Exploring Plant Sesquiterpene Diversity by Generating Chemical Networks

 , , , , , and
, , , , , and
Abstract
:1. Introduction
2. Results
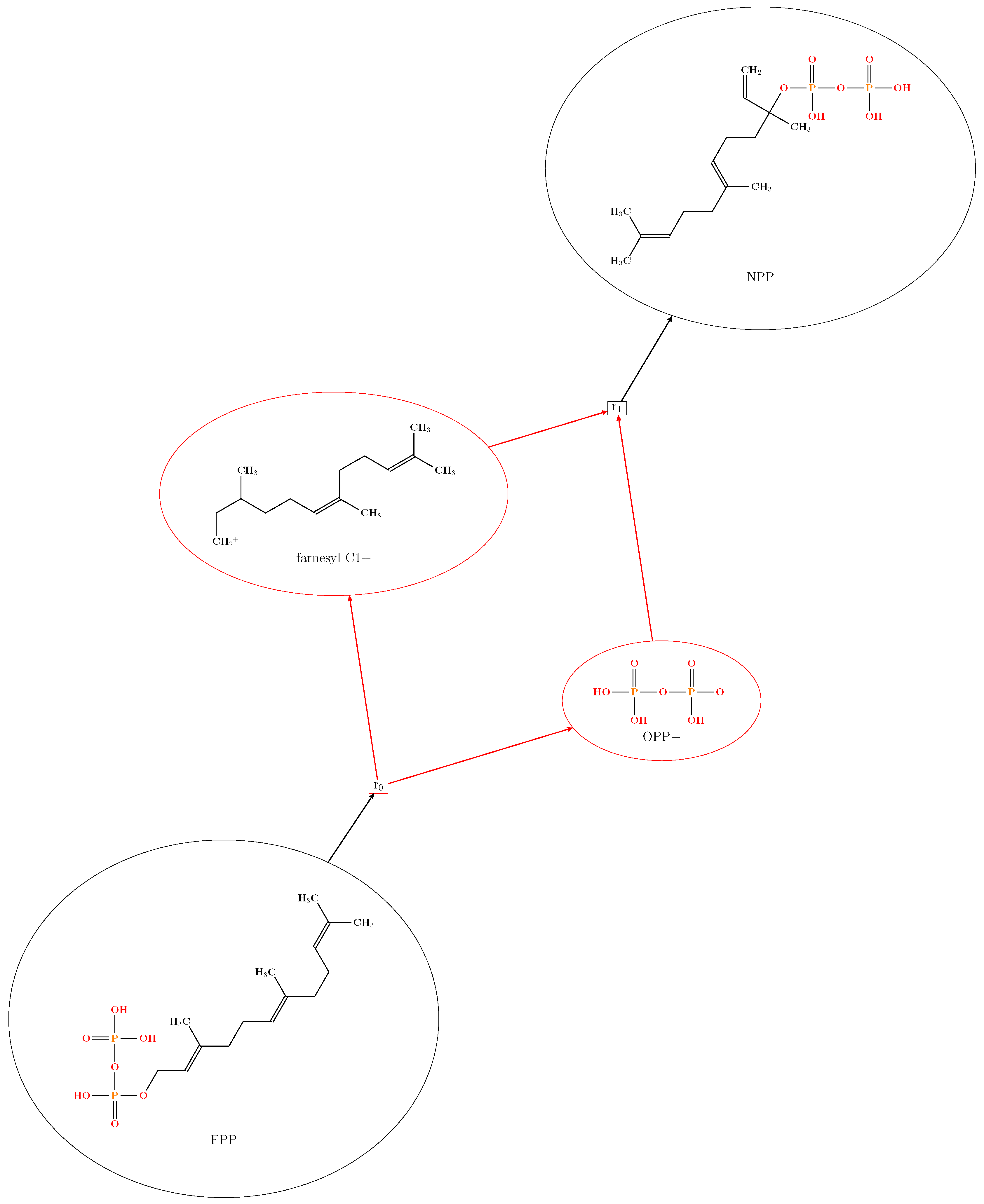
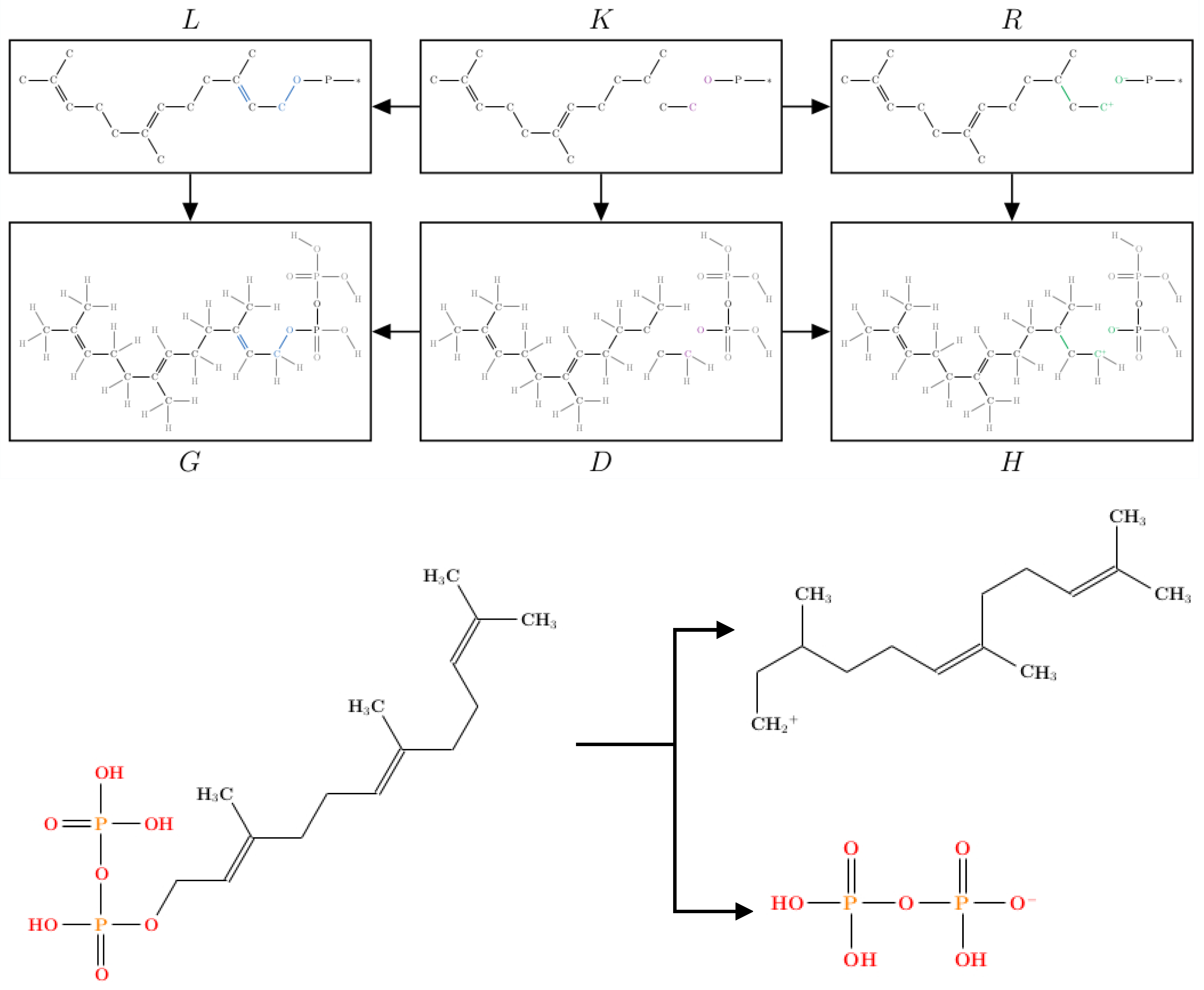
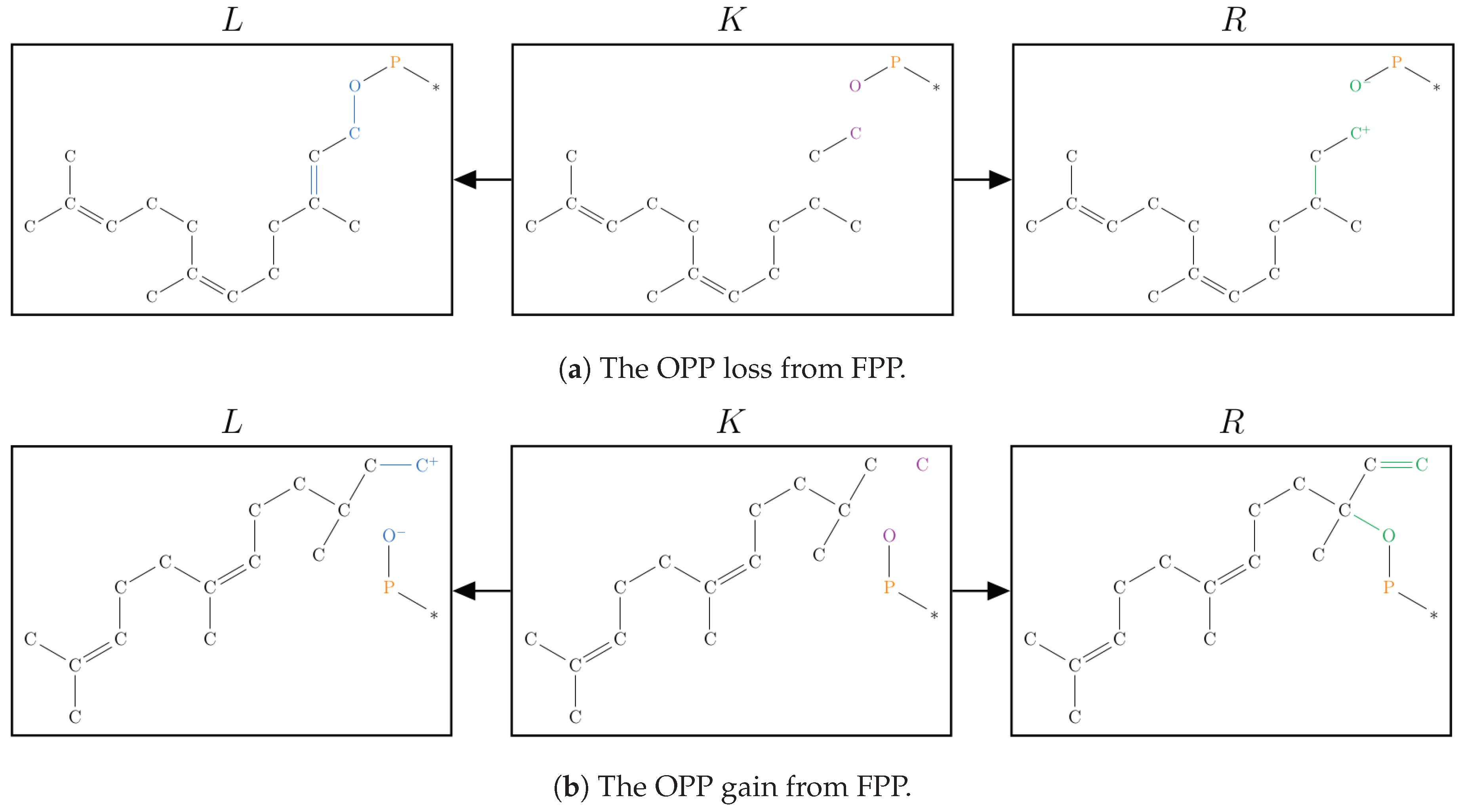
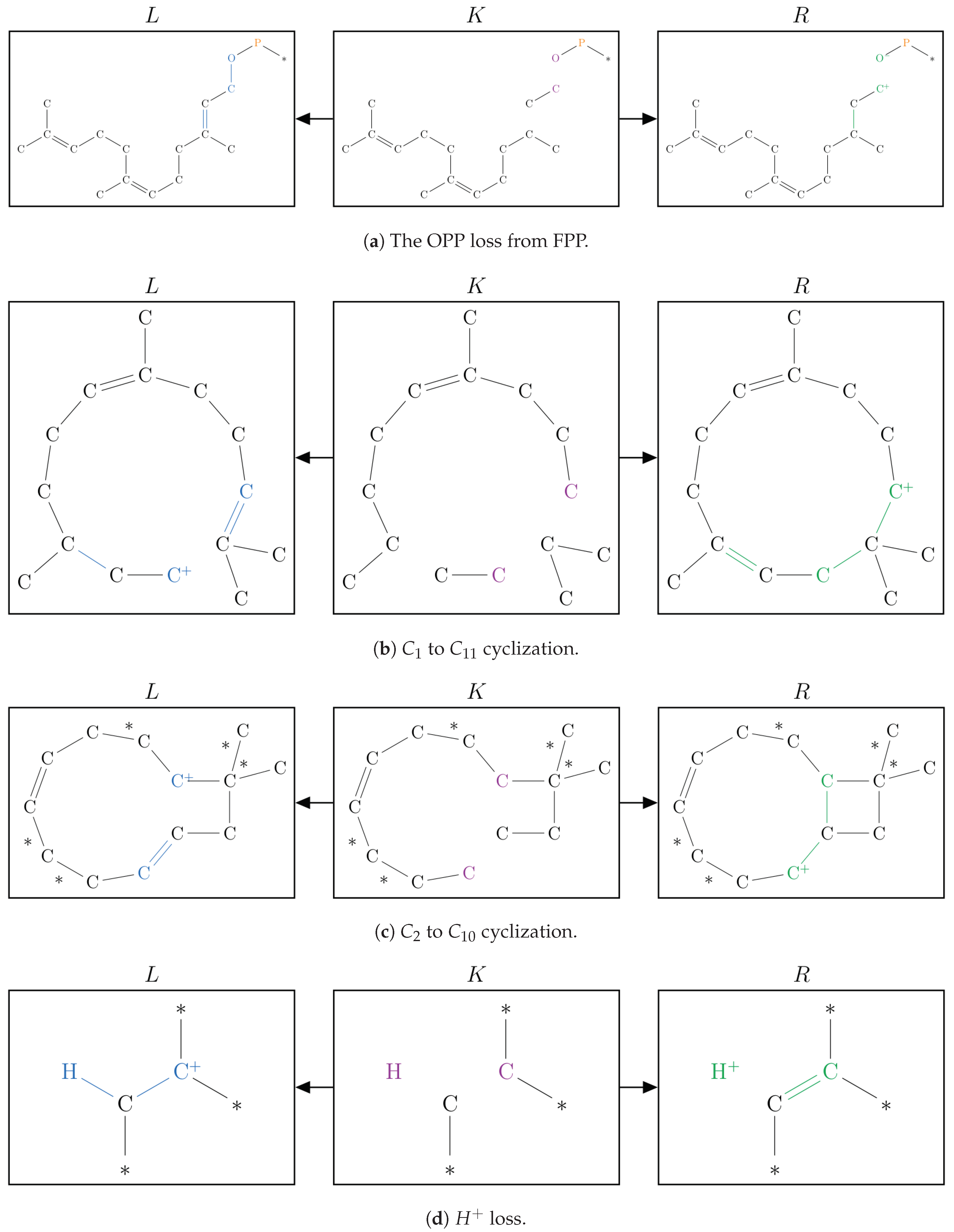
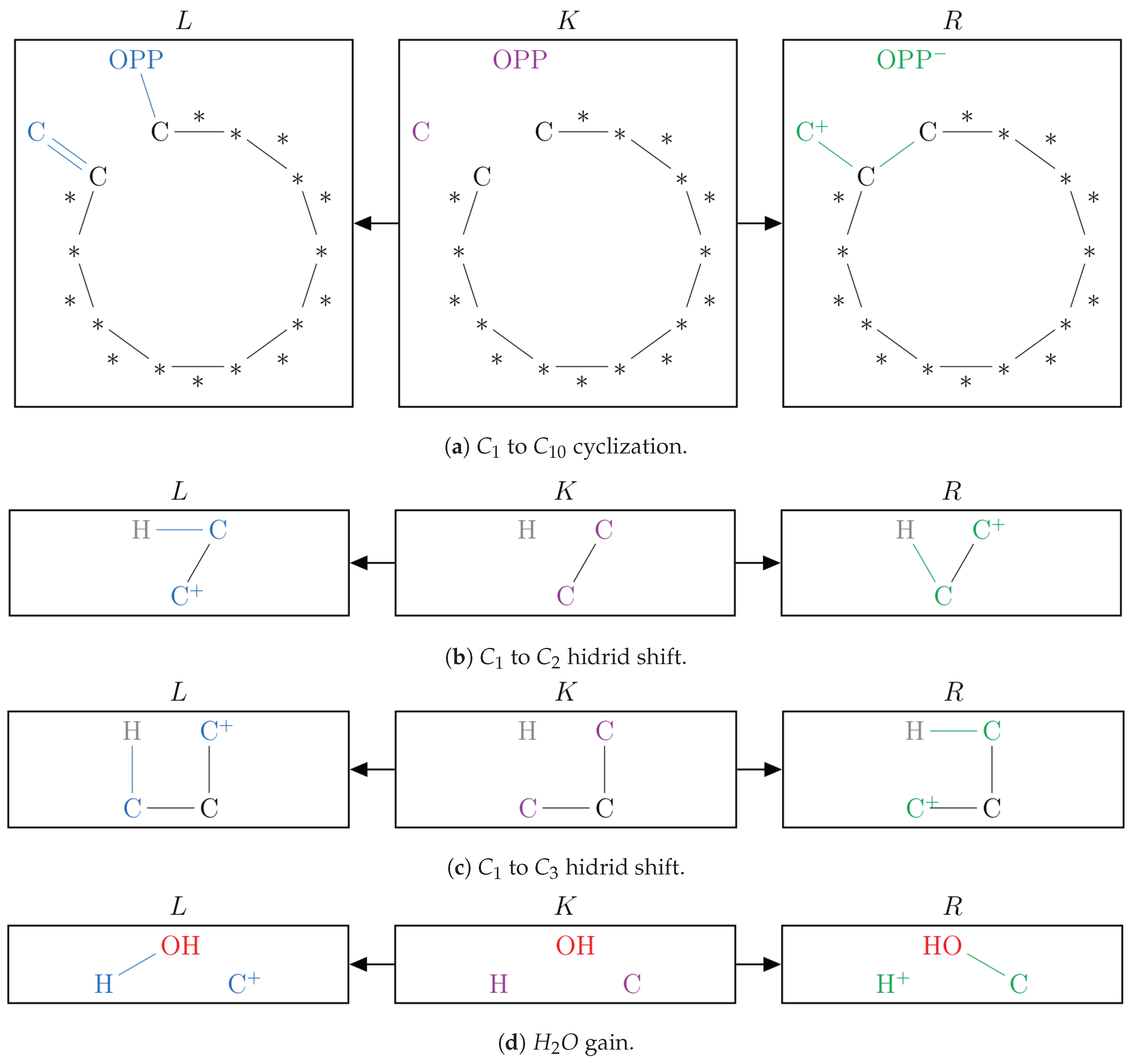
2.1. Protonation-Dependent Diphosphate Cleavage
2.2. Synthesis of -Caryophyllene, -Humulene, -Farnesene, and their Side Products
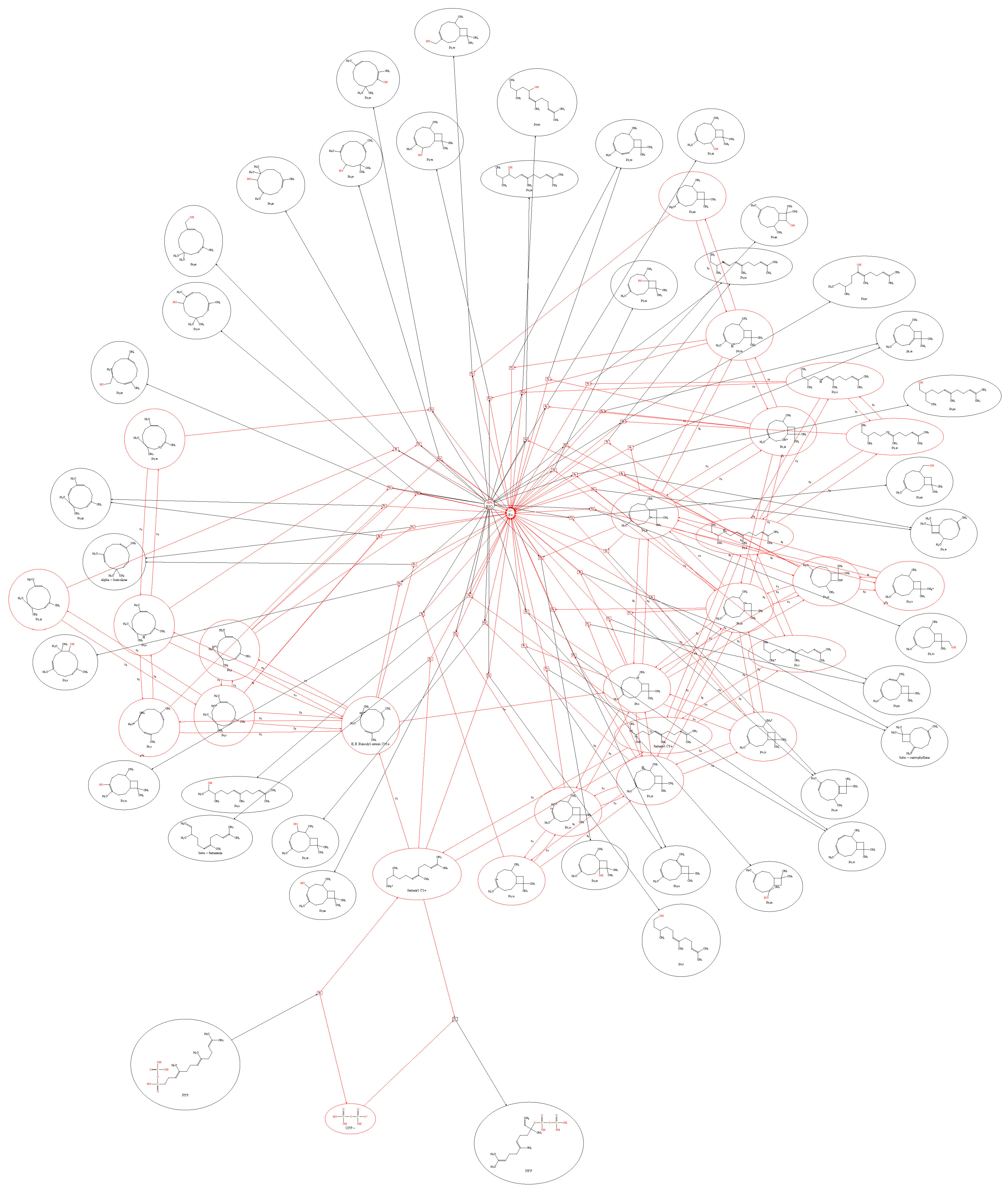
2.3. Large-Scale Exploration of Terpene Space
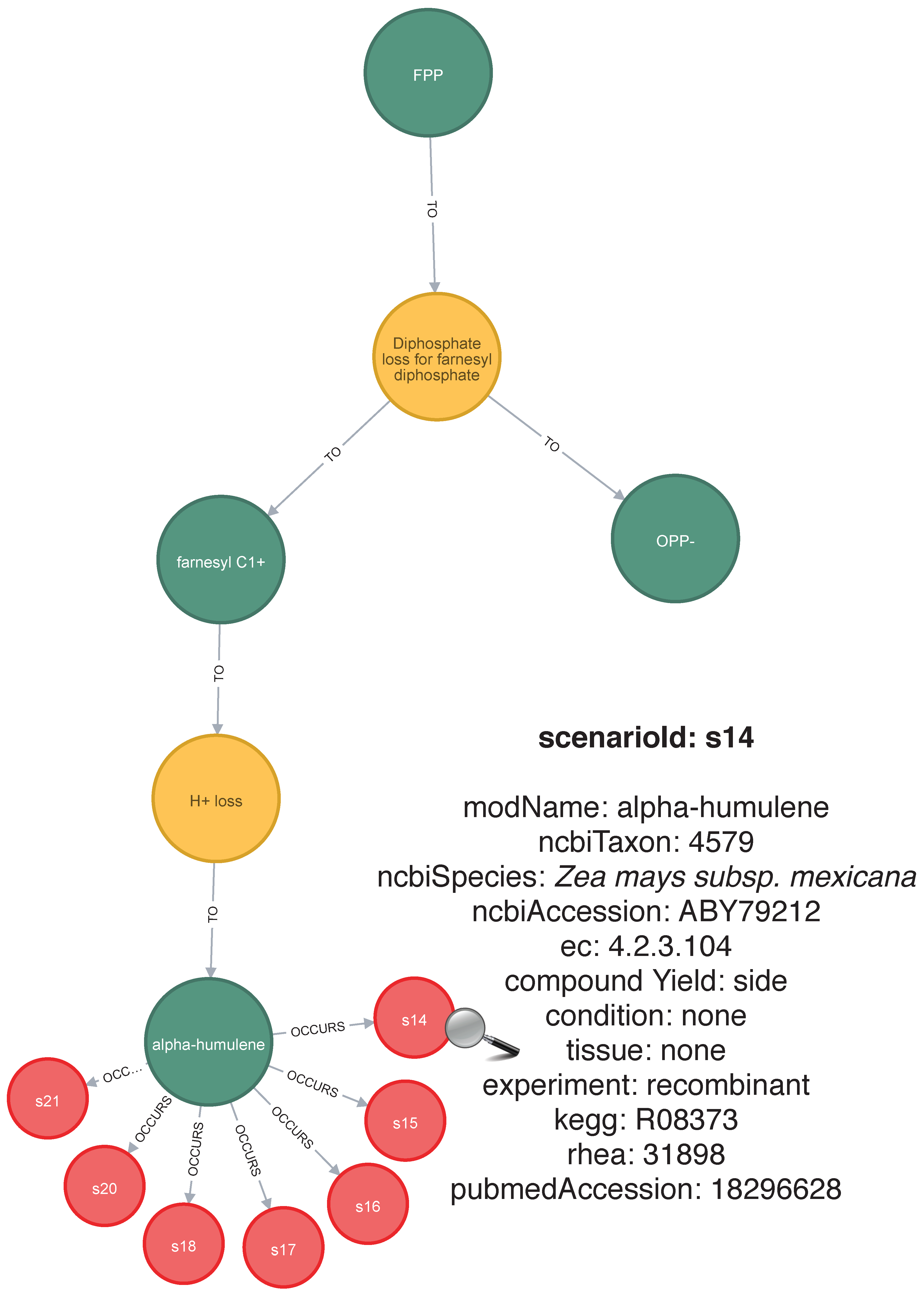
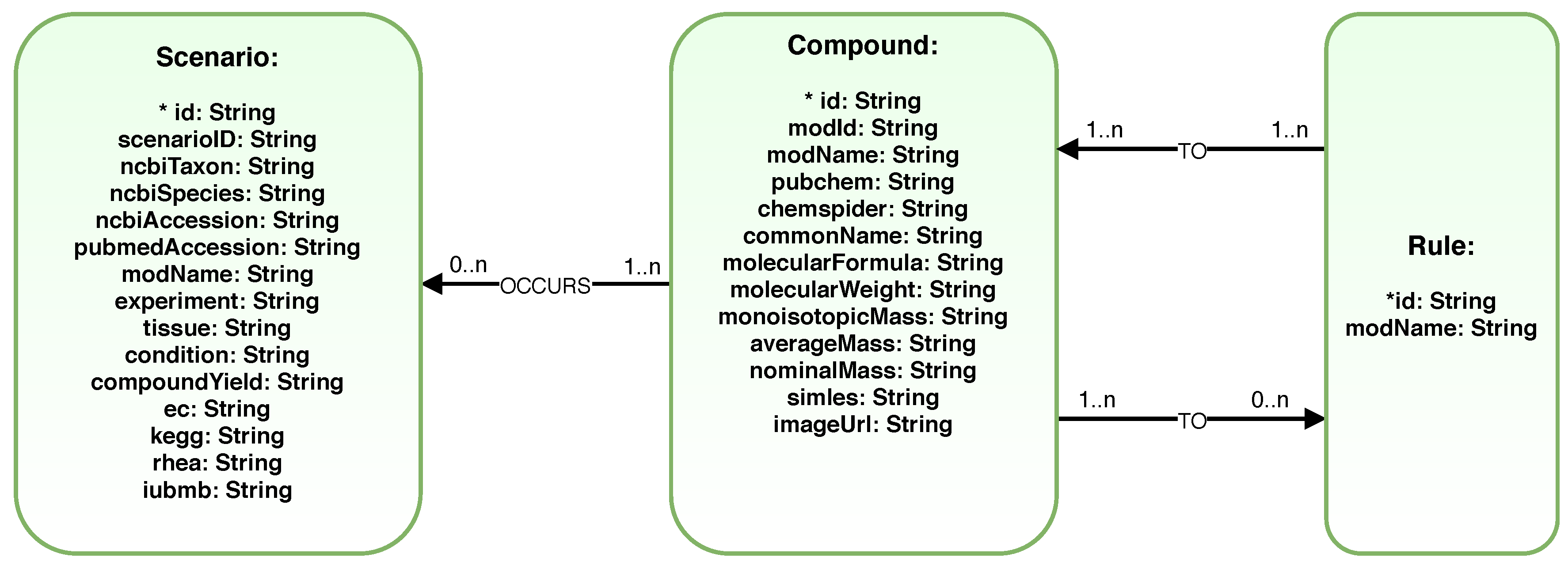
2.4. 2Path-Sesquiterpenes Database
3. Discussion
4. Methods
4.1. Molecules as Undirected Graphs, Graph Transformation, Hypergraphs, and Integer Hyperflows
- (i)
- Reactions may change the number of molecules; hence, both input (substrate) and output (product) graphs are not necessarily connected.
- (ii)
- All atoms are preserved, i.e., a chemical reaction defines a bijection between the vertex sets of input and output graphs.
- (iii)
- Electrons are preserved as well, implying restrictive conditions on the way edges (bonds) can change, corresponding to chemical reaction mechanisms.
4.2. Simulations
4.3. Database Storage
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AAM | Atom to Atom Mapping |
| AFIR | Artificial Force-Induced Reaction |
| CBM | Constraint-based models |
| DBMS | Database Management Systems |
| DMAPP | Dimethylallyl Pyrophosphate |
| DPO | Double pushout graph rewriting |
| FAIR | Findability, Accessibility, Interoperability, and Reuse of digital assets |
| FBA | Flux Balanced Analysis |
| FPP | Farnesyl Diphosphate |
| GGPP | Geranylgeranyl Diphosphate |
| GML | Graph Modeling Language |
| GRRM | Global Reaction Route Mapping |
| GPP | Geranyl Diphosphate |
| ILP | Integer Linear Programming |
| IPP | Isopentenyl Pyrophosphate |
| IUBMB | International Union of Biochemistry and Molecular Biology |
| MEP | Methylerythritol phosphate pathway |
| MVA | Mevalonate pathway |
| NPP | Nerolidyl Diphosphate |
| NoSQL | Not Only Structured Query Language |
| OPP | Diphosphate |
| Portable Document Format | |
| PGDB | Pathway/Genome database |
| STPS | Sesquiterpene Synthases |
| TPS | Terpene Synthase |
References
- Breitmaier, E. Terpenes: Flavors, Fragrances, Pharmaca, Pheromones; Wiley-VCH: Hoboken, NJ, USA, 2006. [Google Scholar] [CrossRef]
- Cheng, A.X.; Xiang, C.Y.; Li, J.X.; Yang, C.Q.; Hu, W.L.; Wang, L.J.; Lou, Y.G.; Chen, X.Y. The rice (E)-β-caryophyllene synthase (OsTPS3) accounts for the major inducible volatile sesquiterpenes. Phytochemistry 2007, 68, 1632–1641. [Google Scholar] [CrossRef]
- Ružička, L. The isoprene rule and the Biogenesis of terpenic compounds. Cell. Mol. Life Sci. 1953, 9, 357–367. [Google Scholar] [CrossRef]
- Vattekkatte, A.; Garms, S.; Brandt, W.; Boland, W. Enhanced structural diversity in terpenoid biosynthesis: Enzymes, substrates and cofactors. Org. Biomol. Chem. 2018, 16, 348–362. [Google Scholar] [CrossRef]
- Wink, M. Biochemistry of Plant Secondary Metabolism; John Willey & Sons Inc.: Hoboken, NJ, USA, 2010; Volume 40. [Google Scholar]
- Chen, F.; Tholl, D.; Bohlmann, J.; Pichersky, E. The family of terpene synthases in plants: A mid-size family of genes for specialized metabolism that is highly diversified throughout the kingdom. Plant J. 2011, 66, 212–229. [Google Scholar] [CrossRef]
- Liu, W.; Feng, X.; Zheng, Y.; Huang, C.H.; Nakano, C.; Hoshino, T.; Bogue, S.; Ko, T.P.; Chen, C.-C.; Cui, Y.; et al. Structure, function and inhibition of ent-kaurene synthase from Bradyrhizobium japonicum. Sci. Rep. 2014, 4, 6214. [Google Scholar] [CrossRef]
- Lesburg, C.A. Crystal Structure of Pentalenene Synthase: Mechanistic Insights on Terpenoid Cyclization Reactions in Biology. Science 1997, 277, 1820–1824. [Google Scholar] [CrossRef] [PubMed]
- Oldfield, E.; Lin, F.Y. Terpene biosynthesis: Modularity rules. Angew. Chem. Int. Ed. 2012, 51, 1124–1137. [Google Scholar] [CrossRef] [PubMed]
- Kempinski, C.; Jiang, Z.; Bell, S.; Chappell, J. Metabolic engineering of higher plants and algae for isoprenoid production. Adv. Biochem. Eng. Biotechnol. 2015, 148, 161–199. [Google Scholar]
- Degenhardt, J.; Köllner, T.G.; Gershenzon, J. Monoterpene and sesquiterpene synthases and the origin of terpene skeletal diversity in plants. Phytochemistry 2009, 70, 1621–1637. [Google Scholar] [CrossRef] [PubMed]
- Schifrin, A.; Khatri, Y.; Kirsch, P.; Thiel, V.; Schulz, S.; Bernhardt, R. A single terpene synthase is responsible for a wide variety of sesquiterpenes in Sorangium cellulosum Soce56. Org. Biomol. Chem. 2016, 14, 3385–3393. [Google Scholar] [CrossRef]
- Tholl, D. Terpene synthases and the regulation, diversity and biological roles of terpene metabolism. Curr. Opin. Plant Biol. 2006, 9, 297–304. [Google Scholar] [CrossRef] [PubMed]
- Christianson, D.W. Structural and Chemical Biology of Terpenoid Cyclases. Chem. Rev. 2017, 117, 11570–11648. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Li, G.; Köllner, T.G.; Jia, Q.; Gershenzon, J.; Chen, F. Positive Darwinian selection is a driving force for the diversification of terpenoid biosynthesis in the genus Oryza. BMC Plant Biol. 2014, 14, 239. [Google Scholar] [CrossRef]
- Tholl, D.; Chen, F.; Petri, J.; Gershenzon, J.; Pichersky, E. Two sesquiterpene synthases are responsible for the complex mixture of sesquiterpenes emitted from Arabidopsis flowers. Plant J. 2005, 42, 757–771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kigathi, R.N.; Weisser, W.W.; Reichelt, M.; Gershenzon, J.; Unsicker, S.B. Plant volatile emission depends on the species composition of the neighboring plant community. BMC Plant Biol. 2019, 19, 58. [Google Scholar] [CrossRef] [PubMed]
- Jeske, L.; Placzek, S.; Schomburg, I.; Chang, A.; Schomburg, D. BRENDA in 2019: A European ELIXIR core data resource. Nucleic Acids Res. 2018, 47, D542–D549. [Google Scholar] [CrossRef] [PubMed]
- Karp, P.D.; Latendresse, M.; Caspi, R. The pathway tools pathway prediction algorithm. Stand. Genom. Sci. 2011, 5, 424. [Google Scholar] [CrossRef]
- Karp, P.D.; Billington, R.; Caspi, R.; Fulcher, C.A.; Latendresse, M.; Kothari, A.; Keseler, I.M.; Krummenacker, M.; Midford, P.E.; Ong, Q.; et al. The BioCyc collection of microbial genomes and metabolic pathways. Briefings Bioinf. 2017. [Google Scholar] [CrossRef]
- Djoumbou-Feunang, Y.; Fiamoncini, J.; Gil-de-la Fuente, A.; Greiner, R.; Manach, C.; Wishart, D.S. BioTransformer: A comprehensive computational tool for small molecule metabolism prediction and metabolite identification. J. Cheminform. 2019, 11, 2. [Google Scholar] [CrossRef]
- Duigou, T.; du Lac, M.; Carbonell, P.; Faulon, J.L. RetroRules: A database of reaction rules for engineering biology. Nucleic Acids Res. 2018, 47, D1229–D1235. [Google Scholar] [CrossRef]
- Maeda, S.; Harabuchi, Y.; Takagi, M.; Taketsugu, T.; Morokuma, K. Artificial Force Induced Reaction (AFIR) Method for Exploring Quantum Chemical Potential Energy Surfaces. Chem. Rec. 2016, 16, 2232–2248. [Google Scholar] [CrossRef] [Green Version]
- Wilkinson, M.D.; Dumontier, M.; Aalbersberg, I.J.; Appleton, G.; Axton, M.; Baak, A.; Blomberg, N.; Boiten, J.W.; da Silva Santos, L.B.; Bourne, P.E.; et al. The FAIR Guiding Principles for scientific data management and stewardship. Sci. Data 2016, 3, 160018. [Google Scholar] [CrossRef] [Green Version]
- Andersen, J.L.; Flamm, C.; Merkle, D.; Stadler, P.F.; Andersen, J.L.; Flamm, C.; Merkle, D.; Stadler, P.F. A software package for chemically inspired graph transformation. In International Conference on Graph Transformation; Springer: Cham, Switzerland, 2016; pp. 73–88. [Google Scholar]
- Andersen, J.L.; Flamm, C.; Merkle, D.; Stadler, P.F. Chemical Transformation Motifs—Modelling Pathways as Integer Hyperflows. IEEE/ACM Trans. Comput. Biol. Bioinform. 2017, 5963, 510–523. [Google Scholar] [CrossRef]
- Smith, W.D. Computational Complexity of Synthetic Chemistry—Basic Facts; Technical Report; NECI: Princeton, NJ, USA, 1997. [Google Scholar]
- Picaud, S.; Olsson, M.E.; Brodelius, M.; Brodelius, P.E. Cloning, expression, purification and characterization of recombinant (+)-germacrene D synthase from Zingiber officinale. Arch. Biochem. Biophys. 2006, 452, 17–28. [Google Scholar] [CrossRef]
- Farzadfar, S.; Zarinkamar, F.; Behmanesh, M.; Hojati, M. Magnesium and manganese interactively modulate parthenolide accumulation and the antioxidant defense system in the leaves of Tanacetum parthenium. J. Plant Physiol. 2016, 202, 10–20. [Google Scholar] [CrossRef]
- Zhang, F.; Chen, N.; Zhou, J.; Wu, R. Protonation-dependent diphosphate cleavage in FPP cyclases and synthases. ACS Catal. 2016, 6, 6918–6929. [Google Scholar] [CrossRef]
- Cane, D.E.; Iyengar, R. The enzymic conversion of farnesyl to nerolidyl pyrophosphate: Role of the pyrophosphate moiety. J. Am. Chem. Soc. 1979, 101, 3385–3388. [Google Scholar] [CrossRef]
- Kollner, T.G.; Held, M.; Lenk, C.; Hiltpold, I.; Turlings, T.C.; Gershenzon, J.; Degenhardt, J. A Maize (E)-beta-Caryophyllene Synthase Implicated in Indirect Defense Responses against Herbivores Is Not Expressed in Most American Maize Varieties. Plant Cell Online 2008, 20, 482–494. [Google Scholar] [CrossRef]
- Irmisch, S.; Krause, S.T.; Kunert, G.; Gershenzon, J.; Degenhardt, J.; Köllner, T.G. The organ-specific expression of terpene synthase genes contributes to the terpene hydrocarbon composition of chamomile essential oils. BMC Plant Biol. 2012, 12, 84. [Google Scholar] [CrossRef]
- Chen, F. Biosynthesis and Emission of Terpenoid Volatiles from Arabidopsis Flowers. Plant Cell Online 2003, 15, 481–494. [Google Scholar] [CrossRef] [Green Version]
- Yu, F.; Okamto, S.; Nakasone, K.; Adachi, K.; Matsuda, S.; Harada, H.; Misawa, N.; Utsumi, R. Molecular cloning and functional characterization of α-humulene synthase, a possible key enzyme of zerumbone biosynthesis in shampoo ginger (Zingiber zerumbet Smith). Planta 2008, 227, 1291–1299. [Google Scholar] [CrossRef] [PubMed]
- Brandizi, M.; Singh, A.; Rawlings, C.; Hassani-Pak, K. Towards FAIRer Biological Knowledge Networks Using a Hybrid Linked Data and Graph Database Approach. J. Integr. Bioinform. 2018, 15. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, W.M.; Wercelens, P.; Walter, M.E.M.; Holanda, M.; Brígido, M. Graph Databases in Molecular Biology. In Brazilian Symposium on Bioinformatics; Springer: Cham, Switzerland, 2018; pp. 50–57. [Google Scholar]
- Smoot, M.E.; Ono, K.; Ruscheinski, J.; Wang, P.L.; Ideker, T. Cytoscape 2.8: New features for data integration and network visualization. Bioinformatics 2010, 27, 431–432. [Google Scholar] [CrossRef] [PubMed]
- Klamt, S.; Haus, U.U.; Theis, F. Hypergraphs and cellular networks. PLoS Comput. Biol. 2009, 5, e1000385. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Dash, S.; Ng, C.Y.; Maranas, C.D. A review of computational tools for design and reconstruction of metabolic pathways. Synth. Syst. Biotechnol. 2017, 2, 243–252. [Google Scholar] [CrossRef] [PubMed]
- Cherdal, S.; Mouline, S. Modelling and Simulation of Biochemical Processes Using Petri Nets. Processes 2018, 6, 97. [Google Scholar] [CrossRef]
- Blazier, A.S.; Papin, J.A. Integration of expression data in genome-scale metabolic network reconstructions. Front. Physiol. 2012, 3 AUG, 299. [Google Scholar] [CrossRef]
- Øyås, O.; Stelling, J. Genome-scale metabolic networks in time and space. Curr. Opin. Syst. Biol. 2018, 8, 51–58. [Google Scholar] [CrossRef]
- Fang, C.; Fernie, A.R.; Luo, J. Exploring the Diversity of Plant Metabolism. Trends Plant Sci. 2018, 24, 83–98. [Google Scholar] [CrossRef]
- Isegawa, M.; Maeda, S.; Tantillo, D.J.; Morokuma, K. Predicting pathways for terpene formation from first principles–routes to known and new sesquiterpenes. Chem. Sci. 2014, 5, 1555–1560. [Google Scholar] [CrossRef]
- Systems, D.C.I. SMARTS—A Language for Describing Molecular Patterns. 2008. Available online: http://www.daylight.com/dayhtml/doc/theory/theory.smarts.html (accessed on 30 January 2019).
- Systems, D.C.I. A Reaction Transform Language. Available online: http://daylight.com/dayhtml/doc/theory/theory.smirks.html (accessed on 30 January 2019).
- Selkov, E.E.; Goryanin, I.I.; Kaimatchnikov, N.P.; Shevelev, E.L.; Yunus, I.A. Factographic data bank on enzymes and metabolic pathways. Stud. Biophys. 1989, 129, 155–164. [Google Scholar]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Caspi, R.; Billington, R.; Fulcher, C.A.; Keseler, I.M.; Kothari, A.; Krummenacker, M.; Latendresse, M.; Midford, P.E.; Ong, Q.; Ong, W.K.; et al. The MetaCyc Database of metabolic pathways. Nucleic Acids Res. 2014, 42, 471–480. [Google Scholar] [CrossRef]
- Fabregat, A.; Korninger, F.; Viteri, G.; Sidiropoulos, K.; Marin-Garcia, P.; Ping, P.; Wu, G.; Stein, L.; D’Eustachio, P.; Hermjakob, H. Reactome graph database: Efficient access to complex pathway data. PLoS Comput. Biol. 2018, 14, 1–13. [Google Scholar] [CrossRef]
- Holliday, G.L.; Andreini, C.; Fischer, J.D.; Rahman, S.A.; Almonacid, D.E.; Williams, S.T.; Pearson, W.R. MACiE: Exploring the diversity of biochemical reactions. Nucleic Acids Res. 2012, 40, D783–D789. [Google Scholar] [CrossRef]
- Bondy, J.A.; Murty, U.S.R. Graph Theory with Applications; Elsevier Science Publishing Co., Inc.: New York, NY, USA, 1976; Volume 290. [Google Scholar]
- Ehrig, H.; Ehrig, K.; Prange, U.; Taenthzer, G. Fundamentals of Algebraic Graph Transformation; Springer: Berlin, Germany, 2006. [Google Scholar] [CrossRef]
- Löwe, M. Algebraic approach to single-pushout graph transformation. Theory Comput. Sci. 1993, 109, 181–224. [Google Scholar] [CrossRef] [Green Version]
- Andersen, J.L.; Flamm, C.; Merkle, D.; Stadler, P.F. Inferring chemical reaction patterns using rule composition in graph grammars. J. Syst. Chem. 2013, 4, 4. [Google Scholar] [CrossRef] [Green Version]
- Zeigarnik, A.V. On Hypercycles and Hypercircuits in Hypergraphs. In Discrete Mathematical Chemistry; DIMACS Series in Discrete Mathematics and Theoretical Computer Science; Hansen, P., Fowler, P.W., Zheng, M., Eds.; American Mathematical Society: Providence, RI, USA, 2000; Volume 51, pp. 377–383. [Google Scholar]
- Andersen, J.L.; Flamm, C.; Merkle, D.; Stadler, P.F. Generic Strategies for Chemical Space Exploration. Int. J. Comput. Biol. Drug Des. 2014, 7, 225–258. [Google Scholar] [CrossRef]
- Himsolt, M. GML: A Portable Graph File Format; Universität Passau: Passau, Germany, 1997. [Google Scholar]
- Minkiewicz, P.; Iwaniak, A.; Darewicz, M. Annotation of peptide structures using SMILES and other chemical codes-practical solutions. Molecules 2017, 22, 2075. [Google Scholar] [CrossRef]
- Van Erven, G.; Silva, W.; Carvalho, R.; Holanda, M. GRAPHED: A graph description diagram for graph databases. In World Conference on Information Systems and Technologies; Springer: Cham, Switzerland, 2018; pp. 1141–1151. [Google Scholar]
- Alcántara, R.; Axelsen, K.B.; Morgat, A.; Belda, E.; Coudert, E.; Bridge, A.; Cao, H.; De Matos, P.; Ennis, M.; Turner, S.; et al. Rhea—A manually curated resource of biochemical reactions. Nucleic Acids Res. 2011, 40, D754–D760. [Google Scholar] [CrossRef]
- McDonald, A.G.; Boyce, S.; Tipton, K.F. ExplorEnz: The primary source of the IUBMB enzyme list. Nucleic Acids Res. 2008, 37, D593–D597. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
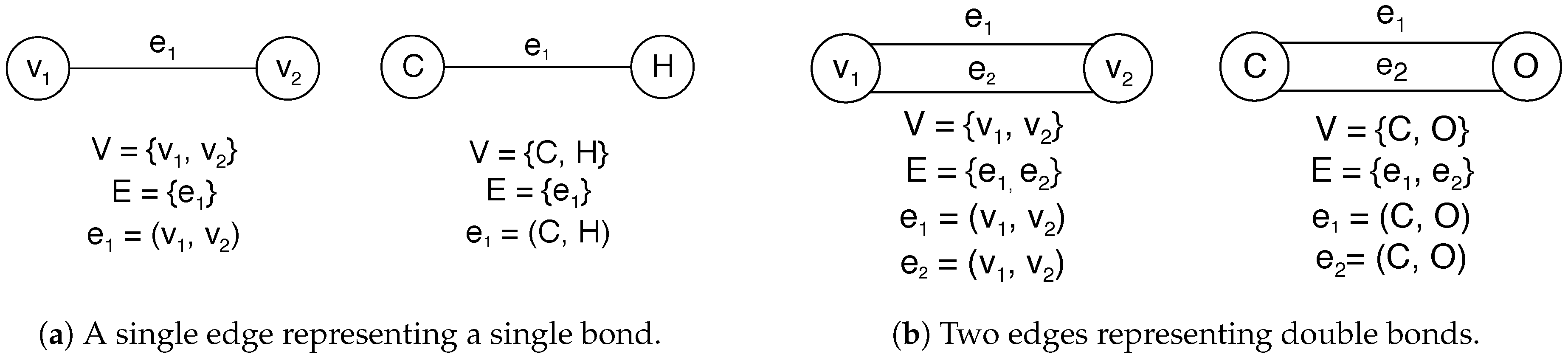{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecules | Reactions | Focus | Storage for results | Biological Evidence | |
|---|---|---|---|---|---|
| 2Path-Sesquiterpenes | Undirected graphs | Graph rewrite rules | Plant sesquiterpenes | Graph database | Scenarios |
| Isegawa et al. [45] | Internal | AFIR/GRRM | Sesquiterpenes | Internal | - |
| RetroRules [22] | SMART | SMIRKS | General | - | RetroRules |
| BioTransformer [21] | SMART | SMIRKS | General | - | MetXBioDB |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Silva, W.M.C.d.; Andersen, J.L.; Holanda, M.T.; Walter, M.E.M.T.; Brigido, M.M.; Stadler, P.F.; Flamm, C. Exploring Plant Sesquiterpene Diversity by Generating Chemical Networks. Processes 2019, 7, 240. https://doi.org/10.3390/pr7040240
Silva WMCd, Andersen JL, Holanda MT, Walter MEMT, Brigido MM, Stadler PF, Flamm C. Exploring Plant Sesquiterpene Diversity by Generating Chemical Networks. Processes. 2019; 7(4):240. https://doi.org/10.3390/pr7040240
Chicago/Turabian StyleSilva, Waldeyr M. C. da, Jakob L. Andersen, Maristela T. Holanda, Maria Emília M. T. Walter, Marcelo M. Brigido, Peter F. Stadler, and Christoph Flamm. 2019. "Exploring Plant Sesquiterpene Diversity by Generating Chemical Networks" Processes 7, no. 4: 240. https://doi.org/10.3390/pr7040240