Enantiomeric Separation of Colchicine and Lacosamide by Nano-LC. Quantitative Analysis in Pharmaceutical Formulations


Abstract
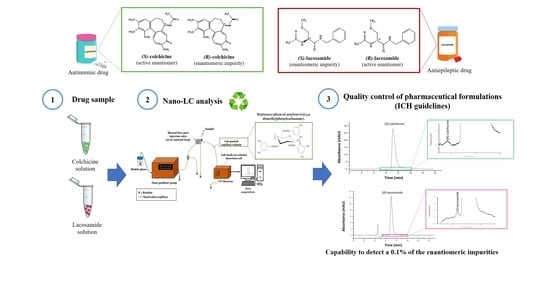
:
1. Introduction
2. Materials and Methods
2.1. Chemicals, Reagents, and Standard Solutions
2.2. Preparation of the Capillary Column
2.3. Nano-LC Analysis
3. Results and Discussion
3.1. Development of a Nano-LC Methodology for the Enantiomeric Separation of Lacosamide and Colchicine
3.2. Analytical Characteristics of the Nano-LC Method Developed
3.3. Analysis of Pharmaceutical Formulations
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Fortuna, A.; Alves, G.; Falcão, A. Chiral chromatographic resolution of antiepileptic drugs and their metabolites: A challenge from the optimization to the application. Biomed. Chromatogr. 2014, 28, 27–58. [Google Scholar] [CrossRef] [PubMed]
- Casado, N.; Valimaña-Traverso, J.; García, M.A.; Marina, M.L. Enantiomeric determination of drugs in pharmaceutical formulations and biological samples by electrokinetic chromatography. Crit. Rev. Anal. Chem. 2019, 1, 1–31. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, A.R.; Maia, A.S.; Cass, Q.B.; Tiritan, M.E. Enantioseparation of chiral pharmaceuticals in biomedical and environmental analyses by liquid chromatography: An overview. J. Chromatogr. B 2014, 968, 8–21. [Google Scholar] [CrossRef] [PubMed]
- Impurities in New Dug Products Q3B (R2). ICH Harmonized Tripartite Guidelines; International Conference of Harmonization (ICH): Brussels, Belgium, 2006. [Google Scholar]
- European Medicines Agency (EMA). Note for Guidance: Investigation of Chiral Active Substances. 1994. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/investigation-chiral-active-substances_en.pdf (accessed on 15 January 2020).
- U.S. Food and Drug Administration (FDA). Guidance, Compliance and Regulatory Information: Development of New Stereoisomeric Drugs. 1992. Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/development-new-stereoisomeric-drugs (accessed on 15 January 2020).
- Sánchez-López, E.; Castro-Puyana, M.; Marina, M.L. Electrophoresis. Capillary Electrophoresis: Chiral Separations. In Encyclopedia of Analytical Science; Worsfold, P., Poole, C., Townshend, A., Miró, M., Eds.; Elsevier: Amsterdam, The Netherlands, 2019; pp. 334–345. [Google Scholar]
- Padró, J.M.; Keunchkarian, S. State-of-the-art and recent developments of immobilized polysaccharide-based chiral stationary phases for enantioseparations by high-performance liquid chromatography (2013–2017). Microchem. J. 2018, 140, 142–157. [Google Scholar] [CrossRef] [Green Version]
- Teixeira, J.; Tiritan, M.E.; Pinto, M.M.M.; Fernandes, C. Chiral stationary phases for liquid chromatography: Recent developments. Molecules 2019, 24, 865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filippou, O.; Bitas, D.; Samanidou, V. Green approaches in sample preparation of bioanalytical samples prior to chromatographic analysis. J. Chromatogr. B 2017, 1043, 44–62. [Google Scholar] [CrossRef] [PubMed]
- Fanali, S. Nano-liquid chromatography applied to enantiomers separation. J. Chromatogr. A 2017, 1486, 20–34. [Google Scholar] [CrossRef] [PubMed]
- Fanali, C.; Asensio-Ramos, M.; Hernández-Borges, J.; Rocco, A.; Fanali, S. Nanoliquid Chromatographic Separations. In Extreme Chromatography; Byrdwell, W.C., Holcapek, M., Eds.; AOCS Press: Urbana, IL, USA, 2011; pp. 301–380. [Google Scholar]
- Valarmathi, R.; Banu, S.F.; Akilandeswari, S.; Senthamarai, R.; Dhharshini, C.S.D. A review on new antiepileptic drug–Lacosamide and its analytical methods. Int. J. Chem. Pharm. Sci. 2013, 2, 181–186. [Google Scholar]
- Parmar, M.D.; Nimavat, K.S.; Vyas, K.B.; Rao, D.V.N.S.; Pande, R.A. A stability-indicating liquid chromatographic method for the quantification of new anti-epileptic drug lacosamide and its intermediates. Int. J. Pharm. Res. Sch. 2012, 1, 40–47. [Google Scholar]
- Chakravarthy, V.K.; Shankar, D.G. HPLC method for determination of lacosamide S (−) enantiomer in bulk and pharmaceutical formulation. Rasayan J. Chem. 2011, 4, 744–752. [Google Scholar]
- Roesner, M.; Capraro, H.G.; Jacobson, A.E.; Atwell, L.; Brossi, A.; Lorio, M.A.; Williams, T.H.; Sik, R.H.; Chignell, C.F. Biological effects of modified colchicines. Improved preparation of 2-demethylcolchicine, 3-demethylcolchicine, and (+)-colchicine and reassignment of the position of the double bond in dehydro-7-deacetamidocolchicines. J. Med. Chem. 1981, 24, 257–261. [Google Scholar] [CrossRef] [PubMed]
- Bhushan, R.; Ali, I. Resolution of racemic mixtures of hyoscyamine and colchicine on impregnated silica gel layers. Chromatographia 1993, 35, 679–680. [Google Scholar] [CrossRef]
- Menéndez-López, N.; Valimaña-Traverso, J.; Castro-Puyana, M.; Salgado, A.; García, M.A.; Marina, M.L. Enantiomeric separation of the antiuremic drug colchicine by electrokinetic chromatography. Method development and quantitative analysis. J. Pharm. Biomed. Anal. 2017, 138, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Shao, H.; Luo, R.; Wang, Q.; Sánchez-López, E.; Fanali, S.; Marina, M.L.; Jiang, Z. A facile and efficient single-step approach for the fabrication of vancomycin functionalized polymer-based monolith as chiral stationary phase for nano-liquid chromatography. J. Chromatogr. A 2018, 1557, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Fernández, V.; Dominguez-Vega, E.; Chankvetadze, B.; Crego, A.L.; García, M.A.; Marina, M.L. Evaluation of new cellulose-based chiral stationary phases Sepapak-2 and Sepapak-4 for the enantiomeric separation of pesticides by nano liquid chromatography and capillary electrochromatography. J. Chromatogr. A 2012, 1234, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Antonucci, V.; Biba, M.; Gong, X.; Ge, Z. Simultaneous enantioseparation of a basic active pharmaceutical ingredient compound and its neutral intermediate using reversed phase and normal phase liquid chromatography with a new type of polysaccharide stationary phase. J. Pharm. Biomed. Anal. 2010, 51, 153–157. [Google Scholar] [CrossRef] [PubMed]
- Gumustas, M.; Ozkan, S.A.; Chankvetadze, B. Separation and elution order of the enantiomers of some β-agonists using polysaccharide-based chiral columns and normal phase eluents by high-performance liquid chromatography. J. Chromatogr. A 2016, 1467, 297–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- European Pharmacopoeia Commision. European Pharmacopoeia, 4th ed.; The European Pharmacopoeia Convention Inc.: Strasbourg, France, 2004; pp. 3843–3849, Supplement 4.6. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| (R)-Lacosamide | (S)-Lacosamide | (S)-Colchicine | (R)-Colchicine | |
|---|---|---|---|---|
| External standard calibration method a | ||||
| Range | 5–100 µg/mL | 2–50 µg/mL | 5–400 µg/mL | 1–15 µg/mL |
| Slope ± t × Sslope | 295 ± 10 | 305 ± 19 | 961 ± 7 | 852 ± 15 |
| Intercept ± t × Sintercept | 237 ± 519 | −175 ± 417 | 276 ± 1216 | 343 ± 126 |
| R2 | 0.996 | 0.995 | 0.999 | 0.999 |
| p-value of ANOVA b | 0.17 | 0.24 | 0.21 | 0.33 |
| Standard additions calibration method c | ||||
| Range | 0–100 µg/mL | 0–50 µg/mL | 0–400 µg/mL | 0–15 µg/mL |
| Slope ± t × Sslope | 324 ± 19 | 256 ± 33 | 958 ± 12 | 865 ± 32 |
| R2 | 0.995 | 0.992 | 0.999 | 0.999 |
| Accuracy d | ||||
| Recovery | 98 ± 8% | 97 ± 3% | 103 ± 4% | 100 ± 2% |
| Precision | ||||
| Instrumental repeatability, RSD (%) e | 2.6 | 7.4 | 2.0 | 0.7 |
| Method repeatability, RSD (%) f | 4.1 | 7.8 | 2.1 | 1.4 |
| Intermediate precision, RSD (%) g | 4.9 | 10.3 | 2.4 | 1.5 |
| LOD h | 2.0 µg/mL | 1.7 µg/mL | 1.0 µg/mL | 1.0 µg/mL |
| LOQ i | 6.7 µg/mL | 5.7 µg/mL | 3.5 µg/mL | 3.5 µg/mL |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Casado, N.; Jiang, Z.; García, M.Á.; Marina, M.L. Enantiomeric Separation of Colchicine and Lacosamide by Nano-LC. Quantitative Analysis in Pharmaceutical Formulations. Separations 2020, 7, 55. https://doi.org/10.3390/separations7040055
Casado N, Jiang Z, García MÁ, Marina ML. Enantiomeric Separation of Colchicine and Lacosamide by Nano-LC. Quantitative Analysis in Pharmaceutical Formulations. Separations. 2020; 7(4):55. https://doi.org/10.3390/separations7040055
Chicago/Turabian StyleCasado, Natalia, Zhengjin Jiang, María Ángeles García, and María Luisa Marina. 2020. "Enantiomeric Separation of Colchicine and Lacosamide by Nano-LC. Quantitative Analysis in Pharmaceutical Formulations" Separations 7, no. 4: 55. https://doi.org/10.3390/separations7040055