
Two Eco-Friendly Chromatographic Methods Evaluated by GAPI for Simultaneous Determination of the Fluoroquinolones Moxifloxacin, Levofloxacin, and Gemifloxacin in Their Pharmaceutical Products
 , and
, and
Abstract
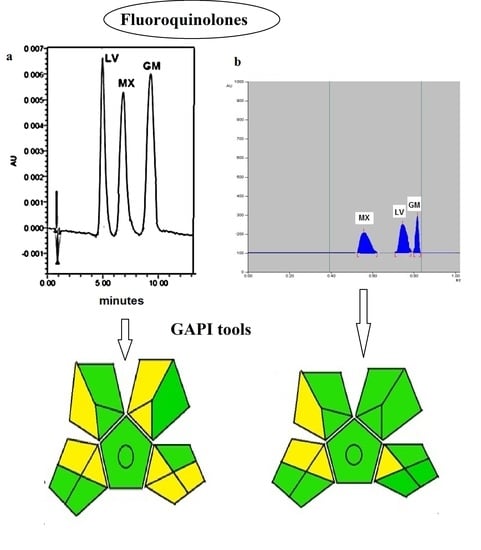
:
1. Introduction
2. Experimental
2.1. Instrumentation
2.2. Chemicals and Reagents
2.3. Pharmaceutical Samples
2.4. Chromatographic Conditions
2.4.1. For HPLC Method
2.4.2. For HTPLC Method
2.5. Standard Solutions and Calibration
2.5.1. For HPLC Method
2.5.2. For HPTLC Method
2.6. Analysis of the Studied Antibiotics in Their Dosage Forms by Proposed Methods
3. Results and Discussion
3.1. Method Development and Optimization
3.2. The Validation of the Methods
3.2.1. Linearity
3.2.2. Detection and Quantitation limits
3.2.3. Selectivity
3.2.4. Accuracy
3.2.5. Precision
3.3. Analysis of Pharmaceutical Products
3.4. Evaluation of the Greenness of the Proposed Analytical Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pharm, T.D.M.; Ziora, Z.M.; Blaskovich, M.A.T. Quinolone antibiotics. Med. Chemcomm. 2019, 10, 1719–1739. [Google Scholar]
- King, D.; Malone, R.; Lilly, S. New Classification and Update on the Quinolone Antibiotics. Am. Fam. Physician 2000, 61, 2741–2748. [Google Scholar] [PubMed]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. Drug Bank 5.0: A major update to the Drug Bank database for 2018. Nucleic Acids Res. 2018, 46, 1074–1082. [Google Scholar] [CrossRef] [PubMed]
- Abdelrady, A.M.; Zaitone, S.A.; Farag, N.E. Cardiotoxic effect of levofloxacin and ciprofloxacin in rats with/without acute myocardial infarction: Impact on cardiac rhythm and cardiac expression of Kv4.3, Kv1.2 and Nav1.5 channels. Biomed. Pharmacother. 2017, 92, 196–206. [Google Scholar] [CrossRef] [PubMed]
- Elbashir, A.; Ebraheem, S.; Elwagee, A.; Aboul-Enein, H. New spectrophotometric methods for the determination of moxifloxacin in pharmaceutical formulations. Acta Chim. Slov. 2013, 60, 159–165. [Google Scholar] [PubMed]
- Kamruzzaman, M.; Alam, A.; Lee, S.; Ragupathy, D.; Kim, Y. Spectrofluorimetric study of the interaction between europium (III) and moxifloxacin in micellar solution and its analytical application. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2012, 86, 375–380. [Google Scholar] [CrossRef] [PubMed]
- Gouda, A.A.; Amin, A.S.; El-Sheikh, R.; Yousef, A.G. Spectrophotometric Determination of Gemifloxacin Mesylate, Moxifloxacin Hydrochloride and Enrofloxacin in Pharmaceutical Formulations Using Acid Dyes. J. Anal. Methods Chem. 2014, 2014, 286379. [Google Scholar] [CrossRef] [PubMed]
- El-Hamshary, M.S.; Fouad, M.A.; Hanafi, R.S.; Al-Easa, H.S.; El-Moghazy, S.M. Screening and optimization of samarium-assisted complexation for the determination of norfloxacin, levofloxacin and lomefloxacin in their corresponding dosage forms employing spectrofluorimetry. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2019, 206, 578–587. [Google Scholar] [CrossRef] [PubMed]
- Wankhede, S.B.; Mahajan, A.M.; Chitlange, S.S. Simultaneous spectrophotometric estimation of Gemifloxacin mesylate and Ambroxol hydrochloride in tablets. Pharma Chem. 2011, 3, 269–273. [Google Scholar]
- Moussa, B.A.; Mahrouse, M.A.; Hassan, M.A. Spectrofluorimetric determination of gemifloxacin mesylate and linezolid in pharmaceutical formulations: Application of quinone-based fluorophores and enhanced native fluorescence. Acta Pharm. 2014, 64, 15–28. [Google Scholar] [CrossRef] [Green Version]
- Ren, Q.; Zhu, X. Methyl-β-Cyclodextrin /Cetyltrimethyl Ammonium Bromide Synergistic Sensitized Fluorescence Method for the Determination of Levofloxacin. J. Fluoresc. 2016, 26, 671–677. [Google Scholar] [CrossRef] [PubMed]
- Ocaña, J.A.; Barragá, F.J.; Callejo, M.; De la Rosa, F. Application of Lanthanide-Sensitised Chemiluminescence to the Determination of Levofloxacin, Moxifloxacin and Trovafloxacin in Tablets. Microchim. Acta 2004, 144, 207–213. [Google Scholar] [CrossRef]
- Kergaravat, S.V.; Gagneten, A.M.; Hernandez, S.R. Development of an electrochemical method for the detection of quinolones: Application to cladoceran ecotoxicity studies. Microchem. J. 2018, 141, 279–286. [Google Scholar] [CrossRef]
- Radi, A.; Wahdan, T.; Anwar, Z.; Mostafa, H. Electrochemical determination of gatifloxacin, moxifloxacin and sparfloxacin fluoroquinolonic antibiotics on glassy carbon electrode in pharmaceutical formulations. Drug Test Anal. 2010, 2, 397–400. [Google Scholar] [CrossRef]
- Vandana, D.; Chaudhary, A.K. A Validated HPTLC Method for Estimation of Moxifloxacin Hydrochloride in Tablets. Pharm. Methods 2010, 1, 54–56. [Google Scholar]
- Khattab, F.I.; Salem, H.; Riad, S.M. Determination of Fluoroquinolone Antibiotics in Industrial Wastewater by High-Pressure Liquid Chromatography and Thin-Layer Chromatography–Densitometric Methods. J. Planar Chromatogr. 2014, 27, 287–293. [Google Scholar] [CrossRef]
- Chepurwar, S.B.; Shirkhedkar, A.A.; Bari, S.B. Validated HPTLC Method for Simultaneous Estimation of Levofloxacin Hemihydrate and Ornidazole in Pharmaceutical Dosage Form. J. Chromatogr. Sci. 2007, 45, 531–536. [Google Scholar] [CrossRef] [Green Version]
- Vaidya, H.; Chorawala, H.; Dedania, Z. Development of Validated HPTLC method for simultaneous determination of levofloxacin hemihydrate and cefpodoxime proxetil in synthetic mixture and tablet dosage form. Pharma Sci. Monit. 2014, 5, 29–34. [Google Scholar]
- Mahmoud, A.M.; Atia, N.N.; El-Shabouri, S.R. Development and Validation of Stability Indicating HPTLC Assay for Determination of Gemifloxacin Mesylate in Dosage Forms. Am. J. Anal. Chem. 2015, 6, 85–97. [Google Scholar] [CrossRef] [Green Version]
- Narayan, U.L.; Garnaik, B.; Patro, S.K. HPTLC Methods for Determination of Gemifloxacin Mesylate in Rabbit Plasma. Br. J. Pharm. Res. 2014, 4, 1707–1714. [Google Scholar] [CrossRef]
- Razzaq, S.N.; Ashfaq, M.; Khan, I.U. Simultaneous determination of dexamethasone and moxifloxacin in pharmaceutical formulations using stability indicating HPLC method. Arab. J. Chem. 2017, 10, 321–328. [Google Scholar] [CrossRef]
- Maddala, V.L.; Ray, P.C.; Bulusu, L.S. Development and Validation of a RP-HPLC method for fast, sensitive and simultaneous determination of few (eight) active pharmaceutical ingredient residues on stainless steel surface in manufactureing plants. Rasayan J. Chem. 2015, 8, 316–320. [Google Scholar]
- Mangukiya, R.P.; Patela, R.A.; Shah, P.A. Chromatographic Methods for Simultaneous Determination of Moxifloxacin Hydrochloride and Difluprednate in Ophthalmic Dosage Form. Acta Chromatogr. 2015, 27, 495–508. [Google Scholar] [CrossRef] [Green Version]
- Belal, F.F.; El-Din, M.K.S.; Saad, S. Micellar liquid chromatographic method for the simultaneous determination of Levofloxacin and Ambroxol in combined tablets: Application to biological fluids. Chem. Cent. J. 2013, 7, 162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bivha, D.; Sujata, M.; Prabhakar, T. Formulation and analysis of levofloxacin hemihydrate by RP-HPLC in bulk and tablet dosage form. Chemistry 2014, 3, 97594519. [Google Scholar]
- Priya, M.V.; Madhavan, P.; Kumar, P. Avalidated RP- HPLC method for the analysis of Moxifloxacin Hydrochloride in pharmaceutical dosage forms and stability studies. J. Chem. Pharm. 2015, 7, 836–841. [Google Scholar]
- EL-Gindy, A.; Emara, S.; Mostafa, A. UV Partial Least-Squares Calibration and Liquid Chromatographic Methods for Direct Quantitation of Levofloxacin in Urine. J. AOAC Int. 2007, 90, 1258–1265. [Google Scholar] [PubMed]
- Durmusa, Z.; Tekkeib, S.E.K.; Kizitasb, M.V. Magnetic Solid Phase ExtractIon for A New HPLC Method for the DeterminatIon of GemifloxacIn in Human Plasma and Breast Milk. J. Chil. Chem. Soc. 2017, 62, 3483–3489. [Google Scholar] [CrossRef] [Green Version]
- Roy, B.; Das, A.; Bhaumik, U.; Sarkar, A.K.; Bose, A.; Mukharjee, J. Determinationof gemifloxacin in different tissues of rat after oral dosing of gemifloxacin mesylate by LC-MS/MS and its application in drug tissue distribution study. J Pharm. Biomed. Anal. 2010, 52, 216–226. [Google Scholar] [CrossRef]
- Ranjane, P.N.; Gandhi, S.V.; Kadukar, S.S. Stability Indicating RP-LC Method for the Determination of Gemifloxacin Mesylate. Chromatographia 2010, 71, 1113–1117. [Google Scholar] [CrossRef]
- Sagirli, O.; Demirci, S.; Önal, A. A very simple high-performance liquid chromatographic method with fluorescence detection for the determination of gemifloxacin in human breast milk. Luminescence 2015, 30, 1326–1329. [Google Scholar] [CrossRef] [PubMed]
- Grunspan, L.D.; Kaiser, M.; Hurtado, F.K.; Costa, T.D.; Tasso, L. HPLC Determination of Gemifloxacin in Different Tissues of Rats Under Normobaric and Hyperbaric Exposure. Chromatographia 2012, 75, 253–262. [Google Scholar] [CrossRef]
- Ganapathy, S.; Raju, G.V.H.; Sankar, D.G. LC Determination of Gemifloxacin in Bulk and Pharmaceutical Formulation. Asian J. Chem. 2009, 21, 6121–6129. [Google Scholar]
- Yabré, M.; Ferey, L.; Gaudin, K. Greening Reversed-Phase Liquid Chromatography Methods Using Alternative Solvents for Pharmaceutical Analysis. Molecules 2018, 23, 1065. [Google Scholar] [CrossRef] [Green Version]
- Attimarad, M.; Ahmed, K.K.M.; Aldhubaib, B.E. High-performance thin layer chromatography: A powerful analytical technique in pharmaceutical drug discovery. Pharm. Methods 2011, 2, 71–75. [Google Scholar] [CrossRef] [Green Version]
- Sprent, P.; Draper, N.R.; Smith, H. Applied Regression Analysis. Biometrics 1981, 37, 863. [Google Scholar] [CrossRef]
- ICH, Q8(R2): Pharmaceutical development. In Proceedings of the International Conference on Harmonization, August 2009.
- Płotka-Wasylka, J. A new tool for the evaluation of the analytical procedure: Green Analytical Procedure. Index. Talanta 2018, 181, 204–209. [Google Scholar] [CrossRef]
- Susdorf, K.; Bevanda, A.M.; Talić, S. Green analytical chemistry: Social dimension and teaching. TrAC Trends Anal. Chem. 2019, 111, 185–196. [Google Scholar] [CrossRef]
- Gałuszka, A.; Migaszewski, Z.; Namieśnik, J. The 12 principles of green analytical chemistry and the Significance mnemonic of green analytical practices. TrAC Trends Anal. Chem. 2013, 50, 78–84. [Google Scholar] [CrossRef]
- Gałuszka, A.; Migaszewski, Z.M.; Konieczka, P. Analytical Eco-Scale for assessing the greenness of analytical procedures. TrAC Trends Anal. Chem. 2012, 37, 61–72. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| (A) HPLC | |||||||
| Compound | Retention Time (min) a | %RSD b of Retention Time | Capacity Factor (K′) | Selectivity (α) c | Resolution (Rs) c | Tailing Factor | Plate Count |
| LV | 4.98 | 1.20 | 4.47 | 1.55 (a1) | 2.10 (b1) | 1.10 | 1841.02 |
| MX | 6.86 | 1.40 | 6.54 | 1.48 (a2) | 2.28 (b2) | 0.99 | 2820.15 |
| GM | 9.34 | 0.44 | 9.26 | 1.20 | 4118.54 | ||
| (B) HPTLC | |||||||
| Compound | Retardation Factor (Rf) | Capacity Factor (K′) | Selectivity (α) c | Resolution (Rs) c | Tailing Factor | ||
| MX | 0.55 | 0.82 | 0.41 (a3) | 1.60 (b3) | 1.3 | ||
| LV | 0.75 | 0.33 | 0.61 (a4) | 1.00 (b4) | 1.2 | ||
| GM | 0.83 | 0.21 | 0.9 | ||||
| HPLC | |||
| Parameters | LV | MX | GM |
| Calibration range (µg mL−1) | 0.1–25.0 | 0.1–25.0 | 0.1–25.0 |
| Detection limit (µg mL−1) | 0.03 | 0.04 | 0.02 |
| Quantitation limit (µg mL−1) | 0.09 | 0.14 | 0.08 |
| Regression equation (Y) a: Slope (b) | 17313.9 | 17405.3 | 12060.3 |
| Standard deviation of the slope (Sb) | 210.9 | 305.098 | 90.17 |
| Relative standard deviation slope (%) | 1.22 | 1.75 | 1.44 |
| Intercept (a) | 1069.84 | 71.59 | 290.6 |
| Correlation coefficient (r) | 0.9999 | 0.9998 | 0.9999 |
| HPTLC | |||
| Parameters | LV | MX | GM |
| Calibration range (µg /band) | 0.05–2.00 | 0.05–2.00 | 0.1–3.0 |
| Detection limit (ng /band) | 15.00 | 14.40 | 43.92 |
| Quantitation limit (ng /band) | 50.01 | 48.00 | 146.40 |
| Regression equation (Y) a: Slope (b) | 20.48 | 31.20 | 22.54 |
| Standard deviation of the slope (Sb) | 0.20 | 0.23 | 0.30 |
| Intercept (a) | −0.93 | 6.23 | 3.47 |
| Correlation coefficient (r) | 0.9997 | 0.9998 | 0.9996 |
| (a) HPTLC Method | %Recovery a ± SD | t-Value b | F-Value b | |
| Proposed | Reported | |||
| Quinabiotic ®® tablets (GM) | 100.88 ± 1.85 | 101.88 ± 0.85 c [19] | 1.30 | 0.98 |
| Monosho ®® tablets(LV) | 98.60 ± 2.10 | 99.79 ± 0.22 c [17] | 1.49 | 0.98 |
| Advancrib ®® tablets (MX) | 101.74 ± 0.85 | 101.2 ± 1.66 c [15] | 0.77 | 0.99 |
| (b) HPLC Method | %Recovery a ± SD | |||
| Proposed | Reported | |||
| Monosho ®® tablets(LV) | 97.3 ± 1.10 | 98.16 ± 0.46 c [25] | 1.91 | 0.98 |
| Advancrib ®® tablets(MX) | 100.96 ± 1.90 | 100.65 ± 1.3 c [26] | 0.36 | 0.99 |
| Quinabiotic ®® tablets(GM) | 98.99 ± 1.00 | 99.20 ± 0.84 c [30] | 0.43 | 1.00 |
| (a) HPLC | ||||||
| Compound | Method | Linearity Range (µg mL−1) | LOD (µg mL−1) | LOQ (µg mL−1) | Matrix | Ref. |
| LV | Proposed | 0.1–25.0 | 0.03 | 0.09 | Dosage form | |
| Reported | 50.0–150.0 | 0.03 | 0.09 | Tablet dosage form | [25] | |
| Reported | 1.0–44.0 | 0.26 | 0.8 | Dosage forms and plasma | [24] | |
| Reported | 0.5–16.5 | 0.03 | 0.09 | Urine | [27] | |
| MX | Proposed | 0.1–25.0 | 0.04 | 0.14 | Dosage form | |
| Reported | 20.0–60.0 | 1.8 | 5.6 | Dosage form | [26] | |
| Reported | 50.0–350.0 | 0.32 | 1.01 | Dosage form | [21] | |
| Reported | 5.0–15.0 | 0.7 | 2.1 | Cleaning validation in factories | [22] | |
| Reported | 1.0–50.0 | 0.84 | 2.56 | Industrial wastewater | [16] | |
| GM | Proposed | 0.1–25.0 | 0.02 | 0.08 | Dosage form | |
| Reported | 5.0–25.0 | 0.171 | 0.518 | Dosage form | [30] | |
| Reported | 0.1–2.5 | 0.63 | 2.1 | Human breast milk | [31] | |
| Reported | 0.2–30.0 | 0.06 | 0.2 | Rat tissues—lung, liver, and kidney | [32] | |
| Reported | 10.34–82.72 | 0.04 | 0.13 | Bulk and pharmaceutical formulation | [33] | |
| (b) HPTLC | ||||||
| Compound | Method | Linearity Range (µg/Band) | LOD (ng/Band) | LOQ (ng/Band) | Matrix | Ref. |
| GM | Proposed | 0.1–3.0 | 43.92 | 146.40 | Dosage form | |
| Reported | 0.1–0.7 | 45.00 | 150.00 | Rabbit plasma | [20] | |
| Reported | 0.002–0.180 | 0.28 | 0.86 | Dosage form | [19] | |
| LV | Proposed | 0.05–2.00 | 14.40 | 48.00 | Dosage form | |
| Reported | 0.05–0.25 | 0.11 | 0.36 | Dosage forms | [17] | |
| Reported | 0.13–0.75 | - | - | Tablet dosage form | [18] | |
| MX | Proposed | 0.05–2.00 | 15.00 | 50.01 | Dosage form | |
| Reported | 9.0–54.0 | - | - | Dosage form | [15] | |
| Reported | 1.2–2.0 | 33.31 | 100.95 | Ophthalmic Dosage form | [23] | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abdel Hameed, E.A.; Abd El-Naby, Z.A.; El Gindy, A.; Saraya, R.E.; Al balawi, A.N.; Zaitone, S.A.; Khairy, G.M. Two Eco-Friendly Chromatographic Methods Evaluated by GAPI for Simultaneous Determination of the Fluoroquinolones Moxifloxacin, Levofloxacin, and Gemifloxacin in Their Pharmaceutical Products. Separations 2022, 9, 330. https://doi.org/10.3390/separations9110330
Abdel Hameed EA, Abd El-Naby ZA, El Gindy A, Saraya RE, Al balawi AN, Zaitone SA, Khairy GM. Two Eco-Friendly Chromatographic Methods Evaluated by GAPI for Simultaneous Determination of the Fluoroquinolones Moxifloxacin, Levofloxacin, and Gemifloxacin in Their Pharmaceutical Products. Separations. 2022; 9(11):330. https://doi.org/10.3390/separations9110330
Chicago/Turabian StyleAbdel Hameed, Eman A., Zaitona A. Abd El-Naby, Alaa El Gindy, Roshdy E. Saraya, Aisha Nawaf Al balawi, Sawsan A. Zaitone, and Gasser M. Khairy. 2022. "Two Eco-Friendly Chromatographic Methods Evaluated by GAPI for Simultaneous Determination of the Fluoroquinolones Moxifloxacin, Levofloxacin, and Gemifloxacin in Their Pharmaceutical Products" Separations 9, no. 11: 330. https://doi.org/10.3390/separations9110330