
UPLC-MS/MS Method for Simultaneous Estimation of Neratinib and Naringenin in Rat Plasma: Greenness Assessment and Application to Therapeutic Drug Monitoring
 , , , ,
, , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. UPLC-MS/MS Instrument and Conditions
2.3. Animal Ethical Approval
2.4. Preparation of Standards and Sample
2.5. Bioanalytical Extraction from the Rat Plasma
2.6. Validation Protocol
2.6.1. Selectivity
2.6.2. Linearity and Sensitivity
2.6.3. Trueness and Precision
2.6.4. Carryover and Dilution Integrity
2.6.5. Matrix Effect and Extraction
2.6.6. Stability Study
2.7. Integrative Application of Multiple Green Metrics Tools
3. Results
3.1. Preparation of the Plasma Samples
3.2. Mobile Phase Selection and Optimization: A Greenness Perspective
3.3. Investigation of Method Greenness
3.4. Optimized LC and MS Conditions
3.5. Validation of Results
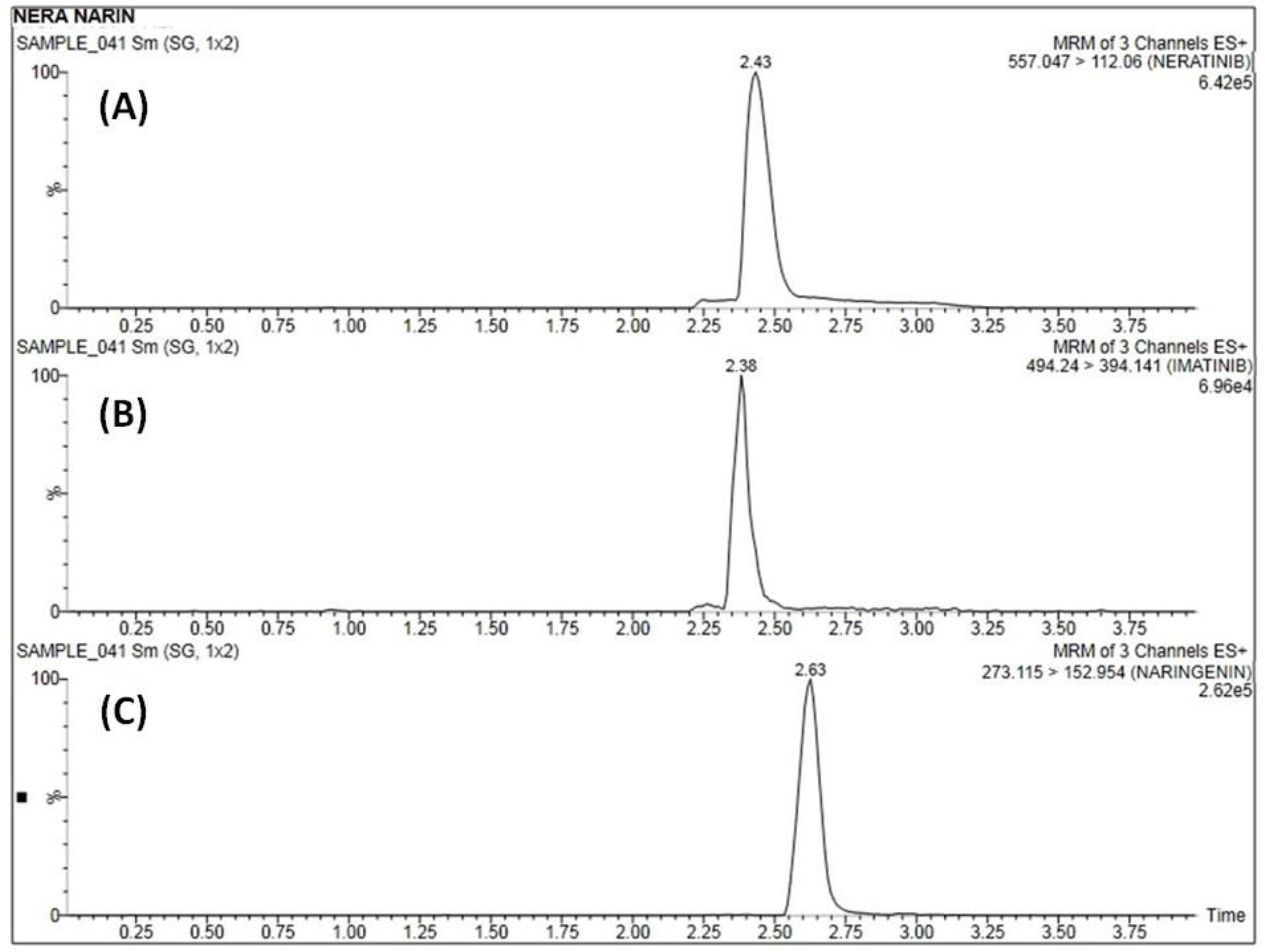
3.5.1. Method Selectivity
3.5.2. Method Linearity and Quantitation Limits
3.5.3. Trueness and Precision
3.5.4. Carryover and Dilution Integrity
3.5.5. Extraction Recovery and Matrix Effect
3.5.6. Stability of the Analytes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Łukasiewicz, S.; Czeczelewski, M.; Forma, A.; Baj, J.; Sitarz, R.; StanisÅ‚awek, A. Breast Cancer-Epidemiology, Risk Factors, Classification, Prognostic Markers, and Current Treatment Strategies-An Updated Review. Cancers 2021, 13, 4287. [Google Scholar] [CrossRef] [PubMed]
- Iancu, G.; Serban, D.; Badiu, C.D.; Tanasescu, C.; Tudosie, M.S.; Tudor, C.; Costea, D.O.; Zgura, A.; Iancu, R.; Vasile, D. Tyrosine kinase inhibitors in breast cancer (Review). Exp. Ther. Med. 2022, 23, 114. [Google Scholar] [CrossRef]
- Schlam, I.; Swain, S.M. HER2-positive breast cancer and tyrosine kinase inhibitors: The time is now. NPJ Breast Cancer 2021, 7, 56. [Google Scholar] [CrossRef] [PubMed]
- Burstein, H.J.; Sun, Y.; Dirix, L.Y.; Jiang, Z.; Paridaens, R.; Tan, A.R.; Awada, A.; Ranade, A.; Jiao, S.; Schwartz, G.; et al. Neratinib, an Irreversible ErbB Receptor Tyrosine Kinase Inhibitor, in Patients With Advanced ErbB2-Positive Breast Cancer. J. Clin. Oncol. 2010, 28, 1301–1307. [Google Scholar] [CrossRef]
- Dhillon, S. Neratinib in Early-Stage Breast Cancer: A Profile of Its Use in the EU. Clin. Drug Investig. 2019, 39, 221–229. [Google Scholar] [CrossRef] [Green Version]
- Kourie, H.R.; Chaix, M.; Gombos, A.; Aftimos, P.; Awada, A. Pharmacodynamics, pharmacokinetics and clinical efficacy of neratinib in HER2-positive breast cancer and breast cancer with HER2 mutations. Expert Opin. Drug Metabol. Toxicol. 2016, 12, 947–957. [Google Scholar] [CrossRef]
- Kanth, M.L.; Kamal, B.R. Development and validation of rp-hplc for estimation of neratinib in bulk and tablet dosage form. Int. J. Pharm. Sci. Drug Res. 2019, 11, 610–614. [Google Scholar] [CrossRef] [Green Version]
- Wani, T.A.; Zargar, S.; Ahmad, A. Ultra Performance Liquid Chromatography Tandem Mass Spectrometric Method Development and Validation for Determination of Neratinib in Human Plasma. S. Afr. J. Chem. 2015, 68, 93–98. [Google Scholar] [CrossRef] [Green Version]
- Alrobaian, M.; Panda, S.S.; Afzal, O.; Kazmi, I.; Alossaimi, M.A.; Al-Abbasi, F.A.; Almalki, W.H.; Soni, K.; Alam, O.; Alam, M.N.; et al. Development of a Validated Bioanalytical UPLC-MS/MS Method for Quantification of Neratinib: A Recent Application to Pharmacokinetic Studies in Rat Plasma. J. Chromatogr. Sci. 2021, 60, 551–558. [Google Scholar] [CrossRef]
- Kiesel, B.F.; Parise, R.A.; Wong, A.; Keyvanjah, K.; Jacobs, S.; Beumer, J.H. LC-MS/MS assay for the quantitation of the tyrosine kinase inhibitor neratinib in human plasma. J. Pharm. Biome. Anal. 2017, 134, 130–136. [Google Scholar] [CrossRef] [Green Version]
- Adaway, J.E.; Keevil, B.G. Therapeutic drug monitoring and LC-MS/MS. J. Chromatogr. B 2011, 883, 33–49. [Google Scholar] [CrossRef]
- Saint-Marcoux, F.; Sauvage, F.o.-L.; Marquet, P. Current role of LC-MS in therapeutic drug monitoring. Anal. Bioanal. Chem. 2007, 388, 1327–1349. [Google Scholar] [CrossRef] [PubMed]
- Shipkova, M.; Svinarov, D. LC-MS/MS as a tool for TDM services: Where are we? Clin. Biochem. 2016, 49, 1009–1023. [Google Scholar] [CrossRef]
- Avataneo, V.; D’Avolio, A.; Cusato, J.; Cant, M.; De Nicolo, A. LC-MS application for therapeutic drug monitoring in alternative matrices. J. Pharm. Biomed. Anal. 2016, 166, 40–51. [Google Scholar] [CrossRef]
- Singh, K.; Bhori, M.; Kasu, Y.A.; Bhat, G.; Marar, T. Antioxidants as precision weapons in war against cancer chemotherapy induced toxicity: Exploring the armoury of obscurity. Saudi Pharm. J. 2018, 26, 177–190. [Google Scholar] [CrossRef] [PubMed]
- Ilghami, R.; Barzegari, A.; Mashayekhi, M.R.; Letourneur, D.; Crepin, M.; Pavon-Djavid, G. The conundrum of dietary antioxidants in cancer chemotherapy. Nut. Rev. 2020, 78, 65–76. [Google Scholar] [CrossRef]
- Wang, R.; Wang, J.; Dong, T.; Shen, J.; Gao, X.; Zhou, J. Naringenin has a chemoprotective effect in MDA-MB231 breast cancer cells via inhibition of caspase-3 and 9 activities. Oncol. Lett. 2019, 17, 1217–1222. [Google Scholar] [PubMed] [Green Version]
- Camargo, C.A.; Gomes-Marcondes, M.C.C.; Wutzki, N.C.; Aoyama, H. Naringin Inhibits Tumor Growth and Reduces Interleukin-6 and Tumor Necrosis Factor α Levels in Rats with Walker 256 Carcinosarcoma. Anticancer Res. 2012, 32, 129. [Google Scholar] [PubMed]
- Jha, D.K.; Shah, D.S.; Talele, S.R.; Amin, P.D. Correlation of two validated methods for the quantification of naringenin in its solid dispersion: HPLC and UV spectrophotometric methods. SN Appl. Sci. 2020, 2, 698. [Google Scholar] [CrossRef] [Green Version]
- Musmade, K.P.; Trilok, M.; Dengale, S.J.; Bhat, K.; Reddy, M.S.; Musmade, P.B.; Udupa, N. Development and validation of liquid chromatographic method for estimation of naringin in nanoformulation. J. Pharm. 2014, 2014, 864901. [Google Scholar] [CrossRef] [Green Version]
- Bhandari, R.; Kuhad, A.; Paliwal, J.K.; Kuhad, A. Development of a new, sensitive, and robust analytical and bio-analytical RP-HPLC method for in-vitro and in-vivo quantification of naringenin in polymeric nanocarriers. J. Anal. Sci. Technol. 2019, 10, 11. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.; Li, P.; Chen, D.; Fang, T.; Li, H.; Su, W. LC/MS/MS quantitation assay for pharmacokinetics of naringenin and double peaks phenomenon in rats plasma. Int. J. Pharm. 2006, 307, 292–299. [Google Scholar] [CrossRef] [PubMed]
- Magdy, M.A.; Farid, N.F.; Anwar, B.H.; Abdelhamid, N.S. Four Greenness evaluations of two chromatographic methods: Application to fluphenazine HCl and nortriptyline HCl pharmaceutical combination in presence of their potential impurities perphenazine and dibenzosuberone. Chromatographia 2002, 85, 1075–1086. [Google Scholar] [CrossRef]
- Gamal, M.; Naguib, I.A.; Panda, D.S.; Abdallah, F.F. Comparative study of four greenness assessment tools for selection of greenest analytical method for assay of hyoscine N-butyl bromide. Anal. Methods 2021, 13, 369–380. [Google Scholar] [CrossRef] [PubMed]
- United States Food and Drug Administration (USFDA) Bioanalytical Method Validation—Guidance for Industry; USFDA: Silver Spring, MD, USA, 2018.
- Chanduluru, H.K.; Sugumaran, A. Assessment of greenness for the determination of voriconazole in reported analytical methods. RSC Adv. 2022, 12, 6683–6703. [Google Scholar] [CrossRef]
- Mohamed, D.; Fouad, M.M. Application of NEMI, Analytical Eco-Scale and GAPI tools for greenness assessment of three developed chromatographic methods for quantification of sulfadiazine and trimethoprim in bovine meat and chicken muscles: Comparison to greenness profile of reported HPLC methods. Microchem. J. 2020, 157, 104873. [Google Scholar]
- Alabbas, A.B.; Alqahtani, S.M.; Panda, S.S.; Alrobaian, M.; Altharawi, A.; Almalki, W.H.; Barkat, M.A.; Rub, R.A.; Rahman, M.; Mir Najib Ullah, S.N.; et al. Development of a validated UPLC-MS/MS method for simultaneous estimation of neratinib and curcumin in human plasma: Application to greenness assessment and routine quantification. J. Chromatogr. Sci. 2022, bmac067. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reagents | Amount | Amount PP (a) | Hazard | Hazard PP | Total PP (b) |
|---|---|---|---|---|---|
| (1) ACN | <10 mL | 1 | Yes | 4 | 4 |
| (2) Acetate buffer | <10 mL | 1 | None | 0 | 0 |
| Instrument | Energy | ||||
| (1) UPLC-MS/MS | 1.5 kWh per sample | 2 | |||
| (2) Ultrasonicator bath | <0.1 kWh per sample | 0 | |||
| (3) Filtration pump | <0.1 kWh per sample | 0 | |||
| (4) Centrifuge Unit | ≤1.5 kWh per sample | 1 | |||
| (5) Occupational hazards | Process Hermetization | 0 | |||
| (6) Waste | <10 mL | 3 | |||
| Overall PPs: | 10 | ||||
| AES total score: | 100 – 10 = 90 | ||||
| Remark: | Excellent greenness | ||||
| Analyte | Nominal Concentration (ng·mL−1) | Intraday (n = 6) | Interday(n = 18) | ||
|---|---|---|---|---|---|
| Trueness (RE, %) | Precision (RSD, %) | Trueness (RE, %) | Precision (RSD, %) | ||
| NER | 10 (LLOQ) | −10.5 | 2.4 | −10.1 | 2.2 |
| 100 (LQC) | −9.2 | 2.1 | −10.2 | 2.5 | |
| 500 (MQC) | −8.7 | 1.4 | −9.1 | 1.2 | |
| 1000 (HQC) | −10.23 | 1.5 | −10.5 | 1.3 | |
| NRN | 10 (LLOQ) | −10.5 | 1.8 | −9.3 | 1.8 |
| 100 (LQC) | −8.76 | 2.8 | −10.6 | 1.6 | |
| 500 (MQC) | −10.36 | 1.4 | −9.91 | 1.26 | |
| 1000 (HQC) | −10.02 | 1.3 | −10.01 | 1.1 | |
| Stability Type | Compound | Concentration (ng·mL−1) | RE, % | RSD, % |
|---|---|---|---|---|
| Bench-top at room temperature (up to 6 h) | NER | 100 (LQC) | −11.75 | 2.74 |
| 1000 (HQC) | −13.74 | 1.44 | ||
| NRN | 100 (LQC) | −11.37 | 2.42 | |
| 1000 (HQC) | −14.43 | 1.22 | ||
| Short term at room temperature (up to 24 h) | NER | 100 (LQC) | −11.27 | 1.47 |
| 1000 (HQC) | −14.45 | 0.99 | ||
| NRN | 100 (LQC) | −12.4 | 1.38 | |
| 1000 (HQC) | −14.33 | 0.57 | ||
| Long-term at −20 °C (14 days) | NER | 100 (LQC) | −11.75 | 1.64 |
| 1000 (HQC) | −13.31 | 2.34 | ||
| NRN | 100 (LQC) | −13.65 | 1.62 | |
| 1000 (HQC) | −14.35 | 1.04 | ||
| Freeze–thaw at −20 °C (up to 3rd cycle) | NER | 100 (LQC) | −12.7 | 1.75 |
| 1000 (HQC) | −11.78 | 1.23 | ||
| NRN | 100 (LQC) | −12.2 | 1.85 | |
| 1000 (HQC) | −13.5 | 0.91 | ||
| Post-preparation at 8 °C | NER | 100 (LQC) | −14.7 | 3.14 |
| 1000 (HQC) | −14.08 | 1.86 | ||
| NRN | 100 (LQC) | −11.6 | 2.77 | |
| 1000 (HQC) | −12.87 | 1.13 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Altharawi, A.; Alqahtani, S.M.; Panda, S.S.; Alrobaian, M.; Alabbas, A.B.; Almalki, W.H.; Alossaimi, M.A.; Barkat, M.A.; Rub, R.A.; Mir Najib Ullah, S.N.; et al. UPLC-MS/MS Method for Simultaneous Estimation of Neratinib and Naringenin in Rat Plasma: Greenness Assessment and Application to Therapeutic Drug Monitoring. Separations 2023, 10, 167. https://doi.org/10.3390/separations10030167
Altharawi A, Alqahtani SM, Panda SS, Alrobaian M, Alabbas AB, Almalki WH, Alossaimi MA, Barkat MA, Rub RA, Mir Najib Ullah SN, et al. UPLC-MS/MS Method for Simultaneous Estimation of Neratinib and Naringenin in Rat Plasma: Greenness Assessment and Application to Therapeutic Drug Monitoring. Separations. 2023; 10(3):167. https://doi.org/10.3390/separations10030167
Chicago/Turabian StyleAltharawi, Ali, Safar M. Alqahtani, Sagar Suman Panda, Majed Alrobaian, Alhumaidi B. Alabbas, Waleed Hassan Almalki, Manal A. Alossaimi, Md. Abul Barkat, Rehan Abdur Rub, Shehla Nasar Mir Najib Ullah, and et al. 2023. "UPLC-MS/MS Method for Simultaneous Estimation of Neratinib and Naringenin in Rat Plasma: Greenness Assessment and Application to Therapeutic Drug Monitoring" Separations 10, no. 3: 167. https://doi.org/10.3390/separations10030167