heterocyclic Enol Ethers. Am. Chem. Soc. 2021, 143, 20501–20512. [Google Scholar] [CrossRef]
- Ke, J.; Lee, W.-C.C.; Wang, X.; Wang, Y.; Wen, X.; Zhang, X.P.J. Metalloradical Activation of In Situ-Generated α-Alkynyldiazomethanes for Asymmetric Radical Cyclopropanation of Alkenes. Am. Chem. Soc. 2022, 144, 2368–2378. [Google Scholar] [CrossRef]
- Yu, M.; Fu, X.J. Visible Light Promoted Hydroxylation of a Si–C(sp3) Bond Catalyzed by Rhodium Porphyrins in Water. Am. Chem. Soc. 2011, 133, 15926–15929. [Google Scholar] [CrossRef]
- Li, G.; Han, A.; Pulling, M.E.; Estes, D.P.; Norton, J.R. Evidence for Formation of a Co–H Bond from (H2O)2Co(dmgBF2)2 under H2: Application to Radical Cyclizations. J. Am. Chem. Soc. 2012, 134, 14662–14665. [Google Scholar] [CrossRef]
- Zhang, Q.; Wang, S.; Zhang, Q.; Xiong, T.; Zhang, Q. Radical Addition-Triggered Remote Migratory Isomerization of Unactivated Alkenes to Difluoromethylene-Containing Alkenes Enabled by Bimetallic Catalysis. ACS Catal. 2022, 12, 527–535. [Google Scholar] [CrossRef]
- Stevens, H.; Duan, P.-C.; Dechert, S.; Meyer, F. Competing H2 versus Intramolecular C–H Activation at a Dinuclear Nickel Complex via Metal–Metal Cooperative Oxidative Addition. J. Am. Chem. Soc. 2020, 142, 6717–6728. [Google Scholar] [CrossRef]
- Ott, J.C.; Bürgy, D.; Guan, H.; Gade, L.H. 3d Metal Complexes in T-shaped Geometry as a Gateway to Metalloradical Reactivity. Acc. Chem. Res. 2022, 55, 857–868. [Google Scholar] [CrossRef]
- Chen, J.; Liang, Y.-J.; Wang, P.-Z.; Li, G.-Q.; Zhang, B.; Qian, H.; Huan, X.-D.; Guan, W.; Xiao, W.-J.; Chen, J.-R. Photoinduced Copper-Catalyzed Asymmetric C–O Cross-Coupling. J. Am. Chem. Soc. 2021, 143, 13382–13392. [Google Scholar] [CrossRef]
- Lee, H.; Ahn, J.M.; Oyala, P.H.; Citek, C.; Yin, H.; Fu, G.C.; Peters, J.C. Investigation of the C–N Bond-Forming Step in a Photoinduced, Copper-Catalyzed Enantioconvergent N–Alkylation: Characterization and Application of a Stabilized Organic Radical as a Mechanistic Probe. J. Am. Chem. Soc. 2022, 144, 4114–4213. [Google Scholar] [CrossRef]
- Sinha, S.K.; Guin, S.; Maiti, S.; Biswas, J.P.; Porey, S.; Maiti, D. Toolbox for Distal C-H Bond Functionalizations in Organic Molecules. Chem. Rev. 2022, 122, 5682–5841. [Google Scholar] [CrossRef]
- Wang, X.; Ke, J.; Zhu, Y.; Deb, A.; Xu, Y.; Zhang, X.P. Asymmetric Radical Process for General Synthesis of Chiral Heteroaryl Cyclopropanes. J. Am. Chem. Soc. 2021, 143, 11121–11129. [Google Scholar] [CrossRef]
- Hu, Y.; Lang, K.; Li, C.; Gill, J.B.; Kim, I.; Lu, H.; Fields, K.B.; Marshall, M.; Cheng, Q.; Cui, X.; et al. Enantioselective Radical Construction of 5-Membered Cyclic Sulfonamides by Metalloradical C–H Amination. J. Am. Chem. Soc. 2019, 141, 18160–18169. [Google Scholar] [CrossRef]
- Juliá, F.; Constantin, T.; Leonori, D. Applications of Halogen-Atom Transfer (XAT) for the Generation of Carbon Radicals in Synthetic Photochemistry and Photocatalysis. Chem. Rev. 2022, 122, 2292–2352. [Google Scholar] [CrossRef]
- Jia, Z.; Zhang, L.; Luo, S. Asymmetric C–H Dehydrogenative Allylic Alkylation by Ternary Photoredox-Cobalt-Chiral Primary Amine Catalysis under Visible Light. J. Am. Chem. Soc. 2022, 144, 10705–10710. [Google Scholar] [CrossRef]
- Hershberger, J.W.; Klingler, R.J.; Kochi, J.K. Electron-transfer catalysis. Radical chain mechanism for the ligand substitution of metal carbonyls. J. Am. Chem. Soc. 1982, 104, 3034–3043. [Google Scholar]
- Chen, T.-R.; Lee, H.-P.; Chen, J.-D.; Chen, K.H.-C. An 18+δ iridium dimer releasing metalloradicals spontaneously. Dalton Trans. 2010, 39, 9458–9461. [Google Scholar] [CrossRef] [PubMed]
- Klein, A.; Vogler, C.; Kaim, W. Organometallics, The δ in 18 + δ Electron Complexes: Importance of the Metal/Ligand Interface for the Substitutional Reactivity of “Re(0)” Complexes (α-diimine-)ReI(CO)3(X). Organometallics 1996, 15, 236–244. [Google Scholar] [CrossRef]
- Mao, F.; Schut, D.M.; Tyler, D.R. Catalysis by 18 + δ compounds. Cyclooligomerization of acetylenes catalyzed by Co(CO)3L2 Organometallics. Organometallics 1996, 15, 4770–4775. [Google Scholar] [CrossRef]
- Powell, D.A.; Ramsden, P.D.; Batey, R.A. Phase-Transfer-Catalyzed Alkylation of Guanidines by Alkyl Halides under Biphasic Conditions: A Convenient Protocol for the Synthesis of Highly Functionalized Guanidines. J. Org. Chem. 2003, 68, 2300–2309. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.-S.; Shen, Z.-L.; Loh, T.-P. Indium (Zinc)-Copper-Mediated Barbier-Type Alkylation Reaction of Nitrones in Water: Synthesis of Amines and Hydroxylamines. Org. Lett. 2009, 11, 1209–1212. [Google Scholar] [CrossRef]
- Mirabdolbaghi, R.; Dudding, T. Expanding the Forefront of Strong Organic Brønsted Acids: Proton-Catalyzed Hydroamination of Unactivated Alkenes and Activation of Au(I) for Alkyne Hydroamination. Org. Lett. 2015, 17, 1930–1933. [Google Scholar] [CrossRef]
- Blieck, R.; Bahri, J.; Taillefer, M.; Monnier, F. Copper-Catalyzed Hydroamination of Terminal Allenes. Org. Lett. 2016, 18, 1482–1485. [Google Scholar] [CrossRef]
- Zhao, X.; She, Y.; Fang, K.; Li, G. CuCl-Catalyzed Ullmann-Type C−N Cross-Coupling Reaction of Carbazoles and 2-Bromopyridine Derivatives. J. Org. Chem. 2017, 82, 1024–1033. [Google Scholar] [CrossRef]
- Yoo, W.-J.; Tsukamoto, T.; Kobayashi, S. Visible Light-Mediated Ullmann-Type C−N Coupling Reactions of Carbazole Derivatives and Aryl Iodides. Org. Lett. 2015, 17, 3640–3642. [Google Scholar] [CrossRef]
- Lee, D.-H.; Taher, A.; Hossain, S.; Jin, M.-J. An Efficient and General Method for the Heck and Buchwald_Hartwig Coupling Reactions of Aryl Chlorides. Org. Lett. 2011, 13, 5540–5543. [Google Scholar] [CrossRef]
- Mitsudo, K.; Shimohara, S.; Mizoguchi, J.; Mandai, H.; Suga, S. Synthesis of Nitrogen-Bridged Terthiophenes by Tandem Buchwald_Hartwig Coupling and Their Properties. Org. Lett. 2012, 14, 2702–2705. [Google Scholar] [CrossRef]
- Chen, T.-R.; Chen, Y.-T.; Chen, Y.-S.; Lee, W.-J.; Lin, Y.-H.; Wang, H.-C. Iridium/graphene nanostructured catalyst for the N-alkylation of amines to synthesize nitrogen-containing derivatives and heterocyclic compounds in a green process. RSC Adv. 2022, 12, 4760–4770. [Google Scholar] [CrossRef]
- Chen, T.-R. Synthesis and characterization of cyclometalated iridium(III) complexes containing benzoxazole derivatives and different ancillary ligands. J. Organomet. Chem. 2008, 693, 3117–3130. [Google Scholar] [CrossRef]
- Thiyagarajan, S.; Gunanathan, C. Direct Catalytic Symmetrical, Unsymmetrical N,N-Dialkylation and Cyclization of Acylhydrazides Using Alcohols. Org. Lett. 2020, 22, 6617–6622. [Google Scholar] [CrossRef]
- Yuan, K.; Jiang, F.; Sahli, Z.; Achard, M.; Roisnel, T.; Bruneau, C. Iridium-Catalyzed Oxidant-Free Dehydrogenative C-H Bond Functionalization: Selective Preparation of N-Arylpiperidines through Tandem Hydrogen Transfers, Angew. Chem. Int. Ed. 2012, 51, 8876–8880. [Google Scholar] [CrossRef]
- Zou, Q.; Wang, C.; Smith, J.; Xue, D.; Xiao, J. Alkylation of Amines with Alcohols and Amines by a Single Catalyst under Mild Conditions. Chem.—Eur. J. 2015, 21, 9656–9661. [Google Scholar] [CrossRef]
- Blank, B.; Kempe, R.J. Catalytic Alkylation of Methyl-N-Heteroaromatics with Alcohols. Am. Chem. Soc. 2010, 132, 924–925. [Google Scholar] [CrossRef]
- Eka Putra, A.; Oe, Y.; Ohta, T. Ruthenium-Catalyzed Enantioselective Synthesis of β-Amino Alcohols from 1,2-Diols by “Borrowing Hydrogen”. Eur. J. Org. Chem. 2013, 2013, 6146–6151. [Google Scholar] [CrossRef]
- Balaraman, E.; Srimani, D.; Diskin-Posner, Y.; Milstein, D. Direct Synthesis of Secondary Amines From Alcohols and Ammonia Catalyzed by a Ruthenium Pincer Complex. Catal. Lett. 2015, 145, 139–144. [Google Scholar] [CrossRef] [Green Version]
- Quintard, A.; Rodriguez, J. A Step into an eco-Compatible Future: Iron- and Cobalt-catalyzed Borrowing Hydrogen Transformation. ChemSusChem 2016, 9, 28–30. [Google Scholar] [CrossRef]
- Santoro, F.; Psaro, R.; Ravasio, N.; Zaccheria, F. N-Alkylation of amines through hydrogen borrowing over a heterogeneous Cu catalyst. RSC Adv. 2014, 4, 2596–2600. [Google Scholar] [CrossRef]
- Clubiey, M.; Henson, T.; Riddington, A.W.; Peck, C.B. Diazepam metabolism in human foetal and adult liver. J. Clin. Pharmacol. 1977, 4, 652–662. [Google Scholar]
- Afanasyev, O.I.; Kuchuk, E.; Usanov, D.L. Chusov, Reductive Amination in the Synthesis of Pharmaceuticals. D. Chem. Rev. 2019, 119, 11857–11911. [Google Scholar] [CrossRef] [PubMed]
- Abbenhuis, R.A.T.M.; Boersma, J.; van Koten, G. Ruthenium-Complex-Catalyzed N-(Cyclo)alkylation of Aromatic Amines with Diols. Selective Synthesis of N-(ω-Hydroxyalkyl)anilines of Type PhNH(CH2)nOH and of Some Bioactive Arylpiperazines. J. Org. Chem. 1998, 63, 4282–4290. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bond Distance (Å) | |||
| Ir(1)-N(1) | 2.157 (7) | Ir(1)-N(2) | 2.049 (7) |
| Ir(1)-C(13) | 2.011 (9) | Ir(1)-C(26) | 2.035 (9) |
| Ir(1)-Cl(5) | 2.457 (3) | Ir(1)-O(5) | 2.093 (5) |
| Ir(2)-N(3) | 2.069 (8) | Ir(2)-N(4) | 2.037 (7) |
| Ir(2)-C(39) | 2.021 (9) | Ir(2)-C(52) | 2.022 (10) |
| Ir(2)-Cl(5) | 2.402 (3) | Ir(2)-O(5) | 2.153 (5) |
| Bond Angles (o) | |||
| Ir(1)-Cl(5)-Ir(2) | 87.76 (11) | Ir(1)-O(5)-Ir(2) | 105.0 (3) |
| C(13)-Ir(1)-C(26) | 92.7 (3) | C(39)-Ir(2)-C(52) | 94.7 (4) |
| N(1)-Ir(1)-N(2) | 95.8 (3) | N(3)-Ir(2)-N(4) | 170.5 (3) |
| C(26)-Ir(1)-N(1) | 171.9 (3) | C(39)-Ir(2)-N(3) | 79.5 (4) |
| C(13)-Ir(1)-Cl(5) | 170.4 (3) | C(39)-Ir(2)-Cl(5) | 173.5 (3) |
| O(5)-Ir(1)-Cl(5) | 83.49 (17) | O(5)-Ir(2)-Cl(5) | 83.62 (17) |
| N(2)-Ir(1)-O(5) | 171.6 (2) | C(52)-Ir(2)-O(5) | 167.7 (3) |
| Ir-Cl | Ir-O | Ir-N | Ir-C | |
|---|---|---|---|---|
| X-ray | 2.457 (3) | 2.153 (5) | 2.157 (7) | 2.035 (9) |
| 2.402 (3) | 2.093 (5) | 2.069 (8) | 2.022 (10) | |
| 2.049 (7) | 2.021 (9) | |||
| 2.037 (7) | 2.011 (9) | |||
| DFT | 2.454 | 2.150 | 2.154 | 2.031 |
| 2.309 | 2.090 | 2.065 | 2.017 | |
| 2.045 | 2.016 | |||
| 2.033 | 2.008 |
 |
|---|
 |
| Amines (1 mmol), alcohols (2 mmol), catalyst (0.018 mmol), without base and solvent, and reaction was carried out at 120 °C for 48 h. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, T.-R.; Chen, Y.-S.; Li, C.-Y.; Lin, Y.-H.; Chen, Y.-T. Spontaneous Release of Metalloradicals and Coordinatively Unsaturated Species in Asymmetric Iridium Dimers to Promote C-N Bond Formation. Inorganics 2022, 10, 237. https://doi.org/10.3390/inorganics10120237
Chen T-R, Chen Y-S, Li C-Y, Lin Y-H, Chen Y-T. Spontaneous Release of Metalloradicals and Coordinatively Unsaturated Species in Asymmetric Iridium Dimers to Promote C-N Bond Formation. Inorganics. 2022; 10(12):237. https://doi.org/10.3390/inorganics10120237
Chicago/Turabian StyleChen, Tsun-Ren, Yi-Sheng Chen, Chia-Ying Li, Yen-Hsing Lin, and Yu-Tung Chen. 2022. "Spontaneous Release of Metalloradicals and Coordinatively Unsaturated Species in Asymmetric Iridium Dimers to Promote C-N Bond Formation" Inorganics 10, no. 12: 237. https://doi.org/10.3390/inorganics10120237