

Innovative Solutions for Food Analysis: Microextraction Techniques in Lipid Peroxidation Product Detection

Abstract
:1. Introduction
2. Gas Diffusion Microextraction
2.1. Alcoholic Beverages
2.2. Oil Samples
2.3. Solid Food
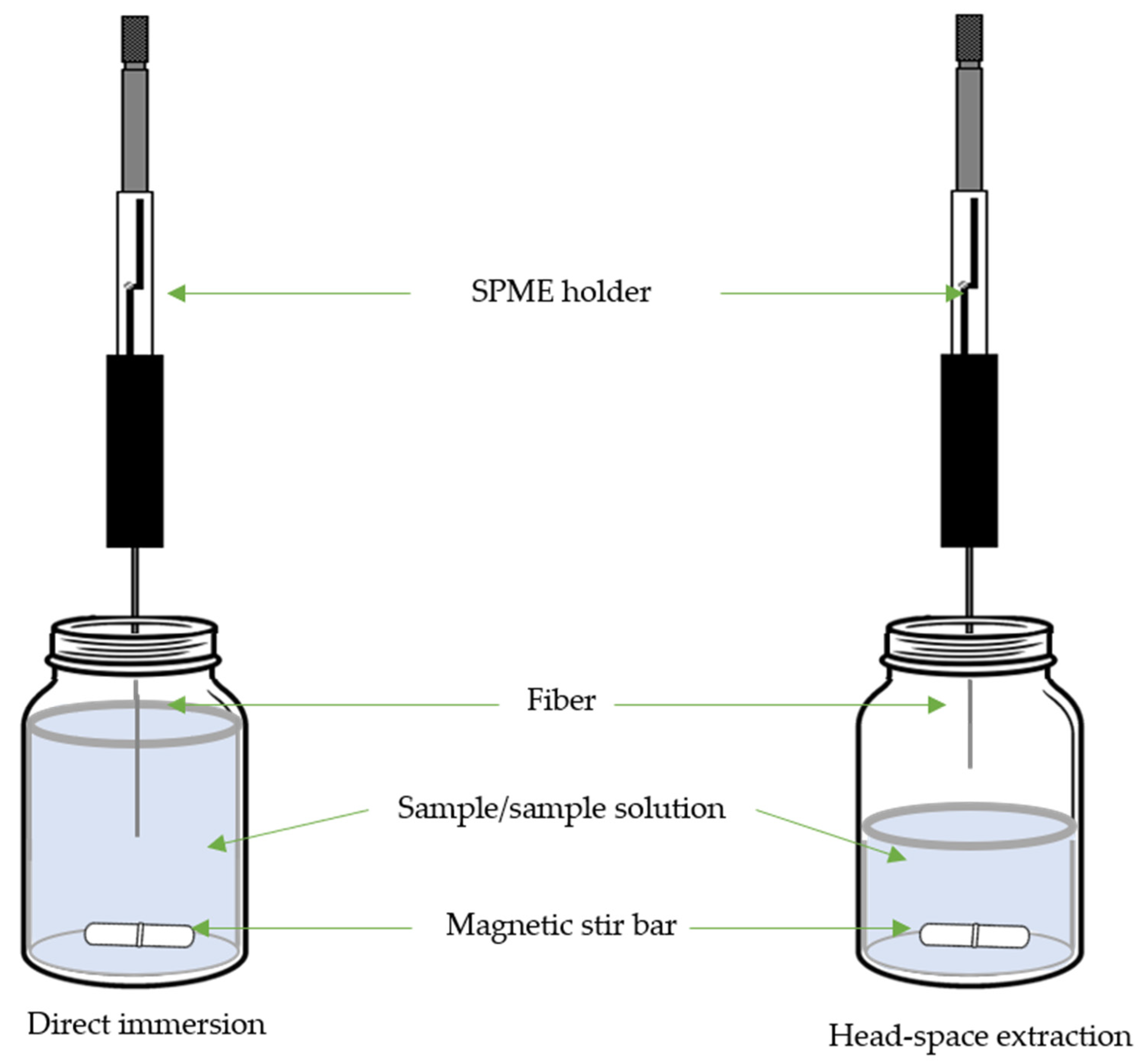
3. Solid-Phase Microextraction
3.1. Oils and High Lipid Content Samples
3.2. High Protein Content Samples
3.3. Cereal and Dairy Products
3.4. Alcoholic Beverages
4. Dispersive Liquid-Liquid Microextraction
4.1. Beverages
4.2. Coffee
4.3. Milk
4.4. Solid Food
4.5. Cereal Products
5. Combined Microextraction Techniques
6. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Yin, H.; Xu, L.; Porter, N.A. Free radical lipid peroxidation: Mechanisms and analysis. Chem. Rev. 2011, 111, 5944–5972. [Google Scholar] [CrossRef] [PubMed]
- Guéraud, F.; Atalay, M.; Bresgen, N.; Cipak, A.; Eckl, P.M.; Huc, L.; Uchida, K. Chemistry and biochemistry of lipid peroxidation products. Free Radic. Res. 2010, 44, 1098–1124. [Google Scholar] [CrossRef] [PubMed]
- Repetto, M.; Semprine, J.; Boveris, A. Lipid peroxidation: Chemical mechanism, biological implications and analytical determination. Lipid Peroxidation 2012, 1, 3–30. [Google Scholar]
- Sun, Y.E.; Wang, W.D.; Chen, H.W.; Li, C. Autoxidation of unsaturated lipids in food emulsion. Crit. Rev. Food Sci. Nutr. 2011, 51, 453–466. [Google Scholar] [CrossRef] [PubMed]
- Pryoir, W.A.; Prier, D.G.; Lightsey, J.W. Kinetic and mechanism studies of peroxy, vitamin e and anti-oxidant free radicals by pulse radiolysis. In Autoxidation in Food and Biological Systems, 1st ed.; Simic, M.G., Karel, M., Eds.; Springer Science & Business Media: Washington, DC, USA, 2013; pp. 17–26. [Google Scholar]
- Zielinski, Z.A.; Pratt, D.A. Lipid Peroxidation: Kinetics, mechanisms, and products. J. Org. Chem. 2017, 82, 2817–2825. [Google Scholar] [CrossRef] [PubMed]
- Islam, F.; Imran, A.; Nosheen, F.; Fatima, M.; Arshad, M.U.; Afzaal, M.; Ijaz, N.; Noreen, R.; Mehta, S.; Biswas, S.; et al. Functional roles and novel tools for improving-oxidative stability of polyunsaturated fatty acids: A comprehensive review. Food Sci Nutr. 2023, 11, 2471–2482. [Google Scholar] [CrossRef]
- Rendón, M.Y.; Salva, T.D.J.G.; Bragagnolo, N. Impact of chemical changes on the sensory characteristics of coffee beans during storage. Food Chem. 2014, 147, 279–286. [Google Scholar] [CrossRef]
- Pegg, R.B.; Shahidi, F. Off flavors and rancidity in foods. In Handbook of Meat, Poultry and Seafood Quality; Nollet, L.M.L., Ed.; Wiley-Blackwell: Oxford, UK, 2012; Part 2; pp. 127–139. [Google Scholar]
- Custodio-Mendoza, J.A.; Aja-Macaya, J.; Valente, I.M.; Rodrigues, J.A.; Almeida, P.J.; Lorenzo, R.A.; Carro, A.M. Determination of malondialdehyde, acrolein and four other products of lipid peroxidation in edible oils by Gas-Diffusion Microextraction combined with Dispersive Liquid-Liquid Microextraction. J. Chromatogr. A 2020, 1627, 461397. [Google Scholar] [CrossRef]
- Rodrigues, F.; Caldeira, M.; Câmara, J.D.S. Development of a dynamic headspace solid-phase microextraction procedure coupled to GC–qMSD for evaluation the chemical profile in alcoholic beverages. Anal. Chim. Acta 2008, 609, 82–104. [Google Scholar] [CrossRef]
- Min, B.; Ahn, D.U. Mechanism of lipid peroxidation in meat and meat products-A review. Food Sci. Biotechnol. 2005, 14, 152–163. [Google Scholar]
- Khan, I.T.; Nadeem, M.; Imran, M.; Ullah, R.; Ajmal, M.; Jaspal, M.H. Antioxidant properties of Milk and dairy products: A comprehensive review of the current knowledge. Lipids Health Dis. 2019, 18, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Carrasco-Pancorbo, A.; Cerretani, L.; Bendini, A.; Segura-Carretero, A.; Del Carlo, M.; Gallina-Toschi, T.; Lercker, G.; Compagnone, D.; Fernandez-Gutierrez, A. Evaluation of the antioxidant capacity of individual phenolic compounds in virgin olive oil. J. Agric. Food Chem. 2005, 53, 8918–8925. [Google Scholar] [CrossRef] [PubMed]
- Wiśniewska, P.; Śliwińska, M.; Dymerski, T.; Wardencki, W.; Namieśnik, J. The analysis of raw spirits–a review of methodology. J. Inst. Brew. 2016, 122, 5–10. [Google Scholar] [CrossRef]
- Hernandes, K.C.; Souza-Silva, É.A.; Assumpção, C.F.; Zini, C.A.; Welke, J.E. Carbonyl compounds and furan derivatives with toxic potential evaluated in the brewing stages of craft beer. Food Addit. Contam. Part A 2020, 37, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Perrone, G.G.; Tan, S.X.; Dawes, I.W. Reactive oxygen species and yeast apoptosis. Biochim. Biophys. Acta Mol. Cell. Res. 2008, 1783, 1354–1368. [Google Scholar] [CrossRef] [PubMed]
- Culleré, L.; Cacho, J.; Ferreira, V. An assessment of the role played by some oxidation-related aldehydes in wine aroma. J. Agric. Food. Chem. 2007, 55, 876–881. [Google Scholar] [CrossRef] [PubMed]
- Amaral, A.B.; Silva, M.V.D.; Lannes, S.C.D.S. Lipid oxidation in meat: Mechanisms and protective factors—A review. Food Sci. Tech. 2018, 38, 1–15. [Google Scholar] [CrossRef]
- Łuczaj, W.; Skrzydlewska, E. DNA damage caused by lipid peroxidation products. Cell Mol. Biol. Lett. 2003, 8, 391–413. [Google Scholar]
- World Health Organization. IARC Monographs on the Identification of Carcinogenic Hazards to Humans. 2023. Available online: https://monographs.iarc.who.int/list-of-classifications/ (accessed on 25 August 2023).
- European Food Safety Authority (EFSA). Opinion of the Scientific Panel on food additives, flavourings, processing aids and materials in contact with food (AFC) related to use of formaldehyde as a preservative during the manufacture and preparation of food additives. EFSA J. 2007, 5, 415. [Google Scholar] [CrossRef]
- Center for Drug Evaluation and Research M7(R2) Addendum: Application of the Principles of the ICH M7 Guideline, U.S. Food and Drug Administration. Available online: https://www.fda.gov/media/157451/download (accessed on 25 August 2023).
- Van Andel, I.; Sleijffers, A.; Schenk, E.; Rambali, B.; Wolterink, G.; Vleeming, W.; van Amsterdam, J.G.C. Adverse Health Effects of Cigarette Smoke: Aldehydes Crotonaldehyde, Butyraldehyde, Hexanal and Malonaldehyde; Dutch National Institute of Public Health and the Environment: Utrecht, The Netherland, 2006; Available online: http://hdl.handle.net/10029/7336 (accessed on 25 August 2023).
- Gomes, R.; Meek, M.K. Acrolein. Concise International Chemical Assessment Document 43. World Health Organization. 2002. Available online: https://www.who.int/ipcs/publications/cicad/en/cicad43.pdf (accessed on 25 August 2023).
- Papastergiadis, A.; Fatouh, A.; Jacxsens, L.; Lachat, C.; Shrestha, K.; Daelman, J.; Kolsteren, P.; Van Langenhove, H.; De Meulenaer, B. Exposure assessment of Malondialdehyde, 4-Hydroxy-2-(E)-Nonenal and 4-Hydroxy-2-(E)-Hexenal through specific foods available in Belgium. Food Chem. Tox. 2014, 73, 51–58. [Google Scholar] [CrossRef]
- Guth, S.; Baum, M.; Cartus, A.T.; Diel, P.; Engel, K.H.; Engeli, B.; Epe, B.; Grune, T.; Haller, D.; Heinz, V.; et al. Evaluation of the genotoxic potential of acrylamide: Arguments for the derivation of a tolerable daily intake (TDI value). Food Chem. Toxicol. 2023, 173, 113632. [Google Scholar] [CrossRef] [PubMed]
- Clark, S.; Winter, C.K. Diacetyl in foods: A review of safety and sensory characteristics. Compr. Rev. Food Sci. Food Saf. 2015, 14, 634–643. [Google Scholar] [CrossRef]
- EFSA Panel on Food Additives and Flavourings (FAF); Younes, M.; Aquilina, G.; Castle, L.; Engel, K.H.; Fowler, P.; Mennes, W. Scientific Opinion on Flavouring Group Evaluation 13 Revision 3 (FGE. 13Rev3): Furfuryl and furan derivatives with and without additional side-chain substituents and heteroatoms from chemical group 14. EFSA J. 2021, 19, 06386. [Google Scholar]
- Kielhorn, J.; Pohlenz-Michel, C.; Schmidt, S.; Mangelsdorf, I. Glyoxal. World Health Organization. 2004. Available online: https://apps.who.int/iris/handle/10665/42867 (accessed on 25 August 2023).
- Renwick, A.G. Structure-based thresholds of toxicological concern—Guidance for application to substances present at low levels in the diet. Toxicol. Appl. Pharmacol. 2005, 207, 585–591. [Google Scholar] [CrossRef] [PubMed]
- Amorati, R.; Valgimigli, L. Advantages and limitations of common testing methods for antioxidants. Free Radic. Res. 2015, 49, 633–649. [Google Scholar] [CrossRef]
- Demirci-Cekic, S.; Özkan, G.; Avan, A.N.; Uzunboy, S.; Çapanoğlu, E.; Apak, R. Biomarkers of oxidative stress and antioxidant defense. J. Pharm. Biomed. Anal. 2022, 209, 114477. [Google Scholar] [CrossRef]
- Grebenteuch, S.; Kroh, L.W.; Drusch, S.; Rohn, S. Formation of secondary and tertiary volatile compounds resulting from the lipid oxidation of rapeseed oil. Foods 2021, 10, 2417. [Google Scholar] [CrossRef]
- Laghrib, F.; Lahrich, S.; El Mhammedi, M.A. Recent Advances in Direct and Indirect Methods for Sensing Carbonyl Compounds Aldehydes in Environment and Foodstuffs. J. Electrochem. Soc. 2019, 166, B1543. [Google Scholar] [CrossRef]
- Li, H.; Li, H.; Liu, X.; Chen, B. Analysis of volatile flavor compounds in top fermented wheat beer by headspace sampling-gas chromatography. Int. J. Agric. Biol. Eng. 2012, 5, 67–75. [Google Scholar]
- Zamani, A.; Fashi, A. Extraction and preconcentration of trace malondialdehyde from lipid-rich foods using ion pair–based solvent bar liquid-phase microextraction. Food Anal. Methods 2019, 12, 1625–1634. [Google Scholar] [CrossRef]
- Zhang, D.; Zhang, J.; Li, M.; Li, W.; Aimaiti, G.; Tuersun, G.; Chu, Q. A novel miniaturised electrophoretic method for determining formaldehyde and acetaldehyde in food using 2-thiobarbituric acid derivatisation. Food Chem. 2011, 129, 206–212. [Google Scholar] [CrossRef]
- Zeb, A.; Ullah, F. A simple spectrophotometric method for the determination of thiobarbituric acid reactive substances in fried fast foods. J. Anal. Methods Chem. 2016, 2016, 9412767. [Google Scholar] [CrossRef] [PubMed]
- Mathew, S.; Grey, C.; Rumpunen, K.; Adlercreutz, P. Analysis of carbonyl compounds in sea buckthorn for the evaluation of triglyceride oxidation, by enzymatic hydrolysis and derivatisation methodology. Food Chem. 2011, 126, 1399–1405. [Google Scholar] [CrossRef]
- Suh, J.H.; Ho, C.T.; Wang, Y. Evaluation of carbonyl species in fish oil: An improved LC–MS/MS method. Food Control 2017, 78, 463–468. [Google Scholar] [CrossRef]
- Serrano, M.; Gallego, M.; Silva, M. Quantitative analysis of aldehydes in canned vegetables using static headspace–gas chromatography–mass spectrometry. J. Chromatogr. A 2017, 1524, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Zelzer, S.; Oberreither, R.; Bernecker, C.; Stelzer, I.; Truschnig-Wilders, M.; Fauler, G. Measurement of total and free malondialdehyde by gas–chromatography mass spectrometry–comparison with high-performance liquid chromatography methology. Free Radic. Res. 2013, 47, 651–656. [Google Scholar] [CrossRef]
- Ghorbani, M.; Aghamohammadhassan, M.; Chamsaz, M.; Akhlaghi, H.; Pedramrad, T. Dispersive solid phase microextraction. TrAC Trends Anal. Chem. 2019, 118, 793–809. [Google Scholar] [CrossRef]
- López-Lorente, Á.I.; Pena-Pereira, F.; Pedersen-Bjergaard, S.; Zuin, V.G.; Ozkan, S.A.; Psillakis, E. The ten principles of green sample preparation. TrAC Trends Anal. Chem. 2022, 148, 116530. [Google Scholar] [CrossRef]
- Atapattu, S.N.; Rosenfeld, J.M. Analytical derivatizations in environmental analysis. J. Chromatogr. 2022, 1678, 463348. [Google Scholar] [CrossRef]
- Kokosa, J.M.; Przyjazny, A. Green microextraction methodologies for sample preparations. Green Anal. Chem. 2022, 3, 100023. [Google Scholar] [CrossRef]
- Pacheco, J.G.; Valente, I.M.; Gonçalves, L.M.; Rodrigues, J.A.; Barros, A.A. Gas-diffusion microextraction. J. Sep. Sci. 2010, 33, 3207–3212. [Google Scholar] [CrossRef] [PubMed]
- Jalili, V.; Barkhordari, A.; Ghiasvand, A. A comprehensive look at solid-phase microextraction technique: A review of reviews. Microchem. J. 2020, 152, 104319. [Google Scholar] [CrossRef]
- Sajid, M. Dispersive liquid-liquid microextraction: Evolution in design, application areas, and green aspects. TrAC Trends Anal. Chem. 2022, 152, 116636. [Google Scholar] [CrossRef]
- More, V.N.; Mundhe, D.G. Microextraction techniques in analysis of drugs. Int. J. Res. Pharm.Chem. 2013, 3, 330–344. [Google Scholar]
- Risheed, C.M.; Fakhre, N.A.; Mohammed, M.I.; Ali, D.K. Hollow Fiber Nano Membrane as Liquid Phase Microextraction for Determination of Enrofloxacin in the Prsence of Florfenicol and Tylosin in Chicken Tissues. Egypt. J. Chem. 2022, 65, 133–141. [Google Scholar] [CrossRef]
- Mirnaghi, F.S.; Goryński, K.; Rodriguez-Lafuente, A.; Boyacı, E.; Bojko, B.; Pawliszyn, J. Microextraction versus exhaustive extraction approaches for simultaneous analysis of compounds in wide range of polarity. J. Chromatogr. A 2013, 1316, 37–43. [Google Scholar] [CrossRef]
- Gonçalves, L.M.; Magalhães, P.J.; Valente, I.M.; Pacheco, J.G.; Dostálek, P.; Sýkora, D.; Rodrigues, J.A.; Barros, A.A. Analysis of aldehydes in beer by gas-diffusion microextraction: Characterization by high-performance liquid chromatography–diode-array detection–atmospheric pressure chemical ionization–mass spectrometry. J. Chromatogr. A 2010, 1217, 3717–3722. [Google Scholar] [CrossRef]
- Ferreira, I.M.; Carvalho, D.O.; da Silva, M.G.; Guido, L.F. Gas-Diffusion Microextraction (GDME) Combined with Derivatization for Assessing Beer Staling Aldehydes: Validation and Application. Foods 2021, 10, 1704. [Google Scholar] [CrossRef]
- Ramos, R.M.; Pacheco, J.G.; Gonçalves, L.M.; Valente, I.M.; Rodrigues, J.A.; Barros, A.A. Determination of free and total diacetyl in wine by HPLC–UV using gas-diffusion microextraction and pre-column derivatization. Food Control 2012, 24, 220–224. [Google Scholar] [CrossRef]
- Cruz, M.P.; Valente, I.M.; Gonçalves, L.M.; Rodrigues, J.A.; Barros, A.A. Application of gas-diffusion microextraction to the analysis of free and bound acetaldehyde in wines by HPLC–UV and characterization of the extracted compounds by MS/MS detection. Anal. Bioanal. Chem. 2012, 403, 1031–1037. [Google Scholar] [CrossRef]
- Ramos, R.M.; Gonçalves, L.M.; Vyskočil, V.; Rodrigues, J.A. Voltammetric determination of trace amounts of diacetyl at a mercury meniscus modified silver solid amalgam electrode following gas-diffusion microextraction. Talanta 2017, 169, 203–208. [Google Scholar] [CrossRef] [PubMed]
- Santos, C.M.; Valente, I.M.; Gonçalves, L.M.; Rodrigues, J.A. Chromatographic analysis of methylglyoxal and other α-dicarbonyls using gas-diffusion microextraction. Analyst 2013, 138, 7233–7237. [Google Scholar] [CrossRef] [PubMed]
- Custodio-Mendoza, J.A.; Valente, I.M.; Ramos, R.M.; Lorenzo, R.A.; Carro, A.M.; Rodrigues, J.A. Analysis of free malondialdehyde in edible oils using gas-diffusion microextraction. J. Food Compos. Anal. 2019, 82, 103254. [Google Scholar] [CrossRef]
- Ferreira, R.C.; Ramos, R.M.; Gonçalves, L.M.; Almeida, P.J.; Rodrigues, J.A. Application of gas-diffusion microextraction to solid samples using the chromatographic determination of α-diketones in bread as a case study. Analyst 2015, 140, 3648–3653. [Google Scholar] [CrossRef] [PubMed]
- Cordeiro, L.; Valente, I.M.; Santos, J.R.; Rodrigues, J.A. Qualitative carbonyl profile in coffee beans through GDME-HPLC-DAD-MS/MS for coffee preliminary characterization. Food Res. Int. 2018, 107, 536–543. [Google Scholar] [CrossRef]
- Câmara, J.S.; Perestrelo, R.; Berenguer, C.V.; Andrade, C.F.; Gomes, T.M.; Olayanju, B.; Pereira, J.A. Green extraction techniques as advanced sample preparation approaches in biological, food, and environmental matrices: A review. Molecules 2022, 27, 2953. [Google Scholar] [CrossRef]
- Jeleń, H.H.; Obuchowska, M.; Zawirska-Wojtasiak, R.; Wasowicz, E. Headspace solid-phase microextraction use for the characterization of volatile compounds in vegetable oils of different sensory quality. J. Agric. Food Chem. 2000, 48, 2360–2367. [Google Scholar] [CrossRef]
- Uchida, T.; Gotoh, N.; Wada, S. Method for analysis of 4-hydroxy-2-(E)-nonenal with solid-phase microextraction. Lipids 2002, 37, 621–626. [Google Scholar] [CrossRef]
- Fujioka, K.; Shibamoto, T. Improved malonaldehyde assay using headspace solid-phase microextraction and its application to the measurement of the antioxidant activity of phytochemicals. J. Agric. Food Chem. 2005, 53, 4708–4713. [Google Scholar] [CrossRef]
- Pastorelli, S.; Valzacchi, S.; Rodriguez, A.; Simoneau, C. Solid-phase microextraction method for the determination of hexanal in hazelnuts as an indicator of the interaction of active packaging materials with food aroma compounds. Food Addit. Contam. 2006, 23, 1236–1241. [Google Scholar] [CrossRef]
- Ma, C.; Ji, J.; Tan, C.; Chen, D.; Luo, F.; Wang, Y.; Chen, X. Headspace solid-phase microextraction coupled to gas chromatography for the analysis of aldehydes in edible oils. Talanta 2014, 120, 94–99. [Google Scholar] [CrossRef]
- Goicoechea, E.; Van Twillert, K.; Duits, M.; Brandon, E.D.; Kootstra, P.R.; Blokland, M.H.; Guillén, M.D. Use of an in vitro digestion model to study the bioaccessibility of 4-hydroxy-2-nonenal and related aldehydes present in oxidized oils rich in omega-6 acyl groups. J. Agric. Food Chem. 2008, 56, 8475–8483. [Google Scholar] [CrossRef]
- Guillén, M.D.; Carton, I.; Salmeron, J.; Casas, C. Headspace composition of cod liver oil and its evolution in storage after opening. First evidence of the presence of toxic aldehydes. Food chem. 2009, 114, 1291–1300. [Google Scholar] [CrossRef]
- Damerau, A.; Kamlang-ek, P.; Moisio, T.; Lampi, A.M.; Piironen, V. Effect of SPME extraction conditions and humidity on the release of volatile lipid oxidation products from spray-dried emulsions. Food Chem. 2014, 157, 1–9. [Google Scholar] [CrossRef]
- Martin-Rubio, A.S.; Sopelana, P.; Guillén, M.D. The key role of ovalbumin in lipid bioaccessibility and oxidation product profile during the in vitro digestion of slightly oxidized soybean oil. Food Func. 2019, 10, 4440–4451. [Google Scholar] [CrossRef]
- Liu, X.; Jin, Q.; Liu, Y.; Huang, J.; Wang, X.; Mao, W.; Wang, S. Changes in volatile compounds of peanut oil during the roasting process for production of aromatic roasted peanut oil. J. Food Sci. 2011, 76, C404–C412. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Xu, X.L.; Ouyang, G.F.; Zhou, G.H. Changes in volatile compounds of traditional Chinese Nanjing water-boiled salted duck during processing. J. Food Sci. 2006, 71, S371–S377. [Google Scholar] [CrossRef]
- Mariutti, L.R.; Nogueira, G.C.; Bragagnolo, N. Solid phase microextraction-gas chromatography for the evaluation of secondary lipid oxidation products in chicken patties during long-term storage. J. Braz. Chem. Soc. 2009, 20, 1849–1855. [Google Scholar] [CrossRef]
- Estévez, M.; Ventanas, S.; Cava, R. Oxidation of lipids and proteins in frankfurters with different fatty acid compositions and tocopherol and phenolic contents. Food Chem. 2007, 100, 55–63. [Google Scholar] [CrossRef]
- Chopin, C.; Kone, M.; Serot, T. Study of the interaction of fish myosin with the products of lipid oxidation: The case of aldehydes. Food Chem. 2007, 105, 126–132. [Google Scholar] [CrossRef]
- Iglesias, J.; Gallardo, J.M.; Medina, I. Determination of carbonyl compounds in fish species samples with solid-phase microextraction with on-fibre derivatization. Food Chem. 2010, 123, 771–778. [Google Scholar] [CrossRef]
- Lopez, A.; Bellagamba, F.; Tirloni, E.; Vasconi, M.; Stella, S.; Bernardi, C.; Pazzaglia, M.; Moretti, V.M. Evolution of food safety features and volatile profile in white sturgeon caviar treated with different formulations of salt and preservatives during a long-term storage time. Foods 2021, 10, 850. [Google Scholar] [CrossRef] [PubMed]
- Domínguez, R.; Purriños, L.; Pérez-Santaescolástica, C.; Pateiro, M.; Barba, F.J.; Tomasevic, I.; Bastianello Campagnol, P.C.; Lorenzo, J.M. Characterization of volatile compounds of dry-cured meat products using HS-SPME-GC/MS technique. Food Anal. Methods 2019, 12, 1263–1284. [Google Scholar] [CrossRef]
- García-Llatas, G.; Lagarda, M.J.; Clemente, G.; Farré, R. Monitoring of headspace volatiles in milk-cereal-based liquid infant foods during storage. Eur. J. Lipid Sci. Technol. 2006, 108, 1028–1036. [Google Scholar] [CrossRef]
- García-Llatas, G.; Lagarda, M.J.; Romero, F.; Abellán, P.; Farré, R. A headspace solid-phase microextraction method of use in monitoring hexanal and pentane during storage: Application to liquid infant foods and powdered infant formulas. Food Chem. 2007, 101, 1078–1086. [Google Scholar] [CrossRef]
- Clarke, H.J.; Mannion, D.T.; O’Sullivan, M.G.; Kerry, J.P.; Kilcawley, K.N. Development of a headspace solid-phase microextraction gas chromatography mass spectrometry method for the quantification of volatiles associated with lipid oxidation in whole milk powder using response surface methodology. Food Chem. 2019, 292, 75–80. [Google Scholar] [CrossRef]
- Majcher, M.A.; Goderska, K.; Pikul, J.; Jeleń, H.H. Changes in volatile, sensory and microbial profiles during preparation of smoked ewe cheese. J. Sci. Food Agric. 2011, 91, 1416–1423. [Google Scholar] [CrossRef]
- Natrella, G.; Faccia, M.; Lorenzo, J.M.; De Palo, P.; Gambacorta, G. Sensory characteristics and volatile organic compound profile of high-moisture mozzarella made by traditional and direct acidification technology. J. Dairy Sci. 2020, 103, 2089–2097. [Google Scholar] [CrossRef]
- Cardinali, F.; Foligni, R.; Ferrocino, I.; Harasym, J.; Orkusz, A.; Milanović, V.; Franciosa, O.; Garofalo, C.; Mannozzi, C.; Mozzon, M.; et al. Microbiological, morpho-textural, and volatile characterization of Portuguese Queijo de Nisa PDO cheese. Food Res. Int. 2022, 162, 112011. [Google Scholar] [CrossRef]
- Vesely, P.; Lusk, L.; Basarova, G.; Seabrooks, J.; Ryder, D. Analysis of aldehydes in beer using solid-phase microextraction with on-fiber derivatization and gas chromatography/mass spectrometry. J. Agric. Food Chem. 2003, 51, 6941–6944. [Google Scholar] [CrossRef]
- Saison, D.; De Schutter, D.P.; Delvaux, F.; Delvaux, F.R. Determination of carbonyl compounds in beer by derivatisation and headspace solid-phase microextraction in combination with gas chromatography and mass spectrometry. J. Chromatogr. A 2009, 1216, 5061–5068. [Google Scholar] [CrossRef] [PubMed]
- Moreira, N.; Meireles, S.; Brandão, T.; de Pinho, P.G. Optimization of the HS-SPME–GC–IT/MS method using a central composite design for volatile carbonyl compounds determination in beers. Talanta 2013, 117, 523–531. [Google Scholar] [CrossRef] [PubMed]
- Hernandes, K.C.; Souza-Silva, É.A.; Assumpção, C.F.; Zini, C.A.; Welke, J.E. Validation of an analytical method using HS-SPME-GC/MS-SIM to assess the exposure risk to carbonyl compounds and furan derivatives through beer consumption. Food Addit. Contam. Part A 2019, 36, 1808–1821. [Google Scholar] [CrossRef] [PubMed]
- Perez Olivero, S.J.; Perez Trujillo, J.P. A new method for the determination of carbonyl compounds in wines by headspace solid-phase microextraction coupled to gas chromatography−ion trap mass spectrometry. J. Agric. Food Chem. 2010, 58, 12976–12985. [Google Scholar] [CrossRef] [PubMed]
- Butkhup, L.; Jeenphakdee, M.; Jorjong, S.; Samappito, S.; Samappito, W.; Chowtivannakul, S. HS-SPME-GC-MS analysis of volatile aromatic compounds in alcohol related beverages made with mulberry fruits. Food Sci. Biotechnol. 2011, 20, 1021–1032. [Google Scholar] [CrossRef]
- Lago, L.O.; Nicolli, K.P.; Marques, A.B.; Zini, C.A.; Welke, J.E. Influence of ripeness and maceration of the grapes on levels of furan and carbonyl compounds in wine—Simultaneous quantitative determination and assessment of the exposure risk to these compounds. Food Chem. 2017, 230, 594–603. [Google Scholar] [CrossRef]
- Ferreira, D.C.; Hernandes, K.C.; Nicolli, K.P.; Souza-Silva, É.A.; Manfroi, V.; Zini, C.A.; Welke, J.E. Development of a method for determination of target toxic carbonyl compounds in must and wine using HS-SPME-GC/MS-SIM after preliminary GC× GC/TOFMS analyses. Food Anal. Methods 2019, 12, 108–120. [Google Scholar] [CrossRef]
- Moreira, N.; Araújo, A.M.; Rogerson, F.; Vasconcelos, I.; De Freitas, V.; de Pinho, P.G. Development and optimization of a HS-SPME-GC-MS methodology to quantify volatile carbonyl compounds in Port wines. Food Chem. 2019, 270, 518–526. [Google Scholar] [CrossRef]
- Piergiovanni, M.; Carlin, S.; Lotti, C.; Vrhovsek, U.; Mattivi, F. Development of a Fully Automated Method HS-SPME-GC-MS/MS for the Determination of Odor-Active Carbonyls in Wines: A “Green” Approach to Improve Robustness and Productivity in the Oenological Analytical Chemistry. J. Agric. Food Chem. 2023. [CrossRef]
- Wardencki, W.; Sowiński, P.; Curyło, J. Evaluation of headspace solid-phase microextraction for the analysis of volatile carbonyl compounds in spirits and alcoholic beverages. J. Chromatogr. A 2003, 984, 89–96. [Google Scholar] [CrossRef]
- López-Vázquez, C.; Orriols, I.; Perelló, M.C.; De Revel, G. Determination of aldehydes as pentafluorobenzyl derivatives in grape pomace distillates by HS-SPME-GC/MS. Food Chem. 2012, 130, 1127–1133. [Google Scholar] [CrossRef]
- Perestrelo, R.; Silva, C.L.; Silva, P.; Medina, S.; Pereira, R.; Câmara, J.S. Untargeted fingerprinting of cider volatiles from different geographical regions by HS-SPME/GC-MS. Microchem. J. 2019, 148, 643–651. [Google Scholar] [CrossRef]
- Yu, J.; Zhou, Z.; Xu, X.; Ren, H.; Gong, M.; Ji, Z.; Liu, S.; Hu, Z.; Mao, J. Differentiating Huangjiu with Varying Sugar Contents from Different Regions Based on Targeted Metabolomics Analyses of Volatile Carbonyl Compounds. Foods 2023, 12, 1455. [Google Scholar] [CrossRef] [PubMed]
- Lim, H.H.; Shin, H.S. In-solution derivatization and detection of glyoxal and methylglyoxal in alcoholic beverages and fermented foods by headspace solid-phase microextraction and gas chromatography–mass spectrometry. J. Food Compos. Anal. 2020, 92, 103584. [Google Scholar] [CrossRef]
- Elisia, I.; Kitts, D.D. Quantification of hexanal as an index of lipid oxidation in human milk and association with antioxidant components. J. Clin. Biochem. Nutr. 2011, 49, 147–152. [Google Scholar] [CrossRef] [PubMed]
- Maire, M.; Rega, B.; Cuvelier, M.E.; Soto, P.; Giampaoli, P. Lipid oxidation in baked products: Impact of formula and process on the generation of volatile compounds. Food Chem. 2013, 141, 3510–3518. [Google Scholar] [CrossRef]
- Fenaille, F.; Visani, P.; Fumeaux, R.; Milo, C.; Guy, P.A. Comparison of mass spectrometry-based electronic nose and solid phase microextraction gas chromatography mass spectrometry technique to assess infant formula oxidation. J. Agric. Food Chem. 2003, 51, 2790–2796. [Google Scholar] [CrossRef]
- Li, X.; He, Y.; Xie, Y.; Zhu, D.; Yang, L.; Wang, S.; Liu, H. Effect of catalase on lipid oxidation and flavor substances of α-instant rice during storage. Food Sci. Tech. 2022, 42, e46822. [Google Scholar] [CrossRef]
- Rezaee, M.; Assadi, Y.; Hosseini, M.R.M.; Aghaee, E.; Ahmadi, F.; Berijani, S. Determination of organic compounds in water using dispersive liquid-liquid microextraction. J. Chromatogr. A 2006, 1116, 1–9. [Google Scholar] [CrossRef]
- Yan, H.; Wang, H. Recent development and applications of dispersive liquid-liquid microextraction. J. Chromatogr. A 2013, 1295, 1–15. [Google Scholar] [CrossRef]
- Jain, R.; Singh, R. Applications of dispersive liquid–liquid micro-extraction in forensic toxicology. TrAC Trends Anal. Chem. 2016, 75, 227–237. [Google Scholar] [CrossRef]
- El-Deen, A.K.; Elmansi, H.; Belal, F.; Magdy, G. Recent advances in dispersion strategies for dispersive liquid-liquid microextraction from green chemistry perspectives. Microchem. J. 2023, 191, 108807. [Google Scholar] [CrossRef]
- Rykowska, I.; Ziemblińska, J.; Nowak, I. Modern approaches in dispersive liquid-liquid microextraction (DLLME) based on ionic liquids: A review. J. Mol. Liq. 2018, 259, 319–339. [Google Scholar] [CrossRef]
- Marcinkowska, R.; Konieczna, K.; Marcinkowski, Ł.; Namieśnik, J.; Kloskowski, A. Application of ionic liquids in microextraction techniques: Current trends and future perspectives. TrAC Trends Anal. Chem. 2019, 119, 115614. [Google Scholar] [CrossRef]
- Xu, X.; Su, R.; Zhao, X.; Liu, Z.; Li, D.; Li, X.; Zhang, H.; Wang, Z. Determination of formaldehyde in beverages using microwave-assisted derivatization and ionic liquid-based dispersive liquid–liquid microextraction followed by high-performance liquid chromatography. Talanta 2011, 85, 2632–2638. [Google Scholar] [CrossRef]
- Galuch, M.B.; Magon, T.F.S.; Silveira, R.; Nicácio, A.E.; Pizzo, J.S.; Bonafe, E.G.; Maldaner, L.; Santos, O.O.; Visentainer, J.V. Determination of acrylamide in brewed coffee by dispersive liquid–liquid microextraction (DLLME) and ultra-performance liquid chromatography tandem mass spectrometry (UPLC-MS/MS). Food Chem. 2019, 282, 120–126. [Google Scholar] [CrossRef]
- Zhang, C.; Cagliero, C.; Pierson, S.A.; Anderson, J.L. Rapid and sensitive analysis of polychlorinated biphenyls and acrylamide in food samples using ionic liquid-based in situ dispersive liquid-liquid microextraction coupled to headspace gas chromatography. J. Chromatogr. A 2017, 1481, 1–11. [Google Scholar] [CrossRef]
- Custodio-Mendoza, J.A.; Caamaño-Fernandez, C.; Lage, M.A.; Almeida, P.J.; Lorenzo, R.A.; Carro, A.M. GC–MS determination of malondialdehyde, acrolein, and 4-hydroxy-2-nonenal by ultrasound-assisted dispersive liquid-liquid microextraction in beverages. Food Chem. 2022, 384, 132530. [Google Scholar] [CrossRef]
- Nascimento, C.F.; Brasil, M.A.; Costa, S.P.; Pinto, P.C.; Saraiva, M.L.M.; Rocha, F.R. Exploitation of pulsed flows for on-line dispersive liquid–liquid microextraction: Spectrophotometric determination of formaldehyde in milk. Talanta 2015, 144, 1189–1194. [Google Scholar] [CrossRef]
- Altunay, N.; Elik, A.; Tuzen, M.; Lanjwani, M.F.; Mogaddam, M.R.A. Determination and extraction of acrylamide in processed food samples using alkanol-based supramolecular solvent-assisted dispersive liquid-liquid microextraction coupled with spectrophotometer: Optimization using factorial design. J. Food Compos. Anal. 2023, 115, 105023. [Google Scholar] [CrossRef]
- Nematollahi, A.; Kamankesh, M.; Hosseini, H.; Hadian, Z.; Ghasemi, J.; Mohammadi, A. Investigation and determination of acrylamide in 24 types of roasted nuts and seeds using microextraction method coupled with gas chromatography–mass spectrometry: Central composite design. J. Food Meas. Charact. 2020, 14, 1249–1260. [Google Scholar] [CrossRef]
- Zokaei, M.; Abedi, A.S.; Kamankesh, M.; Shojaee-Aliababadi, S.; Mohammadi, A. Ultrasonic-assisted extraction and dispersive liquid-liquid microextraction combined with gas chromatography-mass spectrometry as an efficient and sensitive method for determining of acrylamide in potato chips samples. Food chem. 2017, 234, 55–61. [Google Scholar] [CrossRef]
- Nematollahi, A.; Kamankesh, M.; Hosseini, H.; Ghasemi, J.; Hosseini-Esfahani, F.; Mohammadi, A. Investigation and determination of acrylamide in the main group of cereal products using advanced microextraction method coupled with gas chromatography-mass spectrometry. J. Cereal Sci. 2019, 87, 157–164. [Google Scholar] [CrossRef]
- Norouzi, E.; Kamankesh, M.; Mohammadi, A.; Attaran, A. Acrylamide in bread samples: Determining using ultrasonic-assisted extraction and microextraction method followed by gas chromatography-mass spectrometry. J. cereal sci. 2018, 79, 1–5. [Google Scholar] [CrossRef]
- Moreda-Piñeiro, J.; Moreda-Piñeiro, A. Recent advances in combining microextraction techniques for sample pre-treatment. TRAC Trends Anal. Chem. 2015, 71, 265–274. [Google Scholar] [CrossRef]
- Sajid, M.; Płotka-Wasylka, J. Combined extraction and microextraction techniques: Recent trends and future perspectives. TRAC Trends Anal. Chem. 2018, 103, 74–86. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Secondary Product | CAS Number | IARC Category | Tolerable Daily Intake µg/Kg bw/Day | Reference | |
|---|---|---|---|---|---|
| Saturate Carbonyls | Formaldehyde | 50-00-0 | 1 | 150 | [22] |
| Acetaldehyde | 75-07-0 | 2B | 185 a | [23] | |
| Hexanal | 66-25-1 | - | 780 * | [24] | |
| α,β-Unsaturated Carbonyls | Acrolein | 107-02-8 | 2A | 7.5 | [25] |
| 4-hydroxy-2-nonenal | 75899-68-2 | 3 | 1.5 ** | [26] | |
| 4-hydroxy-2-hexenal | 17427-21-3 | 3 | 1.5 ** | [26] | |
| Acrylamide | 79-06-1 | 2A | NE | [27] | |
| Crotonaldehyde | 4170-30-3 | 2B | - | - | |
| Dicarbonyls | Malondialdehyde | 102-52-3 | 3 | 30 ** | [26] |
| Glyoxal | 107-22-2 | - | 200 | [28] | |
| Methylglyoxal | 78-98-8 | 3 | - | - | |
| Diacetyl | 431-03 | - | 900 * | [28] | |
| Furans | Dihydro-2(3H)-furanone | 96-48-0 | 3 | - | - |
| Furfural | 98-01-1 | 3 | 500 | [29] | |
| Target Compound | Sample | GDME | Derivative Reagent | Determination | LOD µg/L or µg/Kg | Recovery % | Ref. | |||
|---|---|---|---|---|---|---|---|---|---|---|
| Mode | Vacceptor solution mL | t min | T °C | |||||||
| 1,3-pentadione Diacetyl | Beer | Immersed | 0.5 | 15 | 40 | O-PDA | HPLC-UV | 3.8–4.6 | - | [48] |
| 2 aldehydes & Furfural | Beer | Immersed | 0.75 | 5 | 30 | DNPH | HPLC-UV | 1.5–12.3 | - | [54] |
| 5 aldehydes | Beer | Suspended | 0.5 | 20 | 40 | HBA | HPLC-DAD | 1.2–1857.7 | >96% | [55] |
| Diacetyl 1 | Wine | Immersed | 0.4 | 20 | 65 | O-PDA | HPLC-UV | 3.8 | - | [56] |
| Acetaldehyde 1 | Wine | Immersed | 1.0 | 15 | 50 | DNPH | HPLC-UV | 800–1100 | - | [57] |
| Diacetyl | Wine & beer | Suspended | 1.0 | 10 | 60 | O-PDA | DPV | 0.053 | - | [58] |
| α-DCC | Wine; black tea & soy sauce | Immersed 2 | 0.5 | 10 | 55 | O-PDA | HPLC-UV | 50–200 | - | [59] |
| MDA | Vegetable oil | Suspended | 0.5 | 30 | 65 | TBA | HPLC-UV/FLD | 250–350 | ≥82% | [60] |
| 4 aldehydes Acrolein & MDA | Vegetable oil | Suspended | 1.0 | 10 | 60 | DPNH | GC-MS | 50–100 | ≥95% | [10] |
| 2 ketones & diacetyl | Ground bread | Suspended | 0.5 | 15 | 65 | O-PDA | HPLC-UV | 6–12 | - | [61] |
| 27 carbonyl compounds 3 | Green & roast coffee beans | Suspended | 0.5 | 16 | 40 | O-PDA | HPLC-DAD | 50–200 | - | [62] |
| Target Compound | Sample | SPME | Derivative Reagent | Determination | LOD µg/L or µg/Kg | Recovery % | Ref. | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Mode | t min | T °C | Fiber | Tdesorption °C | |||||||
| 14 aldehydes & ketones | Vegetable oil | HS | 30 | 20 | DVB/CAR/PDMS | 270 | - | GC-FID & GC-MS | 0.04–2.24 | - | [64] |
| 4-HNE | Oils & porcine liver | DI | 15 | 40 | PDMS/DVB | DNPH | HPLC-SP | 0.001–1.42 | 66–87% | [65] | |
| MDA | Cod liver oil | HS | 10 | RT | PDMS/DVB | 200 | N-MH | GC-NPD | 0.74 | 91% | [66] |
| Hexanal | Hazelnut | HS | 10 | 60 | CAR/PDMS | 300 | - | GC-FID | 8.01 | - | [67] |
| 7 aldehydes | Peanut, soybean and olive oils | HS | 15 | 50 | CAR/PDMS | 250 | - | GC-FID | 4.6–10.2 | 85–110 | [68] |
| 3 α,β-UC | Sunflower oil digestion phases | HS | 60 | 50 | DVB/CAR/PDMS | 250 | - | GC-MS | - | - | [69] |
| 100 carbonyl compounds | Cod liver oil | HS | 60 | 50 | DVB/CAR/PDMS | 220 | - | GC-MS | - | - | [70] |
| 18 VOC | Sunflower oil emulsions | HS | 30 | 50 | DVB/CAR/PDMS | 250 | - | GC-MS | - | - | [71] |
| Aldehydes & 2-pentylfuran | Soybean oils | HS | 55 | 50 | DVB/CAR/PDMS | 250 | - | GC-MS | - | - | [72] |
| VOC | Peanut oil | HS | 40 | 50 | PDMS/DVB | 250 | - | GC-MS | - | - | [73] |
| 4 aldehydes & 1 ketone | Roast & boiled duck | HS | 40 | 45 | CAR/PDMS | 280 | - | GC-MS | - | - | [74] |
| 3 aldehydes | Chicken patties | HS | 10 | 60 | DVB/CAR/PDMS | 250 | - | GC-FID | - | - | [75] |
| Hexanal | Pig sausages | HS | 30 | 50 | DVB/CAR/PDMS | 220 | - | GC-MS | - | - | [76] |
| 2 aldehydes & 2 dialdehydes | Cod | HS | 30 | 50 | CAR/PDMS | 260 | - | GC-FID | - | - | [77] |
| 8 aldehydes | Fish | HS | 15 | 60 | PDMS/DVB | 260 | PFBHA | GC-MS | 1.4–6.1 | 79–102 | [78] |
| 6 aldehydes | Caviar | HS | 30 | 60 | DVB/CAR/PDMS | 250 | - | GC-MS | - | - | [79] |
| 198 VOCs | Dry cured meat | HS | 30 | 37 | 260 | - | GC-MS | - | - | [80] | |
| Aldehydes | Infant formula | HS | 10 | 25 | PDMS/DVB | 250 | - | GC-MS | - | - | [81] |
| 3 aldehydes & pentane | Infant formula | HS | 45 | 37 | CAR/PDMS | 250 | - | GC-FID | 0.02–1.05 | - | [82] |
| 13 Carbonyl compounds | Milk powder | HS | 45 | 43 | 250 | - | GC-MS | 2–6 | - | [83] | |
| VOC | Smoked cheese | HS | 45 | 50 | CAR/PDMS | 260 | - | GC-MS | - | - | [84] |
| VOC | Mozzarella | HS | 15 | 37 | 220 | - | GC-MS | - | - | [85] | |
| VOC | Portuguese cheese | HS | 45 | 50 | DVB/PDMS | 250 | - | GC-MS | - | - | [86] |
| 9 aldehydes | Beer | HS | 60 | 50 | PDMS/DVB | 250 | PFBHA * | GC-MS | - | 89–114 | [87] |
| 41 carbonyl compounds | Beer | HS | 40 | 60 | PDMS/DVB | 250 | PFBHA *,** | GC-MS | 0.003–20,000 | - | [88] |
| 250 carbonyl compounds | Beer | HS | 20 | 45 | PDMS/DVB | 250 | PFBAH ** | GC-ITMS | 0.003–0.510 | 88–114 | [89] |
| 6 carbonyl compound | Beer | HS | 60 | 55 | DVB/CAR/PDMS | 250 | TFEH ** | GC-MS | 0.03–0.5 | 90–105 | [16] |
| 6 carbonyl compound | Craft beer | HS | 60 | 55 | DVB/CAR/PDMS | 250 | TFEH ** | GC-MS | 0.03–0.5 | 90–105 | [90] |
| 18 carbonyl compound | Wine | HS | 45 | 40 | DVB/CAR/PDMS | 250 | - | GC-ITMS | 0.62–129.2 | 19–190 | [91] |
| 80 VOC | Wine | HS | 30 | 40 | DVB/CAR/PDMS | 240 | - | GC-MS | - | - | [92] |
| 6 carbonyl compound | Syrah wines | HS | 45 | 55 | DVB/CAR/PDMS | 250 | TFEH | GCxGC-TOFMS | 0.5–5.2 | 90–106 | [93] |
| 3 aldehydes | Must & wine | HS | 45 | 55 | DVB/CAR/PDMS | 250 | TFEH | GC-qMS | 0.1–0.8 | 90–102 | [94] |
| 38 carbonyl compound | Port wine | HS | 20 | 32 | PDMS/DVB | 250 | PFBHA | GC-MS | 0.006–0.089 | 88–119 | [95] |
| 45 carbonyl compound | Wine | HS | 20 | 40 | PDMS/DVB | 250 | PFBHA | GC-MS/MS | - | 71–146 | [96] |
| 9 aldehydes | Spirits and alcoholic beverages | DI | 15 | 20 | PDMS | 250 | PFBHA | GC-ECD | 0.05–0.5 | - | [97] |
| VOC & SVOC | Beer, wine & whisky | HS | 60 | 30 | PDMS CAR/PDMS DVB/CAR/PDMS | 250 260 260 | - | GC-MS | - | - | [11] |
| 20 aldehydes | Green pomace distillates | HS | 40 | 55 | PDMS/DVB | 250 | PFBHA | GC-MS | 0.0007–0.02 | 76–110 | [98] |
| 107 VOC | Cider | HS | 30 | 50 | DVB/CAR/PDMS | 250 | - | GC-MS | - | - | [99] |
| 53 carbonyl compounds | Huangjiu (alcoholic beverage) | HS | 35 | 45 | DVB/CAR/PDMS | 250 | PFBHA | GC-MS/MS | - | 71–146 | [100] |
| 2 α-DCC | Soybean paste, red pepper past, soy sauce, wine, beer, distilled liquor | HS | 20 | 85 | DVB/CAR/PDMS | 240 | TFEH | GC-MS | 0.7–1.1 | 92–104 | [101] |
| Target Compound | Sample | DLLME | Derivative Reagent | Determ. | LOD µg/L or µg/Kg | Rec. % | Ref. | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Mode | Disperser | Extracting Solvent | T min | T °C | |||||||
| Formaldehyde | Beverages | MW-IL- | ACN | IL 3453W | 1.5 | - | DNPH | HPLC-UV | 0.12 | 85–95 | [112] |
| Acrylamide | Brewed coffee | - | ACN | DCM | - | - | - | UPLC-MS/MS | 900 | 97–106 | [113] |
| PCB and acrylamide | Milk/Coffee | IL | [HeOHMIM][Cl] | [BMIM][NTf2] | - | - | - | HS-GC-ECD-MS | - | - | [114] |
| MDA, acrolein, 4-HNE | Beverages | US | ACN | CH3Cl | 5 | 60 °C | DNPH | GC-MS | 50–200 | 94–102 | [115] |
| Formaldehyde | Milk | IL | MeOH | IL 3453W | 0.75 | 45 °C | ACAC | UV | 100 | 91–103 | [116] |
| Acrylamide | Coffee, chocolate, roasted nuts, French fries, cereals, biscuits, chips, bread, and caramelized fruit | SSA | SUPRAS-2 (SDS/TBABr/AlCl3) | 2 | - | UV | 0.2 | 93–96 | [117] | ||
| Acrylamide | Nuts and seeds | - | PCE | EtOH | 3 | - | Xanthydrol | GC-MS | 0.6 | 95 | [118] |
| Acrylamide | Potato chips | UAE | PCE | EtOH | 2 | - | Xanthydrol | GC-MS | 0.6 | 97 | [119] |
| Acrylamide | Cereal products | - | PCE | EtOH | 1 | - | Xanthydrol | GC-MS | 0.6 | 95 | [120] |
| Acrylamide | Bread | UAE | PCE | MeOH | 1 | - | Xanthydrol | GC-MS | 0.54 | 98 | [121] |
| 4 aldehydes Acrolein & MDA | Vegetable oil | US | ACN | CH3Cl | 5 | 60 | DPNH | GC-MS | 50–100 | ≥95% | 10 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Custodio-Mendoza, J.A.; Ares-Fuentes, A.M.; Carro, A.M. Innovative Solutions for Food Analysis: Microextraction Techniques in Lipid Peroxidation Product Detection. Separations 2023, 10, 531. https://doi.org/10.3390/separations10100531
Custodio-Mendoza JA, Ares-Fuentes AM, Carro AM. Innovative Solutions for Food Analysis: Microextraction Techniques in Lipid Peroxidation Product Detection. Separations. 2023; 10(10):531. https://doi.org/10.3390/separations10100531
Chicago/Turabian StyleCustodio-Mendoza, Jorge A., Ana M. Ares-Fuentes, and Antonia M. Carro. 2023. "Innovative Solutions for Food Analysis: Microextraction Techniques in Lipid Peroxidation Product Detection" Separations 10, no. 10: 531. https://doi.org/10.3390/separations10100531