
Hollow-Fiber Liquid-Phase Microextraction (HF-LPME) Coupled On-Line to Liquid Chromatography for the Determination of the Herbicides 2,4-Dichlorophenoxyacetic Acid and 2-Methyl-4-chlorophenoxyacetic Acid and Their Main Metabolites in Soil Samples


Abstract
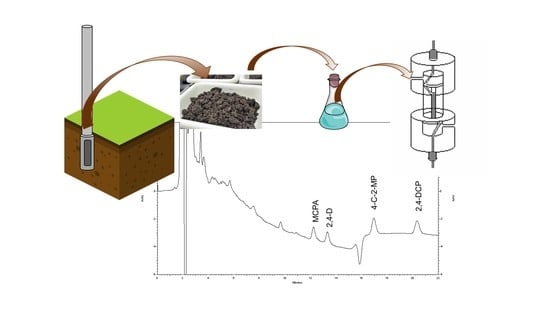
:
1. Introduction
2. Experimental
2.1. Reagents and Solutions
2.2. Instrumentation
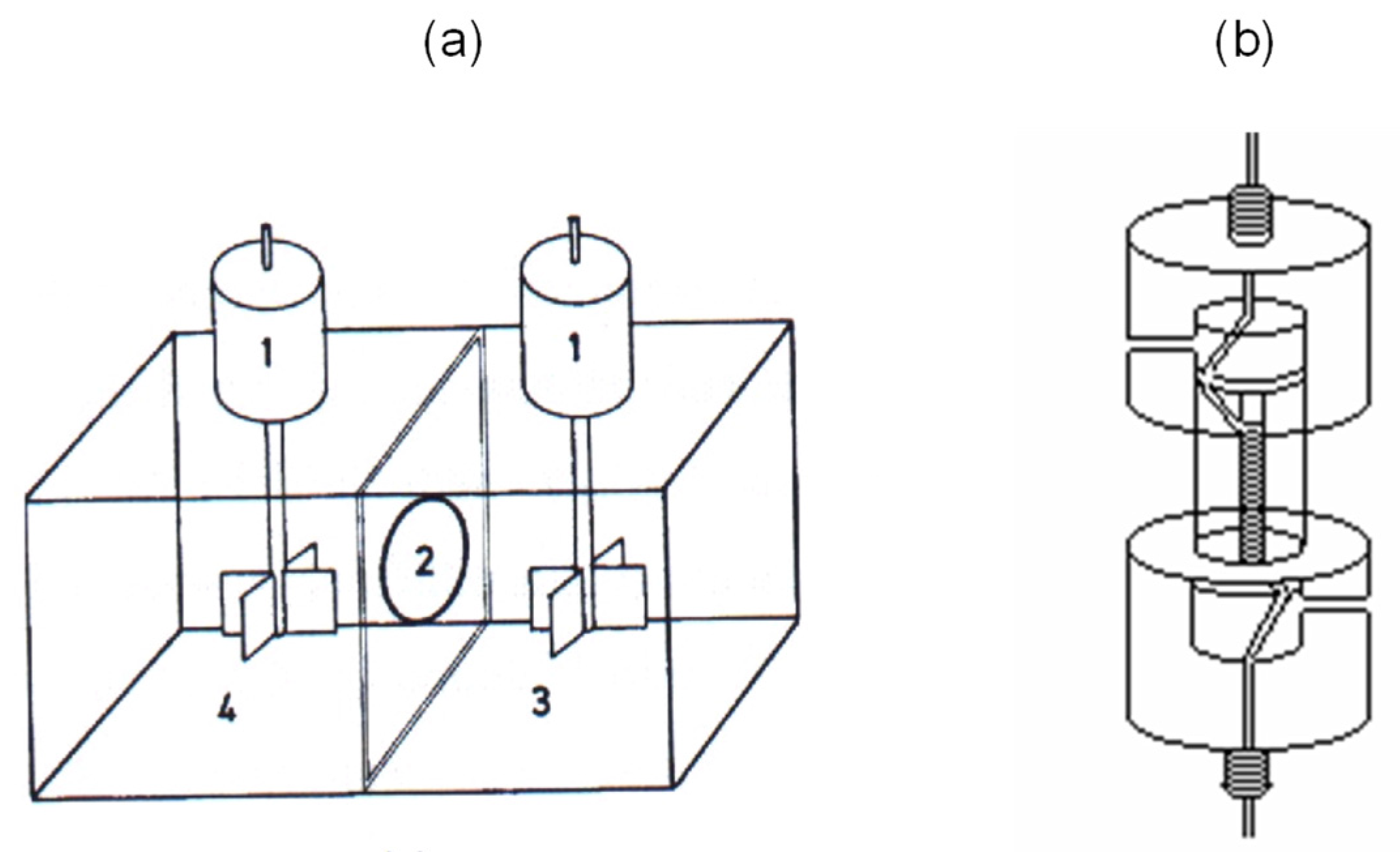
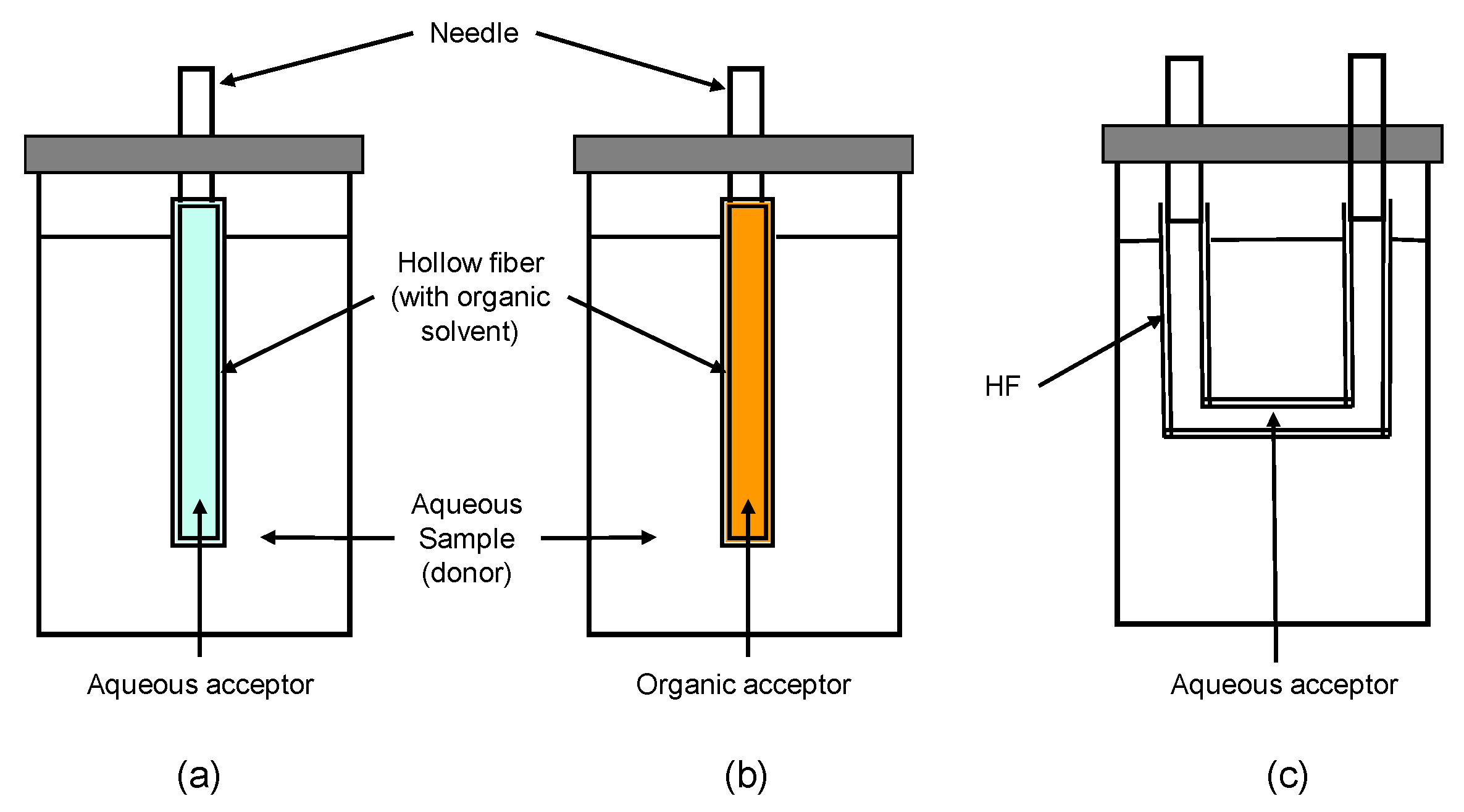
2.3. Membrane Equipment and Procedure
2.4. Soil Samples and Alkaline Extraction
3. Results and Discussion
3.1. Donor (Feed) and Acceptor (Stripping) Phases
3.2. Preliminary Flat-Sheet Membrane Configuration Studies
3.2.1. Selection of the Organic Solvent
3.2.2. Extraction Time and Enrichment Factors
3.3. Hollow-Fiber Configuration: HF-LPME
Sample Clean-Up with HF-LPME
3.4. On-Line HF-LPME Configuration
3.5. Quality Parameters of the On-Line HF-LPME Method
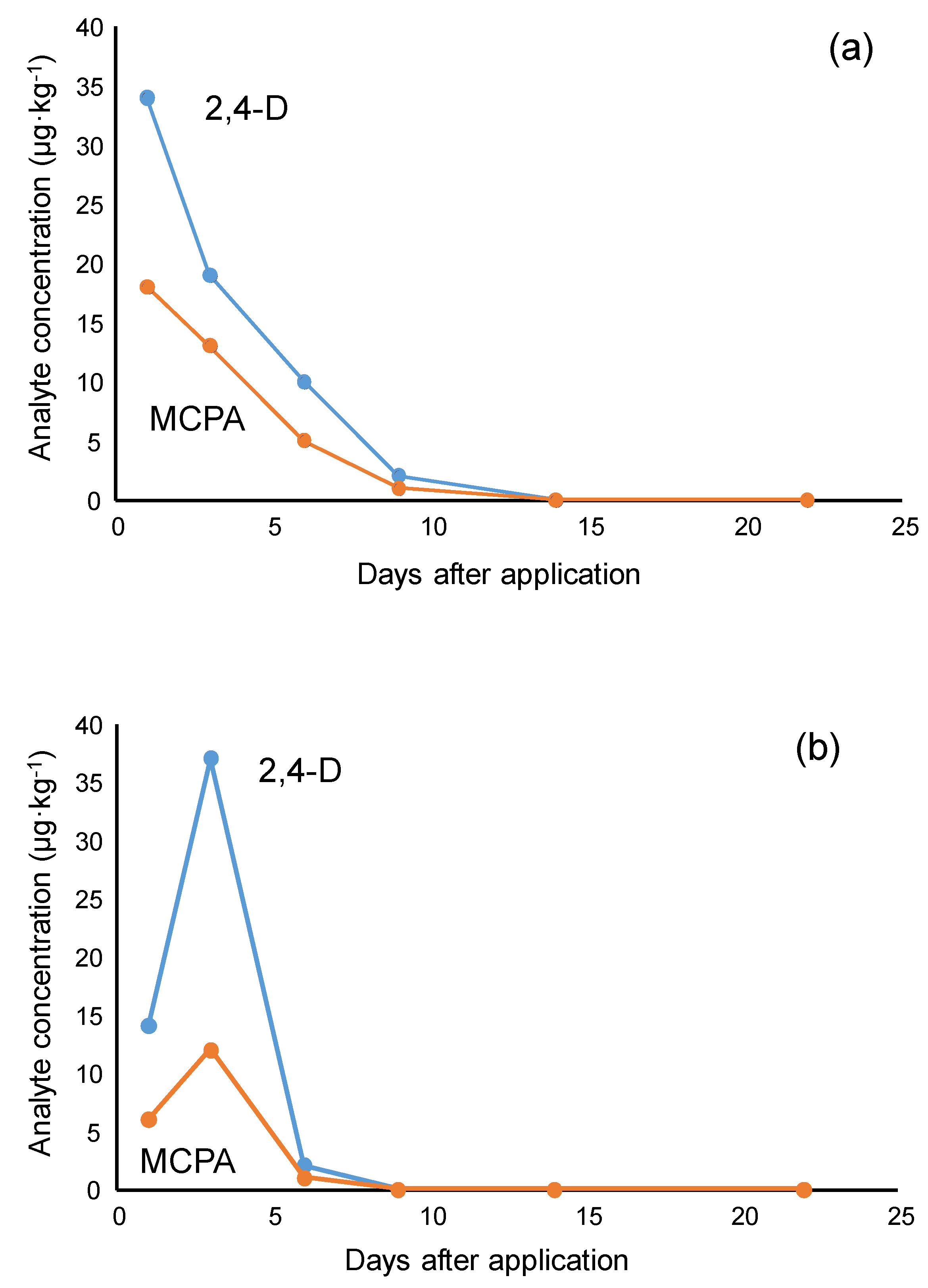
3.6. Analysis of Soil Samples
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- US Environmental Protection Agency. Chlorophenoxy herbicides. In Recognition and Management of Pesticide Poisonings, 6th ed.; Section III, Chapter 10; US-EPA: Washington, DC, USA, 2013; pp. 98–102. Available online: https://www.epa.gov/pesticide-worker-safety/pesticide-poisoning-handbook-complete-document (accessed on 22 March 2023).
- Song, Y. Insight into the mode of action of 2,4-dichlorophenoxyacetic acid (2,4-D) as an herbicide. J. Integr. Plant. Biol. 2014, 56, 106–113. [Google Scholar] [CrossRef]
- Freisthler, M.S.; Robbins, C.R.; Benbrook, C.M.; Young, H.A.; Haas, D.M.; Winchester, P.D.; Perry, M.J. Association between increasing agricultural use of 2,4-D and population biomarkers of exposure: Findings from the National Health and Nutrition Examination Survey, 2001–2014. Environ. Health 2022, 21, 23. [Google Scholar] [CrossRef]
- US Environmental Protection Agency. Pesticides Industry Sales and Usage: 2008–2012 Market Estimates. 2015. Available online: https://www.epa.gov/pesticides/pesticides-industrysales-and-usage-2008-2012-market-estimates (accessed on 1 March 2023).
- Crespin, M.A.; Gallego, M.; Valcarcel, M. Study of the degradation of the herbicides 2,4-D and MCPA at different depths in contaminated agricultural soil. Environ. Sci. Technol. 2001, 35, 4265–4270. [Google Scholar] [CrossRef] [PubMed]
- David, M.D.; Campbell, S.; Li, Q.X. Pressurized fluid extraction of nonpolar pesticides and polar herbicides using in situ derivatization. Anal. Chem. 2000, 72, 3665–3670. [Google Scholar] [CrossRef]
- Mei, X.Y.; Hong, Y.Q.; Chen, G.H. Review on analysis methodology of phenoxy acid herbicide residues. Food Anal. Methods 2016, 9, 1532–1561. [Google Scholar] [CrossRef]
- Armenta, S.; Garrigues, S.; Esteve-Turillas, F.A.; de la Guardia, M. Green extraction techniques in green analytical chemistry. TrAC Trends Anal. Chem. 2019, 116, 248–253. [Google Scholar] [CrossRef]
- Luque-Garcia, J.L.; Morales-Muñoz, S.; Luque de Castro, M.D. Microwave-assisted water extraction of acidic herbicides from soils coupled to continuous filtration, preconcentration, chromatographic separation and UV detection. Chromatographia 2002, 55, 117–122. [Google Scholar] [CrossRef]
- Thorstensen, C.W.; Christiansen, A. Determination of bentazone, dichlorprop, and MCPA in different soils by sodium hydroxide extraction in combination with solid-phase preconcentration. J. Agric. Food Chem. 2001, 49, 4199. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, D.J.; Stearman, G.K.; Wells, M.J.M. Development of an analytical scheme for simazine and 2,4-D in soil and water runoff from ornamental plant nursery plots. J. Agric. Food Chem. 2003, 51, 14–20. [Google Scholar] [CrossRef]
- Moret, S.; Sanchez, J.M.; Salvadó, V.; Hidalgo, M. The evaluation of different sorbents for the preconcentration of phenoxyacetic acid herbicides and their metabolites from soils. J. Chromatogr. A 2005, 1099, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Luque-Garcia, J.L.; Luque de Castro, M.D. Coupling continuous subcritical water extraction, filtration, preconcentration, chromatographic separation and UV detection for the determination of chlorophenoxy acid herbicides in soils. J. Chromatogr. A 2002, 959, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Jönsson, J.A.; Mathiasson, L. Liquid membrane extraction in analytical sample preparation: I. Principles. TrAC Trends Anal. Chem. 1999, 18, 318–325. [Google Scholar] [CrossRef]
- Lord, H.; Pawliszyn, J. Microextraction of drugs. J. Chromatogr. A 2000, 902, 17–63. [Google Scholar] [CrossRef]
- Hassan, J.; Shamsipur, M.; Es’haghi, A.; Fazili, S. Determination of chlorophenoxy acid herbicides in water samples by suspended liquid-phase microextraction-liquid chromatography. Chromatographia 2011, 73, 999–1003. [Google Scholar] [CrossRef]
- Tsai, W.C.; Huang, S.D. Dispersive liquid-liquid-liquid microextraction combined with liquid chromatography for the determination of chlorophenoxy acid herbidices in aqueous samples. J. Chromatogr. A 2009, 1216, 7846–7850. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Xia, L.; Guo, C.; Wu, C.; Chen, G.; Li, G.; Sun, S.; You, J. A sensitive and efficient method for the determination of 8 chlorophenoxy acid herbicides in crops by dispersive liquid-liquid microextraction and HPLC with fluorescence detection and identification by MS. Anal. Methods 2016, 8, 3536–3544. [Google Scholar] [CrossRef]
- Wu, J.; Ee, K.H.; Lee, H.K. Automated dynamic liquid-liquid-liquid microextraction followed by high-performance liquid chromatography-ultraviolet detection for the determination of phenoxy acid herbicides in environmental waters. J. Chromatogr. A 2005, 1082, 121–127. [Google Scholar] [CrossRef]
- Chen, C.C.; Melwanki, M.B.; Huang, S.D. Liquid-liquid-liquid microextraction with automated movement of the acceptor and the donor phase for the extraction of phenoxyacetic acids prior to liquid chromatography detection. J. Chromatogr. A 2006, 1104, 33–39. [Google Scholar] [CrossRef]
- Esrafili, A.; Yamini, Y.; Ghambarian, M.; Moradi, M.; Seidi, S. A novel approach to automation of dynamic hollow fiber liquid-phase microextraction. J. Sep. Sci. 2011, 34, 957–964. [Google Scholar] [CrossRef]
- Ghamari, F.; Bahrami, A.; Yamini, Y.; Shahna, F.G.; Moghimbeigi, A. Development of holow-fiber liquid-phase microextraction method for the determination of urinary trans, trans-muconic acid as a biomarker of benzene exposure. Anal. Chem. Insights 2016, 11, 65–71. [Google Scholar] [CrossRef]
- Liu, K.; He, Y.; Xu, S.; Hu, L.; Luo, K.; Liu, X.; Liu, M.; Zhou, X.; Bai, L. Mechanism of the effect of pH and biochar on the phytotoxicity of the weak acid herbicides imazethapyr and 2,4-D in soil to rice (Oryza sativa) and estimation by chemical methods. Ecotoxicol. Environ. Saf. 2018, 161, 602–609. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Jönsson, J.A.; Mathiasson, L. On-line microporous membrane liquid-liquid extraction for sample pretreament combined with capillary gas chromatography applied to local anaesthesics in blood plasma. Anal. Chem. 1998, 70, 946–953. [Google Scholar] [CrossRef]
- Moral, A.; Caballo, C.; Sicilia, M.D.; Rubio, S. Highly efficient microextraction of chlorophenoxy acid herbicides in natural waters using a decanoic acid-based nanostructured solvent prior to their quantitation by liquid chromatography-mass spectromety. Anal. Chim. Acta 2012, 709, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Lee, H.K. Electro membrane extraction followed by low-density solvent based ultrasound-assisted emulsification microextraction combined with derivatization for determining chlorophenols and analysis by gas chromatography-mass spectrometry. J. Chromatogr. A 2012, 1243, 14–22. [Google Scholar] [CrossRef] [PubMed]
- Moret, S.; Hidalgo, M.; Sanchez, J.M. Development of an Ion-Pairing liquid chromatography method for the determination of phenoxyacetic herbicides and their main metabolites: Application to the analyisis of soil samples. Chromatographia 2006, 63, 109–115. [Google Scholar] [CrossRef]
- Lee, J.; Lee, H.K.; Rasmussen, K.E.; Pedersen-Bjergaad, S. Environmental and bioanalytical applications of hollow fiber membrane liquid-phase microextraction: A review. Anal. Chim. Acta 2008, 624, 253–268. [Google Scholar] [CrossRef] [PubMed]
- Dimitrienko, S.G.; Apyari, V.V.; Tolmacheva, V.V.; Gourbunova, M.V. Liquid-liquid extraction of organic compounds into a single drop of the extractant: Overview of reviews. J. Anal. Chem. 2021, 76, 907–919. [Google Scholar] [CrossRef]
- Gjelstad, A. Three-phase hollow fiber liquid-phase microextraction and parallel artificial liquid membrane extraction. TrAC Trends Anal. Chem. 2019, 113, 25–31. [Google Scholar] [CrossRef]
- Jönsson, J.A.; Mathiasson, L. Liquid membrane extraction in analytical sample preparation: II: Applications. TrAC Trends Anal. Chem. 1999, 18, 325–334. [Google Scholar] [CrossRef]
- Ghambarian, M.; Yamini, Y.; Esrafili, A. Developments in hollow fiber based liquid-phase microextraction: Principles and applications. Microchim. Acta 2012, 177, 271–294. [Google Scholar] [CrossRef]
- Madikizela, L.M.; Pakade, V.E.; Ncube, S.; Tutu, H.; Chimuka, L. Application of hollow fibre-liquid phase microextraction technique for isolation and pre-concentration of pharmaceuticals in water. Molecules 2020, 10, 311. [Google Scholar] [CrossRef] [PubMed]
- Jönsson, J.A.; Mathiasson, L. Membrane-based techniques for sample enrichment. J. Chromatogr. A 2000, 902, 205–225. [Google Scholar] [CrossRef] [PubMed]
- Sandahl, M.; Mathiasson, L.; Jönsson, J.A. Determination of thiophanate-methyl and its metabolites at trace level in spiked natural water using the supported liquid membrane extraction and the microporous membrane liquid-liquid extraction techniques combined on-line with high-performance liquid chromatography. J. Chromatogr. A 2000, 893, 123–131. [Google Scholar]
- Santigosa, E.; Pedersen-Bjergaard, S.; Gimenez-Gomez, P.; Muñoz, M.; Ramos-Payan, M. A rapid and versatile microfluidic method for the simultaneous extraction of polar and non-polar basic pharmaceuticals from human urine. Anal. Chim. Acta 2022, 1208, 338929. [Google Scholar] [CrossRef] [PubMed]
- Chimuka, L.; Mathiasson, L.; Jönsson, J.A. Role of octanol-water partition coefficients in extraction of ionisable organic compounds in a supported liquid membrane with a stagnant acceptor. Anal. Chim. Acta 2000, 416, 77–86. [Google Scholar] [CrossRef]
- Ho, T.S.; Vasskog, T.; Anderssen, T.; Jensen, E.; Rasmussen, K.E.; Pedersen-Bjergaard, E. 25,000-fold pre-concentration in a single step with liquid-phase microextraction. Anal. Chim. Acta 2007, 592, 1–8. [Google Scholar] [CrossRef]
- Di Luccio, M.; Smith, B.D.; Kida, T.; Borges, C.P.; Alves, T.L.M. Separation of fructose from a mixture of sugars using supported liquid membranes. J. Membr. Sci. 2000, 174, 217–224. [Google Scholar] [CrossRef]
- Hou, L.; Lee, H.K. Application of static and dynamic liquid-phase microextraction in the determinatino of polycyclic aromatic hydrocarbons. J. Chromatogr. A 2002, 976, 377–385. [Google Scholar] [CrossRef]
- Sanchez, J.M. Linear calibrations in chromatography: The incorrect use of ordinary least squares for determinations at low levels, and the need to redefine the limit of quantification with this regression model. J. Sep. Sci. 2020, 43, 2708–2717. [Google Scholar] [CrossRef]
- Sanchez, J.M. The inadequate use of the determination coefficient in analytical calibrations: How other parameters can assess the goodness-of-fit more adequately. J. Sep. Sci. 2021, 44, 431–444. [Google Scholar] [CrossRef]
- de Amarante, O.P.; Brito, N.M.; dos Santos, T.C.R.; Nunes, G.S.; Ribeiro, M.L. Determination of 2,4.dichlorophenoxyacetic acid and its major transformation product in soil samples by liquid chormatographic analysis. Talanta 2003, 60, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.E.; Lafond, G.P. Effects of long-term phenoxyalkanoic acid herbicide field applications on the rate of microbial degradation. In Enhanced Biodegradation of Pesticides in the Environment; Rakc, K.D., Coats, J.R., Eds.; ACS Symposium Series: Washington, DC, USA, 1990; Volume 426, pp. 14–22. [Google Scholar]
- US Food and Drug Administration. Bioanalytical Method Validation: Guidance for Industry; FDA: Silver Spring, MD, USA, 2018. Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/bioanalytical-method-validation-guidance-industry (accessed on 18 April 2023).






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Solvent | ||||||||
|---|---|---|---|---|---|---|---|---|
| p-Value | DHE | DHE:Decane (1:1) | Isopropylbenzene | Decane | Decaline | Dodecane (with 4% Dodecanol) | Dodecanol (n = 1) | |
| 2-MP | 0.015 | 73 (8) | 53 (3) | 50 (2) | 0 | 13 (1) | 19 (1) | 13 |
| 2-CP | 0.014 | 80 (6) | 64 (3) | 62 (4) | 48 (4) | 36 (1) | 43 (1) | 16 |
| 4-CP | 0.004 | 76 (1) | 55 (6) | 56 (5) | 0 | 0 | 25 (3) | 3 |
| MCPA | 0.015 | 72 (1) | 60 (5) | 58 (6) | 0 | 0 | 44 (4) | 13 |
| 2,4-D | <0.001 | 67 (1) | 50 (3) | 49 (4) | 0 | 0 | 35 (3) | 11 |
| 4-C-2-MP | 0.171 | 86 (2) | 79 (7) | 77 (5) | 36 (4) | 28 (1) | 57 (6) | 6 |
| 2,4-DCP | 0.042 | 78 (1) | 71 (4) | 70 (3) | 62 (6) | 56 (1) | 65 (1) | 0 |
| Soil Code | Analyte Concentration (µg kg soil−1) | ||||||
|---|---|---|---|---|---|---|---|
| 2-MP | 2-CP | 4-CP | MCPA | 2,4-D | 4-C-2-MP | 2,4-DCP | |
| B1 | d | nd | 1.4 | 3.5 | 6.8 | 1.4 | 1.2 |
| B2 | nd | 1.0 | nd | 1.2 | 2.9 | nd | nd |
| D1 | nd | d | nd | 2.4 | 3.7 | nd | nd |
| D2 | nd | d | nd | 2.3 | 7.2 | nd | nd |
| F1 | nd | d | d | 1.0 | 2.0 | nd | nd |
| F2 | nd | 1.0 | d | nd | nd | nd | nd |
| J1 | 1.6 | 1.1 | nd | d | d | nd | nd |
| J2 | 2.7 | 1.4 | nd | nd | nd | nd | nd |
| J3 | 1.3 | d | nd | nd | nd | nd | nd |
| L1 | 1.8 | d | nd | d | d | nd | nd |
| L2 | d | d | nd | nd | nd | nd | nd |
| L3 | d | d | nd | nd | nd | nd | nd |
| N1 | 1.4 | d | nd | nd | nd | nd | nd |
| N2 | 1.3 | nd | nd | nd | nd | nd | nd |
| P1 | 1.4 | nd | nd | nd | nd | nd | nd |
| P2 | 1.1 | nd | nd | nd | nd | nd | nd |
| T1 | nd | nd | nd | nd | d | nd | nd |
| T2 | nd | 1.8 | nd | nd | 2.8 | nd | 1.3 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moret, S.; Hidalgo, M.; Sanchez, J.M. Hollow-Fiber Liquid-Phase Microextraction (HF-LPME) Coupled On-Line to Liquid Chromatography for the Determination of the Herbicides 2,4-Dichlorophenoxyacetic Acid and 2-Methyl-4-chlorophenoxyacetic Acid and Their Main Metabolites in Soil Samples. Separations 2023, 10, 273. https://doi.org/10.3390/separations10050273
Moret S, Hidalgo M, Sanchez JM. Hollow-Fiber Liquid-Phase Microextraction (HF-LPME) Coupled On-Line to Liquid Chromatography for the Determination of the Herbicides 2,4-Dichlorophenoxyacetic Acid and 2-Methyl-4-chlorophenoxyacetic Acid and Their Main Metabolites in Soil Samples. Separations. 2023; 10(5):273. https://doi.org/10.3390/separations10050273
Chicago/Turabian StyleMoret, Sònia, Manuela Hidalgo, and Juan M. Sanchez. 2023. "Hollow-Fiber Liquid-Phase Microextraction (HF-LPME) Coupled On-Line to Liquid Chromatography for the Determination of the Herbicides 2,4-Dichlorophenoxyacetic Acid and 2-Methyl-4-chlorophenoxyacetic Acid and Their Main Metabolites in Soil Samples" Separations 10, no. 5: 273. https://doi.org/10.3390/separations10050273