
Multiple Heart-Cutting Two-Dimensional HPLC-UV Achiral–Chiral Analysis of Branched-Chain Amino Acids in Food Supplements under Environmentally Friendly Conditions
 , , , , , , and
, , , , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Extraction of the AA Pool from the Food Supplement
2.3. Achiral–Chiral HPLC Analysis
3. Results and Discussion
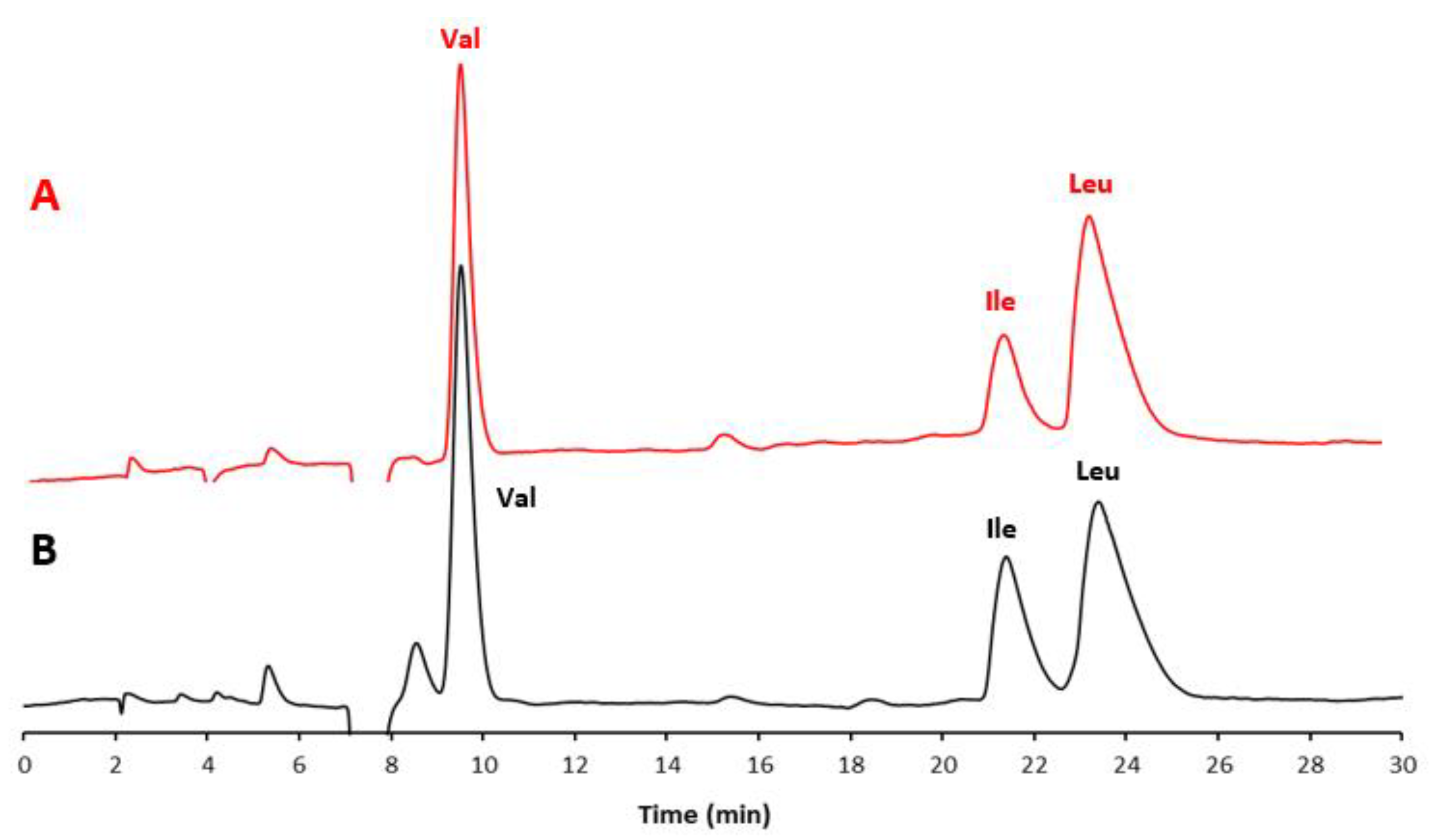
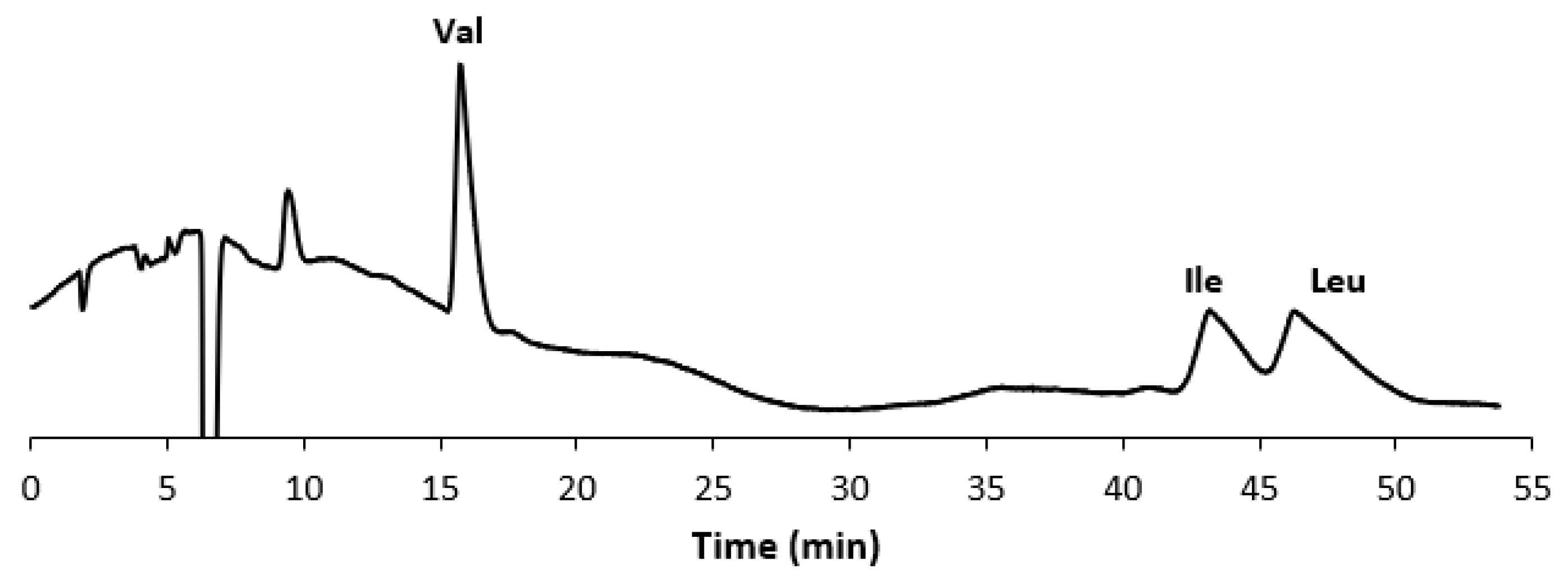
3.1. Development of the Achiral IP-RP-HPLC Method
3.2. Validation of the Optimized Achiral IP-RP-HPLC Method and Quantitative Analysis of the AAs in the Food Supplement
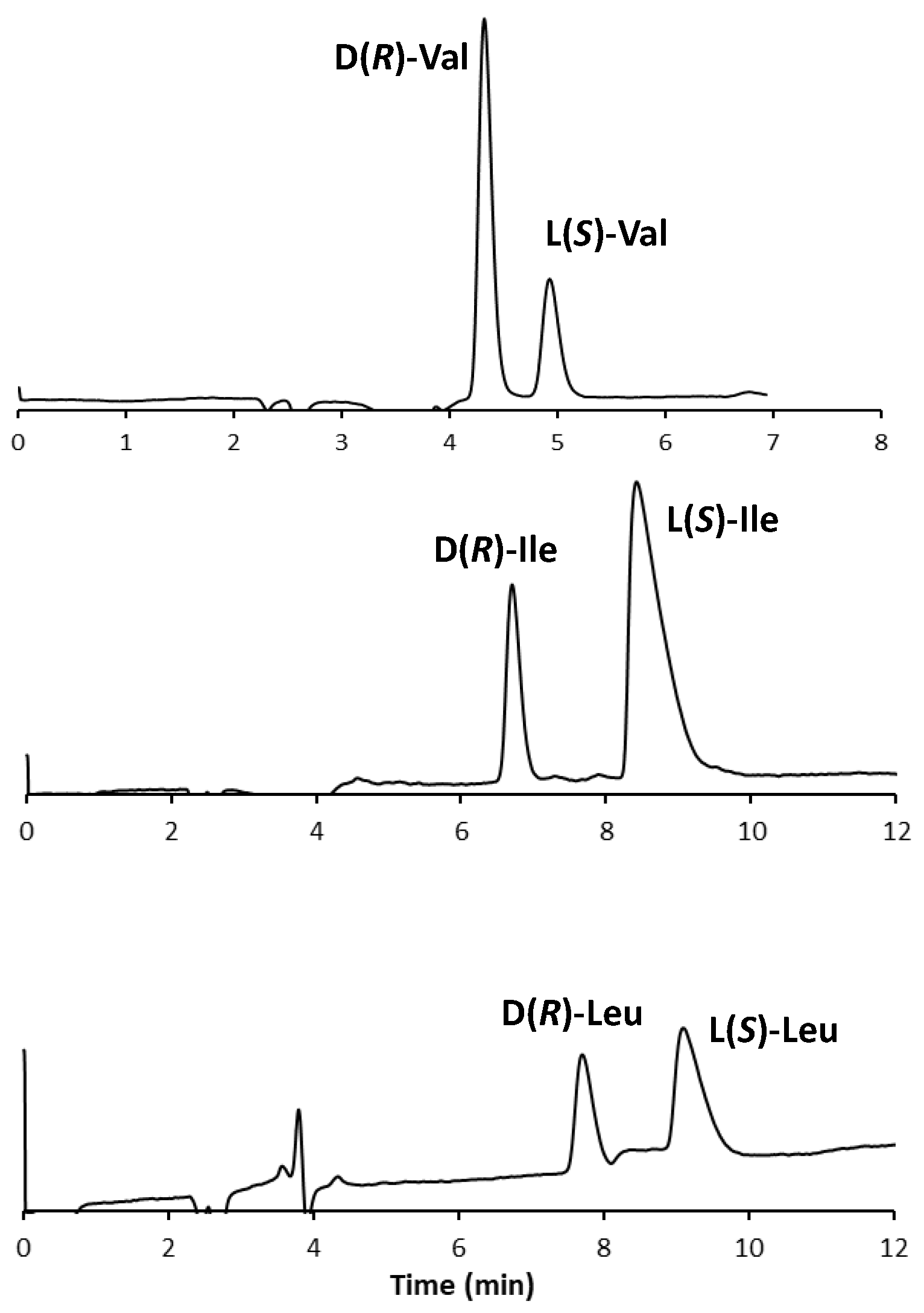
3.3. Optimization of the Experimental Conditions for the CLEC Analysis
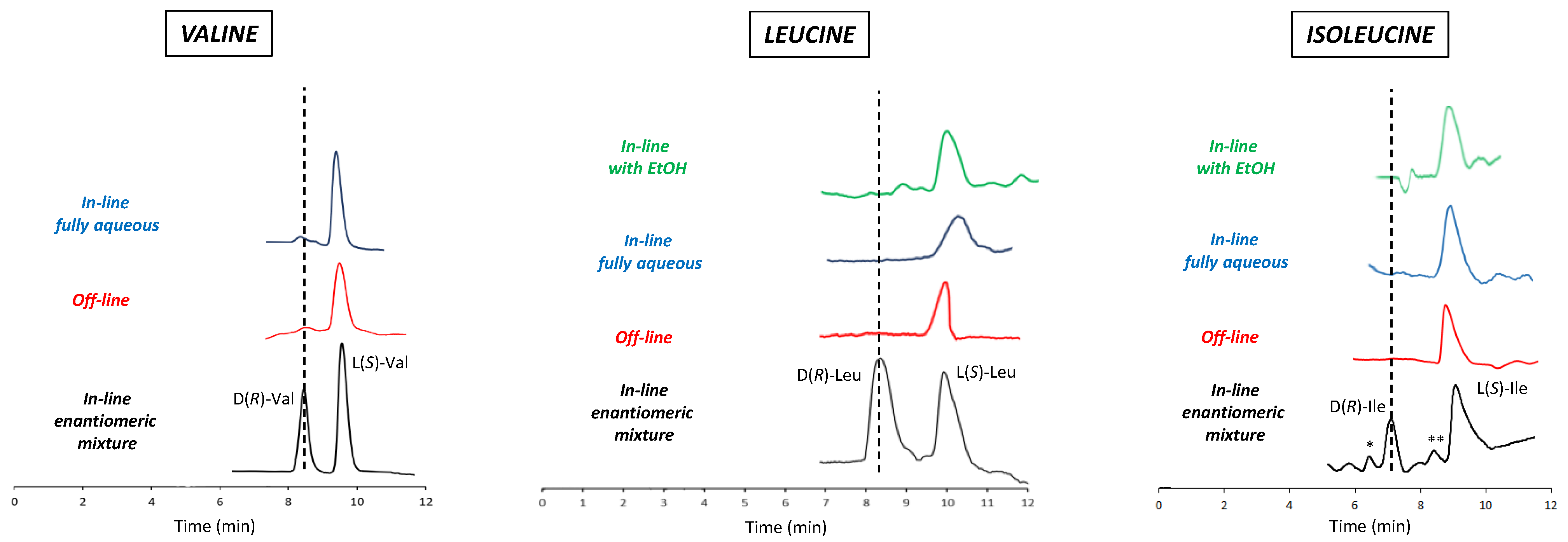
3.4. mLC-LC Achiral–Chiral Analysis of BCAAs in Food Supplements
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Płotka, J.; Tobiszewski, M.; Sulej, A.M.; Kupska, M.; Górecki, T.; Namieśnik, J. Green chromatography. J. Chromatogr. A 2013, 1307, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Rocha, F.R.P.; Nóbrega, J.A.; Filho, O.F. Flow analysis strategies to greener analytical chemistry. An overview. Green Chem. 2001, 3, 216–220. [Google Scholar] [CrossRef]
- Keith, L.H.; Gron, L.U.; Young, J.L. Green Analytical Methodologies. Chem. Rev. 2007, 107, 2695–2708. [Google Scholar] [CrossRef] [PubMed]
- Pereira, A.; David, F.; Vanhoenacker, G.; Sandra, K.; Sandra, P. Green Chromatography (Part 1): Introduction and Liquid Chromatography. LCGC Eur. 2010, 23, 242–259. [Google Scholar]
- Shaaban, H. New insights into liquid chromatography for more eco-friendly analysis of pharmaceuticals. Anal. Bioanal. Chem. 2016, 408, 6929–6944. [Google Scholar] [CrossRef] [PubMed]
- Anastas, P.; Eghbali, N. Green Chemistry: Principles and Practice. Chem. Soc. Rev. 2010, 39, 301–312. [Google Scholar] [CrossRef]
- Anastas, P.T.; Warner, J.C. Green Chemistry: Theory and Practice; Oxford University Press: New York, NY, USA, 1998. [Google Scholar]
- Horváth, I.T.; Anastas, P.T. Innovations and Green Chemistry. Chem. Rev. 2007, 107, 2169–2173. [Google Scholar] [CrossRef] [Green Version]
- Yabré, M.; Ferey, L.; Somé, I.T.; Gaudin, K. Greening Reversed-Phase Liquid Chromatography Methods Using Alternative Solvents for Pharmaceutical Analysis. Molecules 2018, 23, 1065. [Google Scholar] [CrossRef] [Green Version]
- Dogan, A.; Eylem, C.C.; Akduman, N.E.B. Application of green methodology to pharmaceutical analysis using eco-friendly ethanol-water mobile phases. Microchem. J. 2020, 157, 104895. [Google Scholar] [CrossRef]
- Shen, Y.; Chen, B.; van Beek, T.A. Alternative solvents can make preparative liquid chromatography greener. Green Chem. 2015, 17, 4073–4081. [Google Scholar] [CrossRef]
- Eldin, A.B.; Ismaiel, O.A.; Hassan, W.E.; Shalaby, A.A. Green analytical chemistry: Opportunities for pharmaceutical quality control. J. Anal. Chem. 2016, 71, 861–871. [Google Scholar] [CrossRef]
- Stoll, D.R.; Carr, P.W. Two-Dimensional Liquid Chromatography: A State of the Art Tutorial. Anal. Chem. 2017, 89, 519–531. [Google Scholar] [CrossRef]
- León-González, M.E.; Rosales-Conrado, N.; Perez-Arribas, L.V.; Guillén-Casla, V. Two-dimensional liquid chromatography for direct chiral separations: A review. Biomed. Chromatogr. 2014, 28, 59–83. [Google Scholar] [CrossRef]
- Zhang, K.; Li, Y.; Tsang, M.; Chetwyn, N.P. Analysis of pharmaceutical impurities using multi-heartcutting 2D LC coupled with UV-charged aerosol MS detection. J. Sep. Sci. 2013, 36, 2986–2992. [Google Scholar] [CrossRef]
- Karongo, R.; Ge, M.; Geibel, C.; Horak, J.; Lämmerhofer, M. Enantioselective multiple heart cutting online two-dimensional liquid chromatography-mass spectrometry of all proteinogenic amino acids with second dimension chiral separations in one-minute time scales on a chiral tandem column. Anal. Chim. Acta 2021, 1180, 338858. [Google Scholar] [CrossRef]
- Woiwode, U.; Reischl, R.J.; Buckenmaier, S.; Lindner, W.; Lämmerhofer, M. Imaging Peptide and Protein Chirality via Amino Acid Analysis by Chiral × Chiral Two-Dimensional Correlation Liquid Chromatography. Anal. Chem. 2018, 90, 7963–7971. [Google Scholar] [CrossRef]
- Malik, A. Multidimensional Chromatography Edited by Luigi Mondello (Universita Degli Studi Di Messina), Alastair C. Lewis, and Keith D. Bartle (University of Leeds). J. Wiley & Sons. J. Am. Chem. Soc. 2002, 124, 13959–13960. [Google Scholar]
- Blahová, E.; Jandera, P.; Cacciola, F.; Mondello, L. Two-dimensional and serial column reversed-phase separation of phenolic antioxidants on octadecyl-, polyethyleneglycol-, and pentafluorophenylpropyl-silica columns. J. Sep. Sci. 2006, 29, 555–566. [Google Scholar] [CrossRef]
- Ouyang, Y.; Zeng, Y.; Rong, Y.; Song, Y.; Shi, L.; Chen, B.; Yang, X.; Xu, N.; Linhardt, R.J.; Zhang, Z. Profiling Analysis of Low Molecular Weight Heparins by Multiple Heart-Cutting Two Dimensional Chromatography with Quadruple Time-of-Flight Mass Spectrometry. Anal. Chem. 2015, 87, 8957–8963. [Google Scholar] [CrossRef]
- Pursch, M.; Buckenmaier, S. Loop-Based Multiple Heart-Cutting Two-Dimensional Liquid Chromatography for Target Analysis in Complex Matrices. Anal. Chem. 2015, 87, 5310–5317. [Google Scholar] [CrossRef]
- Piendl, S.K.; Geissler, D.; Weigelt, L.; Belder, D. Multiple Heart-Cutting Two-Dimensional Chip-HPLC Combined with Deep-UV Fluorescence and Mass Spectrometric Detection. Anal. Chem. 2020, 92, 3795–3803. [Google Scholar] [CrossRef] [PubMed]
- Hamase, K.; Homma, H.; Takigawa, Y.; Fukushima, T.; Santa, T.; Imai, K. Regional distribution and postnatal changes of d-amino acids in rat brain. Biochim. Biophys. Acta 1997, 1334, 214–222. [Google Scholar] [CrossRef] [PubMed]
- Hamase, K.; Inoue, T.; Morikawa, A.; Konno, R.; Zaitsu, K. Determination of free d-proline and d-leucine in the brains of mutant mice lacking d-amino acid oxidase activity. Anal. Biochem. 2001, 298, 253–258. [Google Scholar] [CrossRef] [PubMed]
- Welsch, T.; Schmidtkunz, C.; Müller, B.; Meier, F.; Chlup, M.; Köhne, A.; Lämmerhofer, M.; Lindner, W. A comprehensive chemoselective and enantioselective 2D-HPLC set-up for fast enantiomer analysis of a multicomponent mixture of derivatized amino acids. Anal. Bioanal. Chem. 2007, 388, 1717–1724. [Google Scholar] [CrossRef] [PubMed]
- Hamase, K.; Morikawa, A.; Ohgusu, T.; Lindner, W.; Zaitsu, K. Comprehensive analysis of branched aliphatic d-amino acids in mammals using an integrated multi-loop two-dimensional column-switching high-performance liquid chromatographic system combining reversed-phase and enantioselective columns. J. Chromatogr. A 2007, 1143, 105–111. [Google Scholar] [CrossRef]
- Ianni, F.; Sardella, R.; Lisanti, A.; Gioiello, A.; Goga, B.T.C.; Lindner, W.; Natalini, B. Achiral–chiral two-dimensional chromatography of free amino acids in milk: A promising tool for detecting different levels of mastitis in cows. J. Pharm. Biomed. Anal. 2015, 116, 40–46. [Google Scholar] [CrossRef]
- Woiwode, U.; Neubauer, S.; Lindner, W.; Buckenmaier, S.; Lämmerhofer, M. Enantioselective multiple heartcut two-dimensional ultra-high-performance liquid chromatography method with a Coreshell chiral stationary phase in the second dimension for analysis of all proteinogenic amino acids in a single run. J. Chromatogr. A 2018, 1562, 69–77. [Google Scholar] [CrossRef]
- Napolitano-Tabares, P.I.; Negrín-Santamaría, I.; Gutiérrez-Serpa, A.; Pino, V. Recent efforts to increase greenness in chromatography. Curr. Opin. Green Sustain. Chem. 2021, 32, 100536. [Google Scholar] [CrossRef]
- Petritis, K.; Chaimbault, P.; Elfakir, C.; Dreux, M. Ion-pair reversed-phase liquid chromatography for determination of polar underivatized amino acids using perfluorinated carboxylic acids as ion pairing agent. J. Chromatogr. A 1999, 833, 147–155. [Google Scholar] [CrossRef]
- Pearson, J.D.; McCroskey, M.C. Perfluorinated acid alternatives to trifluoroacetic acid for reversed-phase high-performance liquid chromatography. J. Chromatogr. A 1996, 746, 277–281. [Google Scholar] [CrossRef]
- Petritis, K.; De Person, M.; Elfakir, C.; Dreux, M. Validation of an Ion-Interaction Chromatography Analysis of Underivatized Amino Acids in Commercial Preparation Using Evaporative Light Scattering Detection. Chromatographia 2004, 60, 293–298. [Google Scholar] [CrossRef]
- Varfaj, I.; Carotti, A.; Mangiapelo, L.; Cossignani, L.; Taticchi, A.; Macchiarulo, A.; Ianni, F.; Sardella, R. Environmentally Sustainable Achiral and Chiral Chromatographic Analysis of Amino Acids in Food Supplements. Molecules 2022, 27, 7724. [Google Scholar] [CrossRef]
- Davankov, V.A.; Kurganov, A.A.; Ponomareva, T.M. Enantioselectivity of complex formation in ligand-exchange chromatographic systems with chiral stationary and/or chiral mobile phases. J. Chromatogr. A 1988, 452, 309–316. [Google Scholar] [CrossRef]
- Davankov, V.A.; Bochkov, A.S.; Kurganov, A.A.; Roumeliotis, P.; Unger, K.K. Separation of unmodified α-amino acid enantiomers by reverse phase HPLC. Chromatographia 1980, 13, 677–685. [Google Scholar] [CrossRef]
- Davankov, V.A.; Kurganov, A.A. The role of achiral sorbent matrix in chiral recognition of amino acid enantiomers in ligand-exchange chromatography. Chromatographia 1983, 17, 686–690. [Google Scholar] [CrossRef]
- Ianni, F.; Lisanti, A.; Marinozzi, M.; Camaioni, E.; Pucciarini, L.; Massoli, A.; Sardella, R.; Concezzi, L.; Natalini, B. Hydrophobic Amino Acid Content in Onions as Potential Fingerprints of Geographical Origin: The Case of Rossa da Inverno sel. Rojo Duro. Molecules 2018, 23, 1259. [Google Scholar] [CrossRef] [Green Version]
- Ianni, F.; Sechi, P.; La Mantia, A.; Pucciarini, L.; Camaioni, E.; Cenci Goga, B.T.; Sardella, R.; Natalini, B. The Relationships between Somatic Cells and Isoleucine, Leucine and Tyrosine Content in Cow Milk. Appl. Sci. 2019, 9, 349. [Google Scholar] [CrossRef] [Green Version]
- Chaimbault, P.; Petritis, K.; Elfakir, C.; Dreux, M. Ion-pair chromatography on a porous graphitic carbon stationary phase for the analysis of twenty underivatized protein amino acids. J. Chromatogr. A 2000, 870, 245–254. [Google Scholar] [CrossRef]
- Johnson, L.D. Research and Evaluation of Organic Hazardous Air Pollutant Source Emission Test Methods. J. Air Waste Manag. Assoc. 1996, 46, 1135–1148. [Google Scholar] [CrossRef] [Green Version]
- Brettschneider, F.; Jankowski, V.; Günthner, T.; Salem, S.; Nierhaus, M.; Schulz, A.; Zidek, W.; Jankowski, J. Replacement of acetonitrile by ethanol as solvent in reversed phase chromatography of biomolecules. J. Chromatogr. B 2010, 878, 763–768. [Google Scholar] [CrossRef]
- Shibue, M.; Mant, C.; Hodges, R. Effect of anionic ion-pairing reagent hydrophobicity on selectivity of peptide separations by reversed-phase liquid chromatography. J. Chromatogr. A 2005, 1080, 68–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eid, S.M.; Farag, M.A.; Bawazeer, S. Underivatized Amino Acid Chromatographic Separation: Optimized Conditions for HPLC-UV Simultaneous Quantification of Isoleucine, Leucine, Lysine, Threonine, Histidine, Valine, Methionine, Phenylalanine, Tryptophan, and Tyrosine in Dietary Supplements. ACS Omega 2022, 7, 31106–31114. [Google Scholar] [CrossRef] [PubMed]
- Arruda, C.; Aldana Mejía, J.A.; Pena Ribeiro, V.; Gambeta Borges, C.H.; Gomes Martins, C.H.; Sola Veneziani, R.C.; Ambrósio, S.R.; Bastos, J.K. Occurrence, chemical composition, biological activities and analytical methods on Copaifera genus—A review. Biomed. Pharmacother. 2019, 109, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Vakili, H.; Talebpour, Z.; Haghighi, F. Development, validation, and uncertainty measurement of HPLC-DAD method for determination of some free amino acids in infant formula and medical food products for inborn errors of metabolism. Food Chem. 2022, 390, 133204. [Google Scholar] [CrossRef] [PubMed]
- Natalini, B.; Sardella, R.; Pellicciari, R. O-Benzyl-(S)-Serine, a New Chiral Selector for Ligand-Exchange Chromatography of Amino Acids. Curr. Anal. Chem. 2005, 1, 85–92. [Google Scholar] [CrossRef]
- Wernicke, R. Separation of Underivatised Amino Acid Enantiomers by Means of a Chiral Solvent-Generated Phase. J. Chromatogr. Sci. 1985, 23, 39–48. [Google Scholar] [CrossRef]
- Weinstein, S. Resolution of D- and L-Amino Acids by HPLC using Copper Complexes of N,N-Dialkyl-α-amino Acids as Novel Chiral Additives. Angew. Chem.-Int. Ed. Engl. 1982, 21, 218. [Google Scholar] [CrossRef]
- Sardella, R.; Macchiarulo, A.; Carotti, A.; Ianni, F.; Rubiño, M.E.G.; Natalini, B. Chiral mobile phase in ligand-exchange chromatography of amino acids: Exploring the copper(II) salt anion effect with a computational approach. J. Chromatogr. A 2012, 1269, 316–324. [Google Scholar] [CrossRef]
- Ianni, F.; Sardella, R.; Lisanti, A.; Giacchè, N.; Conti, P.; Pinto, A.; Tamborini, L.; Natalini, B. Use of an o-Benzyl-(S)-Serine Containing Eluent for the Efficient Ligand-Exchange Chromatography-Based Enantioseparation of Constrained Glutamate Receptor Ligands. Anal. Lett. 2015, 48, 383–395. [Google Scholar] [CrossRef]
- Davankov, V.A. Chiral selectors with chelating properties in liquid chromatography: Fundamental reflections and selective review of recent developments. J. Chromatogr. A 1994, 666, 55–76. [Google Scholar] [CrossRef]
- Remelli, M.; Fornasari, P.; Pulidori, F. Study of retention, efficiency and selectivity in chiral ligand-exchange chromatography with a dynamically coated stationary phase. J. Chromatogr. A 1997, 761, 79–89. [Google Scholar] [CrossRef]
- Remelli, M.; Pozzati, G.; Conato, C. Direct chiral resolution of underivatized amino acids on a stationary phase dynamically modified with the ion-exchanger Nτ-decyl-L-spinacine. J. Sep. Sci. 2015, 38, 894–900. [Google Scholar] [CrossRef]
- Galaverna, G.; Pantò, F.; Dossena, A.; Marchelli, R.; Bigi, F. Chiral Separation of Unmodified α-Hydroxy Acids by Ligand Exchange HPLC Using Chiral Copper(II) Complexes of (S)-Phenylalaninamide as Additives to the Eluent. Chirality 1995, 7, 331–336. [Google Scholar] [CrossRef]
- Hyun, M.H.; Han, S.C.; Lee, C.W.; Lee, Y.K. Preparation and application of a new ligand exchange chiral stationary phase for the liquid chromatographic resolution of α-amino acid enantiomers. J. Chromatogr. A 2002, 950, 55–63. [Google Scholar] [CrossRef]
- Tobiszewski, M.; Marć, M.; Gałuszka, A.; Namieśnik, J. Green Chemistry Metrics with Special Reference to Green Analytical Chemistry. Molecules 2015, 20, 10928–10946. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Compound | Chromatographic Parameters | ||
|---|---|---|---|---|
| k | α | Rs | ||
| Val | 3.85 | |||
| a | Ile | 9.98 | 2.59 | 12.90 |
| Leu | 10.90 | 1.10 | 1.29 | |
| Val | 4.84 | |||
| b | Ile | 13.21 | 2.73 | 13.47 |
| Leu | 14.79 | 1.12 | 1.38 | |
| Val | 6.74 | |||
| c | Ile | 19.90 | 2.95 | 20.23 |
| Leu | 21.45 | 1.08 | 1.08 | |
| Val | 6.97 | |||
| d | Ile | 20.91 | 2.99 | 13.58 |
| Leu | 22.48 | 1.08 | 0.85 | |
| Val | 11.63 | |||
| e | Ile | 35.09 | 3.02 | 24.11 |
| Leu | 38.95 | 1.11 | 1.85 | |
| Val | 7.47 | |||
| f | Ile | 22.37 | 2.99 | 18.16 |
| Leu | 25.97 | 1.16 | 2.92 | |
| Val | 4.78 | |||
| g | Ile | 14.35 | 3.00 | 18.48 |
| Leu | 15.75 | 1.10 | 1.45 | |
| Compound | Theoretical Conc. (mg/mL) | Intraday Precision (RSD%) | Intraday Accuracy (Recovery%) | Interday Precision (RSD%) | Interday Accuracy (Recovery%) |
|---|---|---|---|---|---|
| Val | 0.25 | 2.4 | 95.1 | 4.8 | 97.9 |
| 1.3 | 99.1 | ||||
| 0.6 | 99.2 | ||||
| 0.70 | 0.5 | 95.7 | 3.0 | 96.6 | |
| 1.1 | 96.2 | ||||
| 3.7 | 97.2 | ||||
| Ile | 0.17 | 1.7 | 100.4 | 4.3 | 100.3 |
| 1.8 | 104.6 | ||||
| 0.9 | 104.7 | ||||
| 0.70 | 0.2 | 100.5 | 1.3 | 101.2 | |
| 0.1 | 99.5 | ||||
| 0.7 | 100.5 | ||||
| Leu | 0.17 | 1.7 | 99.3 | 1.9 | 98.7 |
| 1.5 | 99.2 | ||||
| 2.1 | 99.0 | ||||
| 0.70 | 0.5 | 99.8 | 1.1 | 99.3 | |
| 0.9 | 98.3 | ||||
| 0.72 | 99.7 |
| Entry | Compound | Chromatographic Parameters | ||||
|---|---|---|---|---|---|---|
| k2 | α | N1 | N2 | Rs | ||
| Val | 0.44 | 1.34 | 7455 | 5670 | 1.61 | |
| a | Ile | 1.27 | 1.26 | 7010 | 4889 | 2.31 |
| Leu | 1.30 | 1.25 | 3507 | 2168 | 1.58 | |
| Val | 0.58 | 1.43 | 5661 | 3623 | 1.93 | |
| b | Ile | 1.80 | 1.33 | 4014 | 2250 | 2.31 |
| Leu | 1.74 | 1.31 | 2665 | 1802 | 1.90 | |
| Val | 0.59 | 1.50 | 5170 | 3569 | 2.16 | |
| c | Ile | 2.01 | 1.37 | 4268 | 4549 | 3.29 |
| Leu | 1.93 | 1.35 | 1406 | 941 | 1.57 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Varfaj, I.; Moretti, S.; Ianni, F.; Barola, C.; Abualzulof, G.W.A.; Carotti, A.; Cossignani, L.; Galarini, R.; Sardella, R. Multiple Heart-Cutting Two-Dimensional HPLC-UV Achiral–Chiral Analysis of Branched-Chain Amino Acids in Food Supplements under Environmentally Friendly Conditions. Separations 2023, 10, 45. https://doi.org/10.3390/separations10010045
Varfaj I, Moretti S, Ianni F, Barola C, Abualzulof GWA, Carotti A, Cossignani L, Galarini R, Sardella R. Multiple Heart-Cutting Two-Dimensional HPLC-UV Achiral–Chiral Analysis of Branched-Chain Amino Acids in Food Supplements under Environmentally Friendly Conditions. Separations. 2023; 10(1):45. https://doi.org/10.3390/separations10010045
Chicago/Turabian StyleVarfaj, Ina, Simone Moretti, Federica Ianni, Carolina Barola, Ghaid W. A. Abualzulof, Andrea Carotti, Lina Cossignani, Roberta Galarini, and Roccaldo Sardella. 2023. "Multiple Heart-Cutting Two-Dimensional HPLC-UV Achiral–Chiral Analysis of Branched-Chain Amino Acids in Food Supplements under Environmentally Friendly Conditions" Separations 10, no. 1: 45. https://doi.org/10.3390/separations10010045