
A Review on Artificial Intelligence Enabled Design, Synthesis, and Process Optimization of Chemical Products for Industry 4.0
 ,
,
Abstract
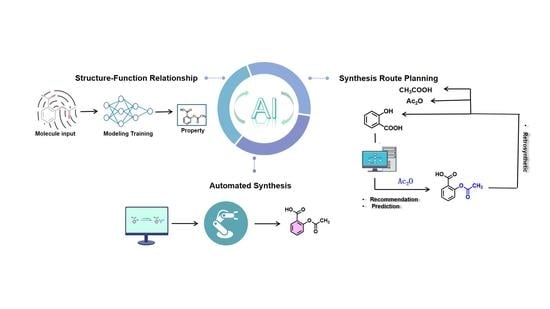
:
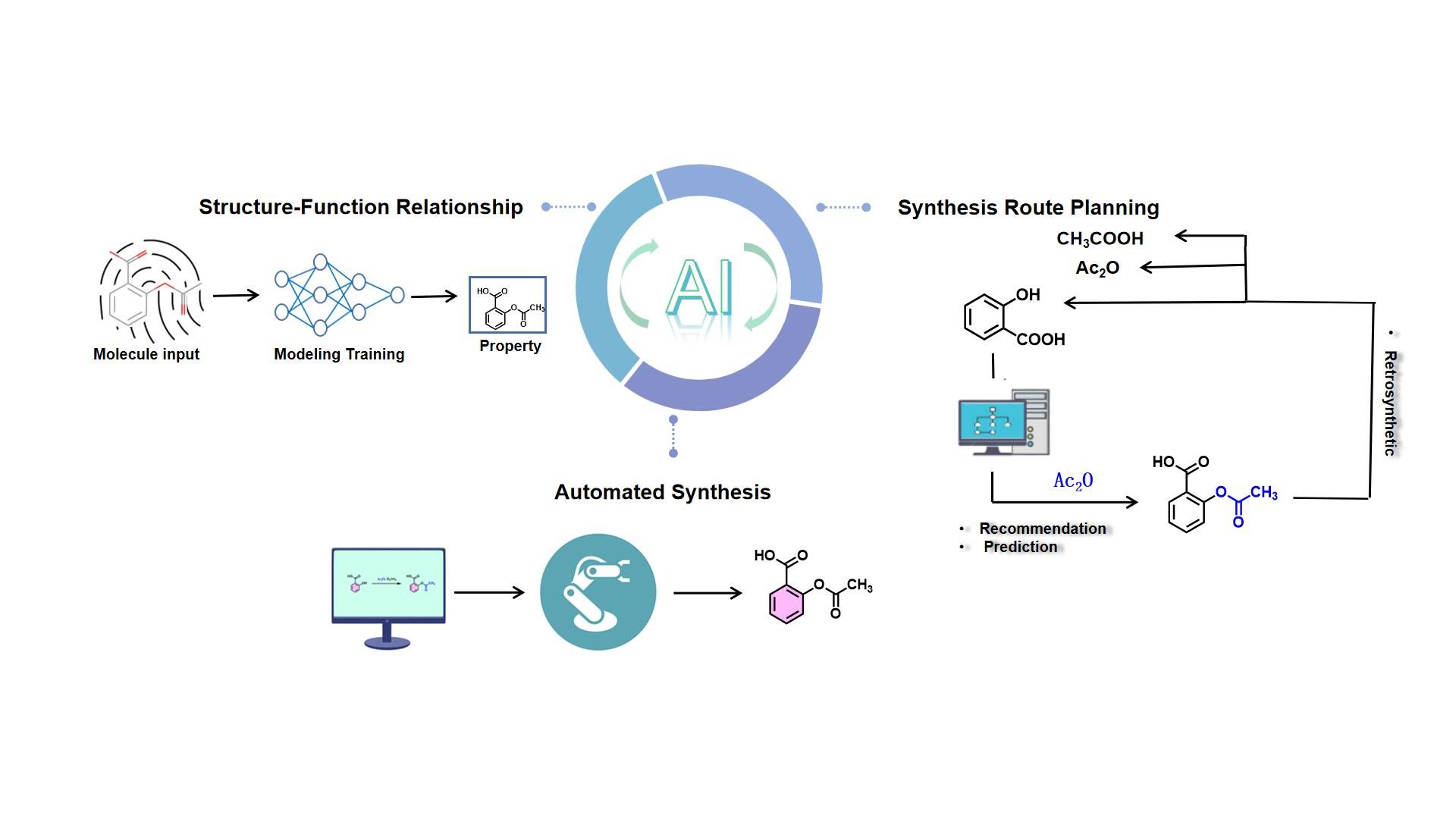{kind=link}
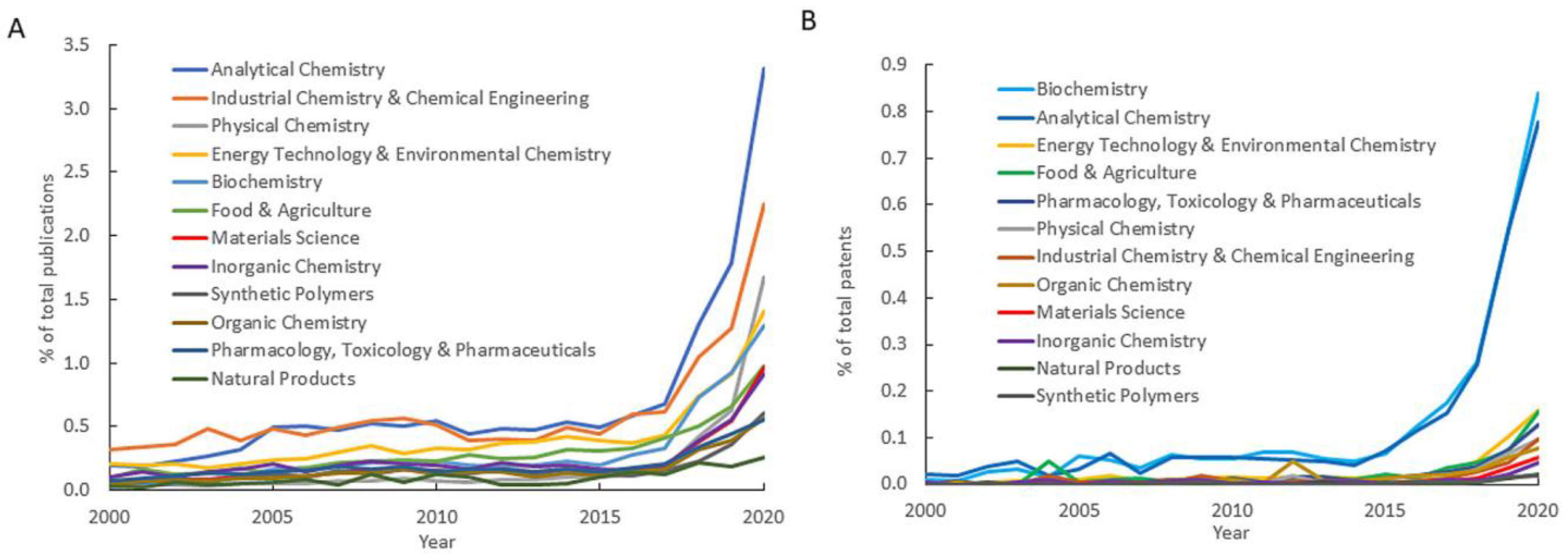{kind=link}
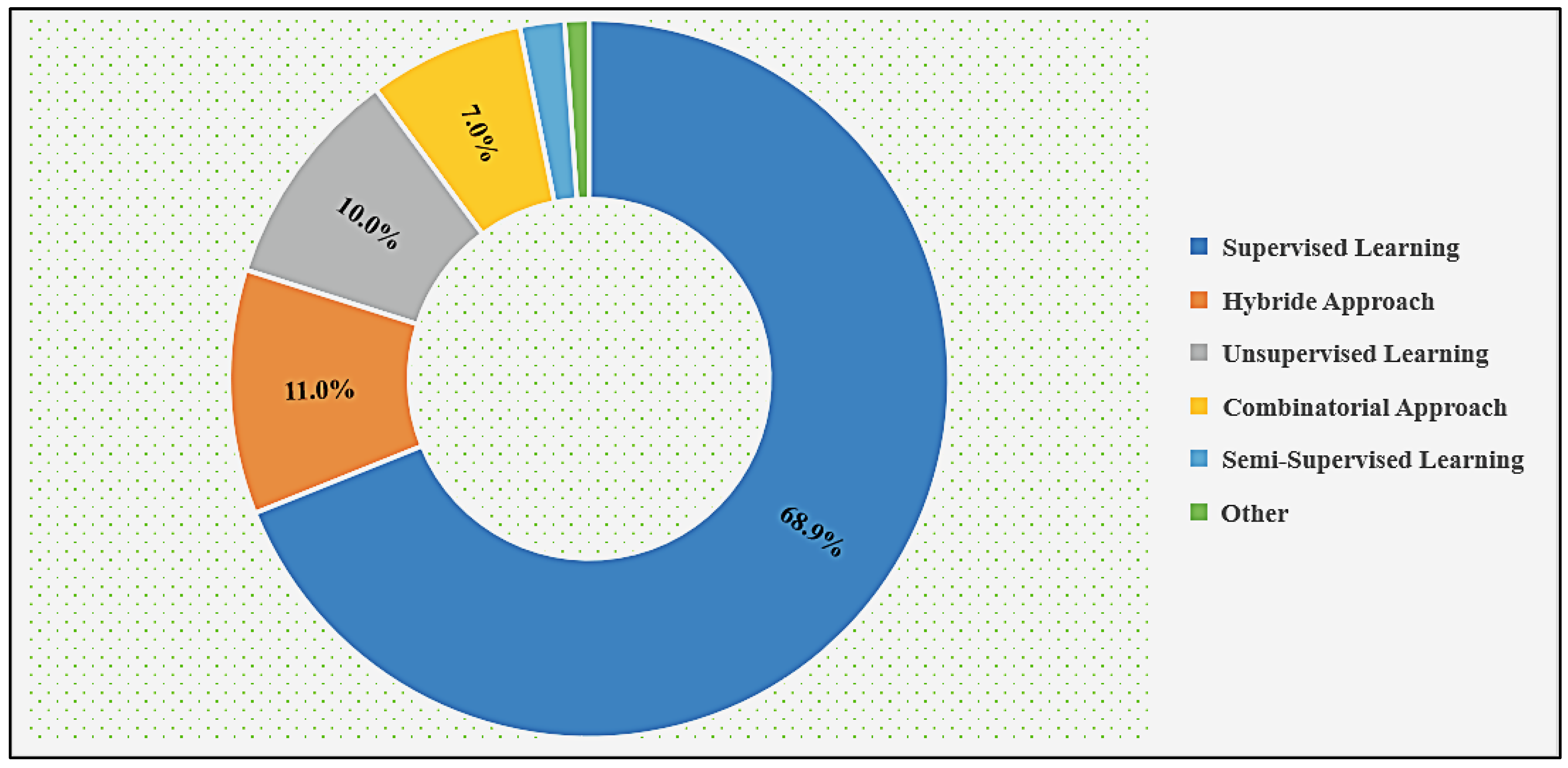{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
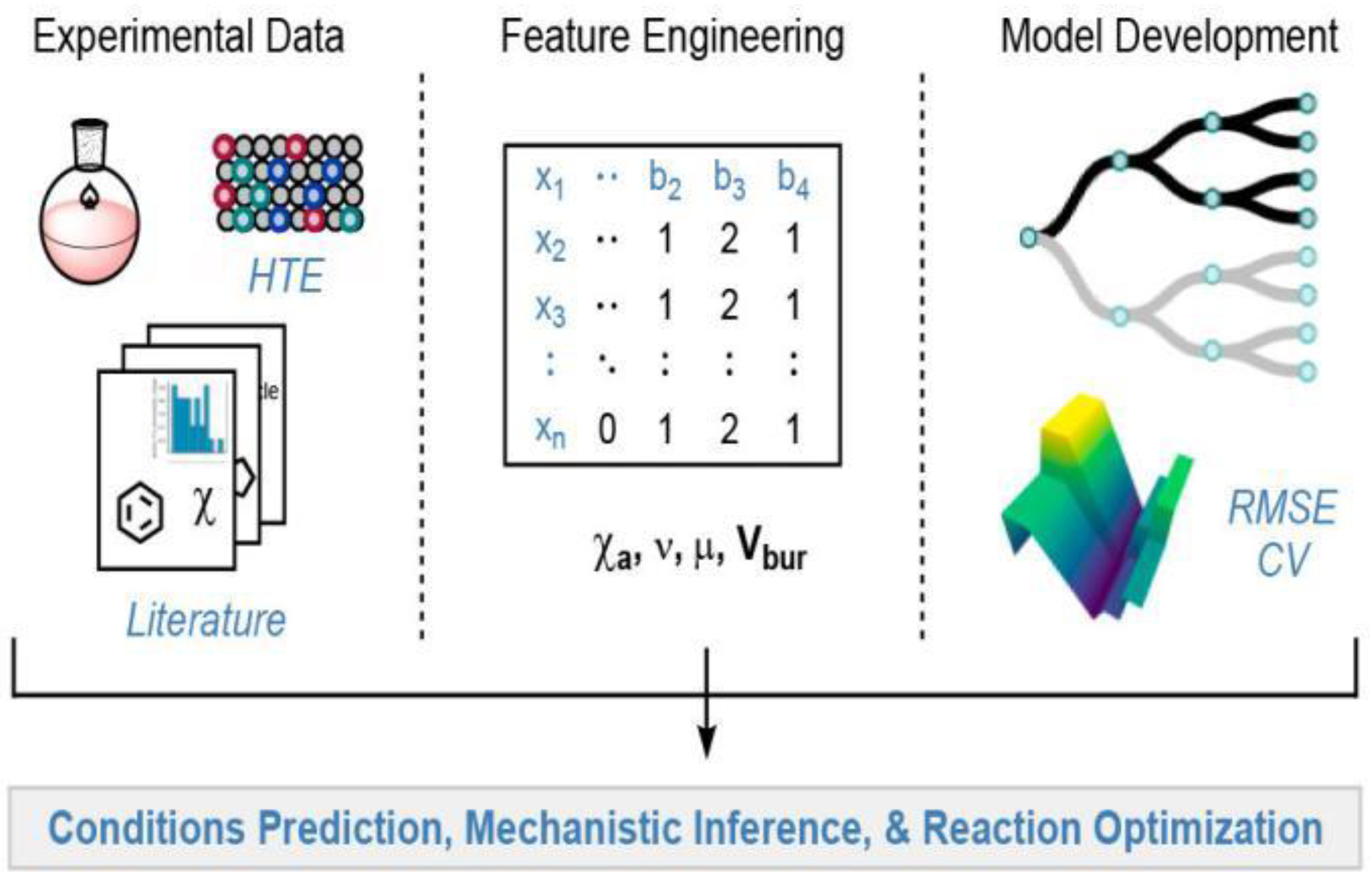{kind=link}
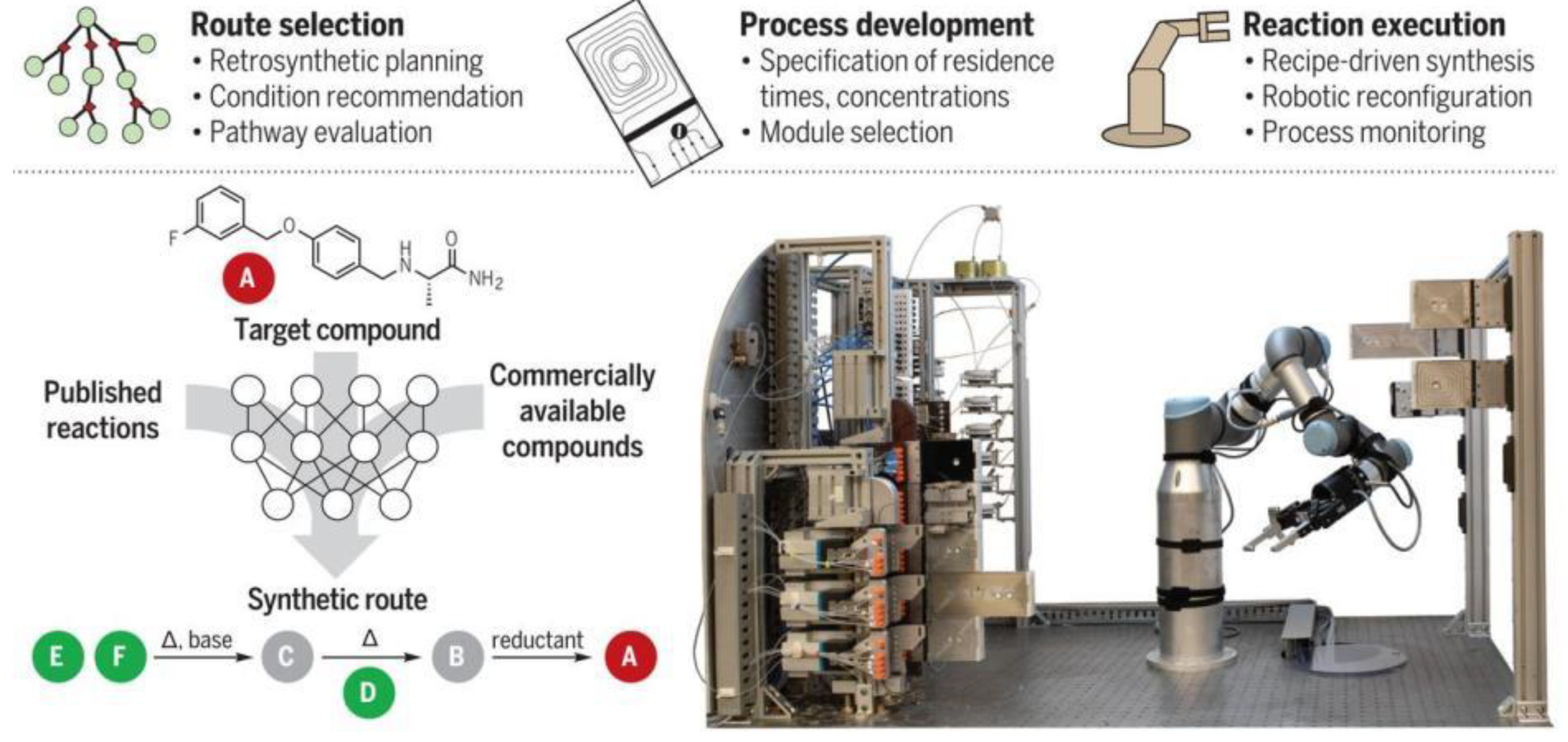{kind=link}
{kind=link}
1. Introduction
2. AI Enabled Chemical Process Intensification
2.1. AI for Structure-Function Relationship Analysis
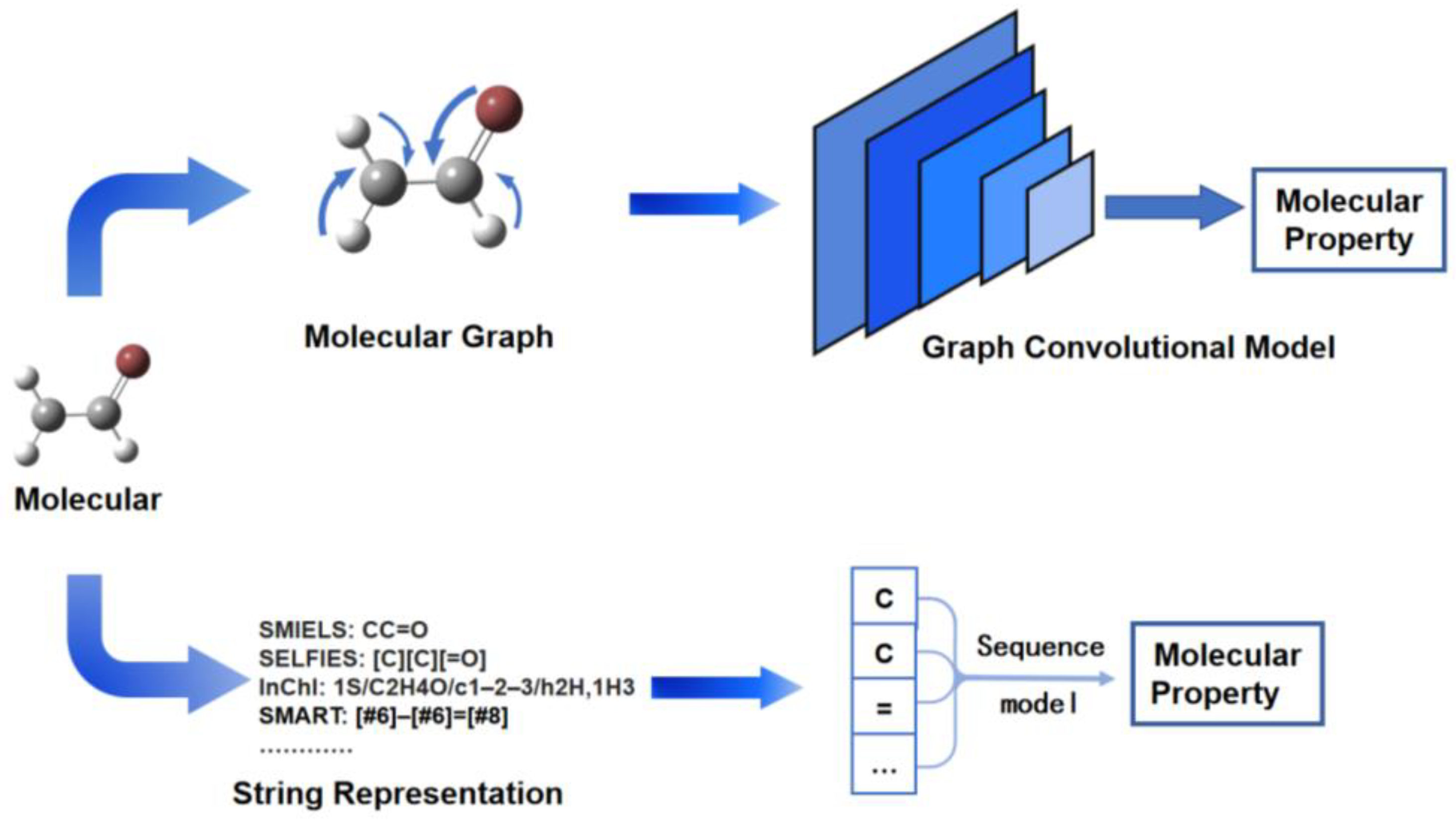
2.1.1. Molecular Property Prediction
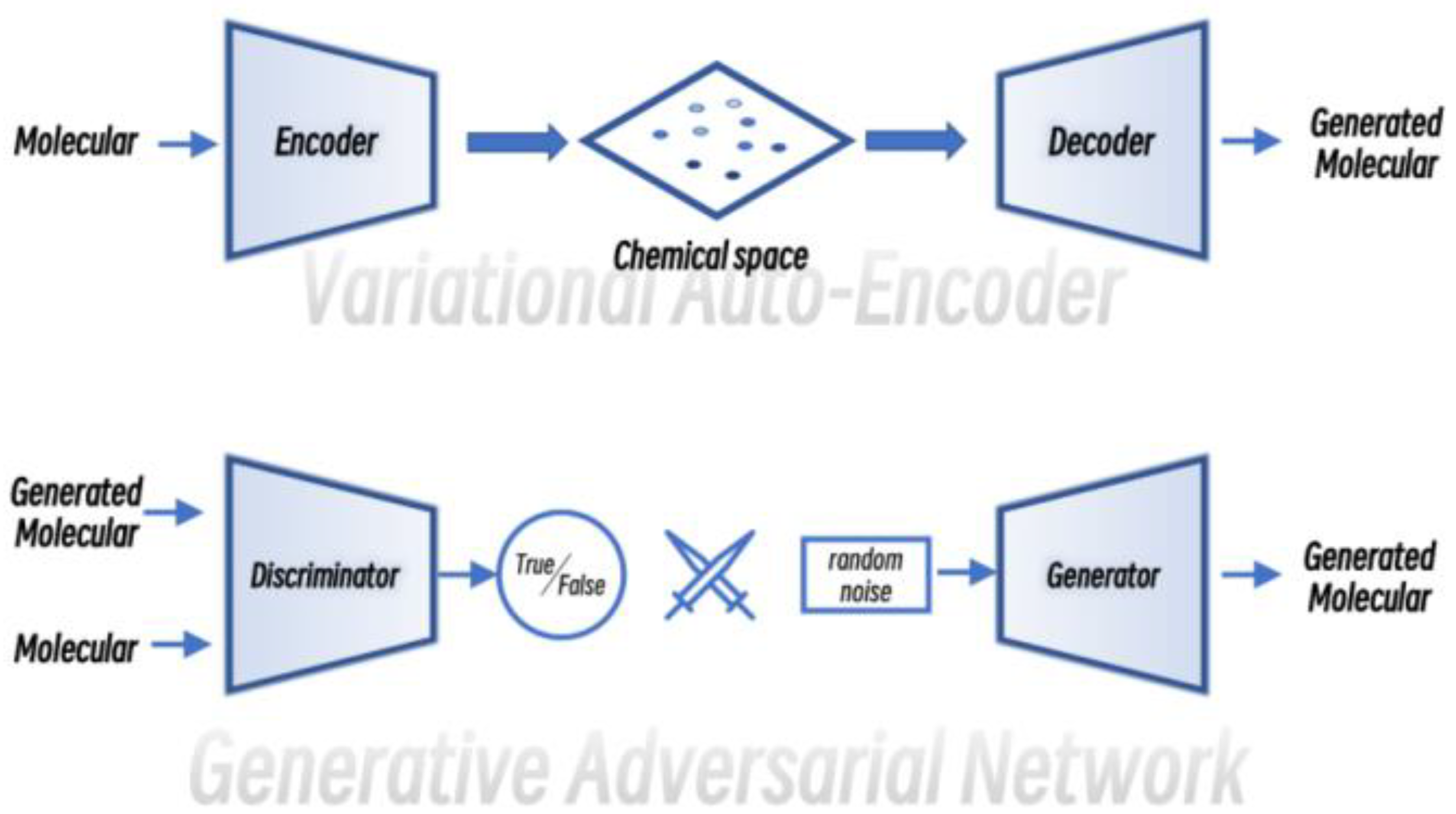
2.1.2. Molecular Design
2.2. AI for Synthetic Route Planning
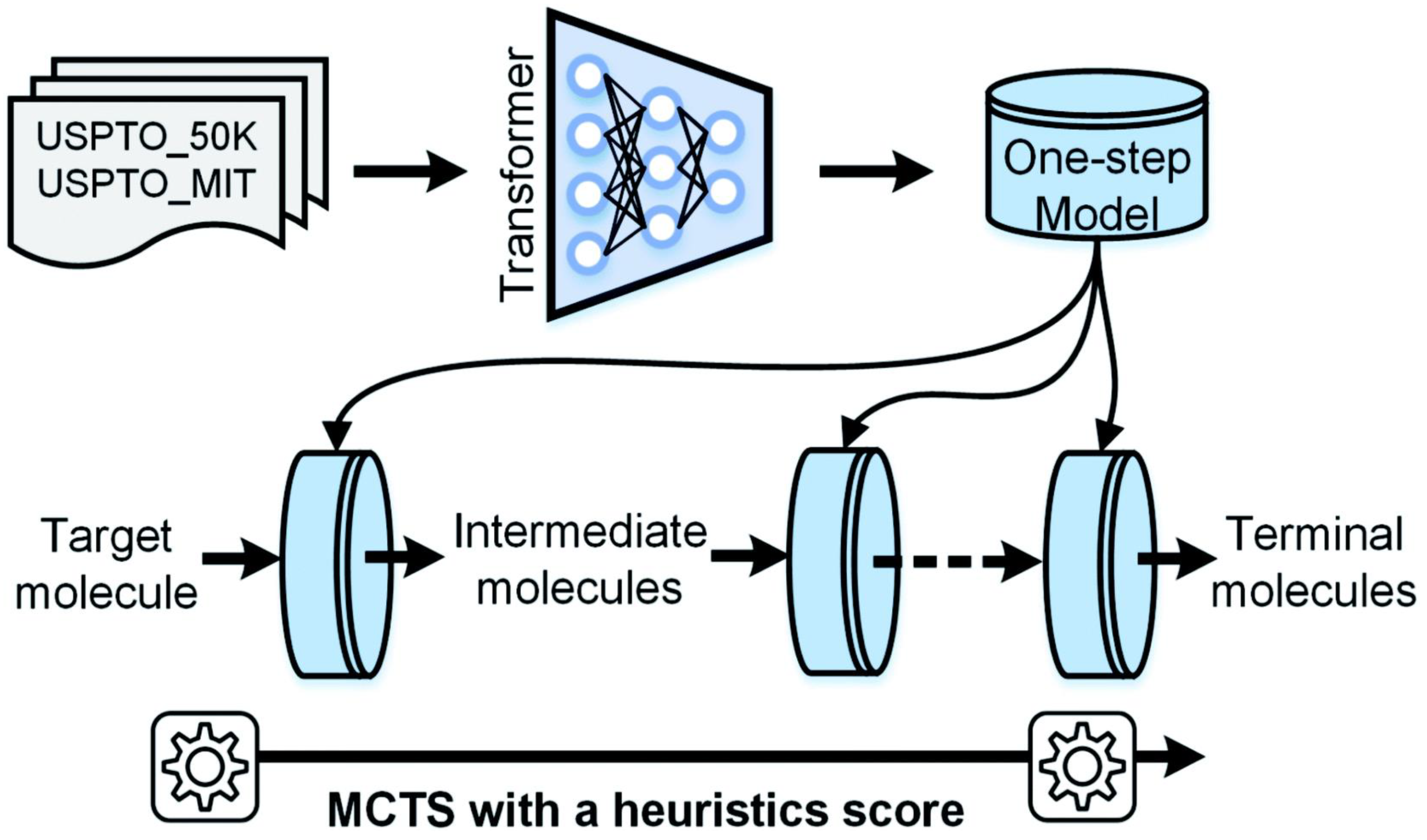
2.2.1. Retrosynthetic Planning
2.2.2. Forward Reaction Prediction
2.2.3. Condition Recommendation
2.3. AI for Automated Synthesis
2.3.1. Robotic Lab Platform
2.3.2. Automated Synthesis
3. Conclusions and Outlook
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Brown, D.G.; Wobst, H.J.; Kapoor, A.; Kenna, L.A.; Southall, N. Clinical development times for innovative drugs. Nat. Rev. Drug Discov. 2021, 21, 793–794. [Google Scholar] [CrossRef] [PubMed]
- Baum, Z.J.; Yu, X.; Ayala, P.Y.; Zhao, Y.; Watkins, S.P.; Zhou, Q. Artificial Intelligence in Chemistry: Current Trends and Future Directions. J. Chem. Inf. Model. 2021, 61, 3197–3212. [Google Scholar] [CrossRef] [PubMed]
- Mowbray, M.; Vallerio, M.; Perez-Galvan, C.; Zhang, D.; Chanona, A.D.R.; Navarro-Brull, F.J. Industrial data science—a review of machine learning applications for chemical and process industries. React. Chem. Eng. 2022, 7, 1471–1509. [Google Scholar] [CrossRef]
- Venkatasubramanian, V. The promise of artificial intelligence in chemical engineering: Is it here, finally? AIChE J. 2018, 65, 466–478. [Google Scholar] [CrossRef] [Green Version]
- Paul, D.; Sanap, G.; Shenoy, S.; Kalyane, D.; Kalia, K.; Tekade, R.K. Artificial intelligence in drug discovery and development. Drug Discov. Today 2020, 26, 80–93. [Google Scholar] [CrossRef]
- Trinh, C.; Meimaroglou, D.; Hoppe, S. Machine Learning in Chemical Product Engineering: The State of the Art and a Guide for Newcomers. Processes 2021, 9, 1456. [Google Scholar] [CrossRef]
- Mitchell, J.B.O. Machine learning methods in chemoinformatics. WIREs Comput. Mol. Sci. 2014, 4, 468–481. [Google Scholar] [CrossRef] [Green Version]
- Griffen, E.J.; Dossetter, A.G.; Leach, A.G.; Montague, S. Can we accelerate medicinal chemistry by augmenting the chemist with Big Data and artificial intelligence? Drug Discov. Today 2018, 23, 1373–1384. [Google Scholar] [CrossRef]
- Vamathevan, J.; Clark, D.; Czodrowski, P.; Dunham, I.; Ferran, E.; Lee, G.; Li, B.; Madabhushi, A.; Shah, P.; Spitzer, M.; et al. Applications of machine learning in drug discovery and development. Nat. Rev. Drug Discov. 2019, 18, 463–477. [Google Scholar] [CrossRef]
- Elton, D.C.; Boukouvalas, Z.; Fuge, M.; Chung, P.W. Deep learning for molecular design—A review of the state of the art. Mol. Syst. Des. Eng. 2019, 4, 828–849. [Google Scholar] [CrossRef]
- Segler, M.H.S.; Preuss, M.; Waller, M.P. Planning chemical syntheses with deep neural networks and symbolic AI. Nature 2018, 555, 604–610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishida, S.; Terayama, K.; Kojima, R.; Takasu, K.; Okuno, Y. Prediction and Interpretable Visualization of Retrosynthetic Re-actions Using Graph Convolutional Networks. J. Chem. Inf. Model. 2019, 59, 5026–5033. [Google Scholar] [PubMed]
- Gajewska, E.P.; Szymkuc, S.; Dittwald, P.; Startek, M.; Popik, O.; Mlynarski, J.; Grzybowski, B.A. Algorithmic Discovery of Tactical Combinations for Advanced Organic Syntheses. Chem 2020, 6, 280–293. [Google Scholar]
- Zheng, S.; Rao, J.; Zhang, Z.; Xu, J.; Yang, Y. Predicting Retrosynthetic Reactions Using Self-Corrected Transformer Neural Networks. J. Chem. Inf. Model. 2019, 60, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.A.; Yang, Q.; Sresht, V.; Bolgar, P.; Hou, X.; Klug-McLeod, J.L.; Butler, C.R. Molecular Transformer unifies reaction prediction and retrosynthesis across pharma chemical space. Chem. Commun. 2019, 55, 12152–12155. [Google Scholar] [CrossRef]
- Coley, C.W.; Jin, W.; Rogers, L.; Jamison, T.F.; Jaakkola, T.S.; Green, W.H.; Barzilay, R.; Jensen, K.F. A graph-convolutional neural network model for the prediction of chemical reactivity. Chem. Sci. 2018, 10, 370–377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, H.; Struble, T.J.; Coley, C.W.; Wang, Y.; Green, W.H.; Jensen, K.F. Using Machine Learning to Predict Suitable Conditions for Organic Reactions. ACS Cent. Sci. 2018, 4, 1465–1476. [Google Scholar]
- Walker, E.; Kammeraad, J.; Goetz, J.; Robo, M.T.; Tewari, A.; Zimmerman, P.M. Learning to Predict Reaction Conditions: Relationships between Solvent, Molecular Structure, and Catalyst. J. Chem. Inf. Model. 2019, 59, 3645–3654. [Google Scholar] [CrossRef]
- Maser, M.R.; Cui, A.Y.; Ryou, S.; DeLano, T.J.; Yue, Y.; Reisman, S.E. Multilabel Classification Models for the Prediction of Cross-Coupling Reaction Conditions. J. Chem. Inf. Model. 2021, 61, 156–166. [Google Scholar] [CrossRef]
- Venkatasubramanian, V.; Mann, V. Artificial intelligence in reaction prediction and chemical synthesis. Curr. Opin. Chem. Eng. 2022, 36, 100749. [Google Scholar] [CrossRef]
- Lu, C.; Liu, Q.; Wang, C.; Huang, Z.; Lin, P.; He, L. Molecular property prediction: A multilevel quantum interactions modeling perspective. In Proceedings of the AAAI Conference on Artificial Intelligence, Honolulu, HI, USA, 27 January–1 February 2019; pp. 1052–1060. [Google Scholar]
- Gilmer, J.; Schoenholz, S.S.; Riley, P.F.; Vinyals, O.; Dahl, G.E. Neural Message Passing for Quantum Chemistry. In Proceedings of the 34th International Conference on Machine Learning, Proceedings of Machine Learning Research, Sydney, NSW, Australia, 6–11 August 2017; pp. 1263–1272. [Google Scholar]
- Blomberg, M.R.A.; Borowski, T.; Himo, F.; Liao, R.-Z.; Siegbahn, P.E.M. Quantum Chemical Studies of Mechanisms for Metalloenzymes. Chem. Rev. 2014, 114, 3601–3658. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Swanson, K.; Jin, W.; Coley, C.; Eiden, P.; Gao, H.; Guzman-Perez, A.; Hopper, T.; Kelley, B.; Mathea, M.; et al. Analyzing Learned Molecular Representations for Property Prediction. J. Chem. Inf. Model. 2019, 59, 3370–3388. [Google Scholar] [CrossRef] [PubMed]
- Weininger, D. SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules. J. Chem. Inf. Model. 1988, 28, 31–36. [Google Scholar] [CrossRef]
- Vaswani, A.; Shazeer, N.; Parmar, N.; Uszkoreit, J.; Jones, L.; Gomez, A.N.; Kaiser, Ł.; Polosukhin, I. Attention is all you need. arXiv 2017. [Google Scholar] [CrossRef]
- Honda, S.; Shi, S.; Hiroki, R. Ueda. SMILES Transformer: Pre-trained Molecular Fingerprint for Low Data Drug Discovery. arXiv 2019, arXiv:1911.04738. [Google Scholar] [CrossRef]
- Schwaller, P.; Vaucher, A.C.; Laino, T.; Reymond, J.-L. Prediction of chemical reaction yields using deep learning. Mach. Learn. Sci. Technol. 2021, 2, 015016. [Google Scholar] [CrossRef]
- Chithrananda, S.; Grand, G.; Ramsundar, B. Chemberta: Large-scale self- supervised pretraining for molecular property prediction. arXiv 2020, arXiv:2010.09885. [Google Scholar] [CrossRef]
- Su, A.; Zhang, C.; She, Y.-B.; Yang, Y.-F. Exploring Deep Learning for Metalloporphyrins: Databases, Molecular Representations, and Model Architectures. Catalysts 2022, 12, 1485. [Google Scholar] [CrossRef]
- Jo, J.; Kwak, B.; Choi, H.-S.; Yoon, S. The message passing neural networks for chemical property prediction on SMILES. Methods 2020, 179, 65–72. [Google Scholar] [CrossRef]
- Mouchlis, V.D.; Afantitis, A.; Serra, A.; Fratello, M.; Papadiamantis, A.G.; Aidinis, V.; Lynch, I.; Greco, D.; Melagraki, G. Ad-vances in De Novo Drug Design: From Conventional to Machine Learning Methods. Int. J. Mol. Sci. 2021, 22, 1676. [Google Scholar] [CrossRef]
- Gómez-Bombarelli, R.; Wei, J.N.; Duvenaud, D.K.; Hernandez-Lobato, J.M.; Sánchez-Lengeling, B.; Sheberla, D.; Aguilera-Iparraguirre, J.; Hirzel, T.D.; Adams, R.P.; Aspuru-Guzik, A. Automatic Chemical Design Using a Data-Driven Continuous Representation of Molecules. ACS Central Sci. 2018, 4, 268–276. [Google Scholar] [CrossRef] [PubMed]
- Segler, M.H.S.; Kogej, T.; Tyrchan, C.; Waller, M.P. Generating Focused Molecule Libraries for Drug Discovery with Recurrent Neural Networks. ACS Central Sci. 2017, 4, 120–131. [Google Scholar] [CrossRef] [PubMed]
- De Cao, N.; Kipf, T. MolGAN: An implicit generative model for small molecular graphs. arXiv 2018, arXiv:1805.11973. [Google Scholar] [CrossRef]
- Flam-Shepherd, D.; Wu, T.; Aspuru-Guzik, A. Graph deconvolutional generation. arXiv 2020, arXiv:2002.07087. [Google Scholar] [CrossRef]
- Brown, N.; Fiscato, M.; Segler, M.H.; Vaucher, A.C. GuacaMol: Benchmarking Models for de Novo Mo-lecular Design. J. Chem. Inf. Model. 2019, 59, 1096–1108. [Google Scholar] [CrossRef]
- Krenn, M.; Hase, F.; Nigam, A.K.; Friederich, P.; Aspuru-Guzik, A. Self- referencing embedded strings (SELFIES): A 100% robust molecular string representation. Mach. Learn. Sci. Technol. 2020, 1, 045024. [Google Scholar] [CrossRef]
- Dybowski, R. Interpretable machine learning as a tool for scientific discovery in chemistry. New J. Chem. 2020, 44, 20914–20920. [Google Scholar] [CrossRef]
- Krishnan, K.; Kassab, R.; Agajanian, S.; Verkhivker, G. Interpretable Machine Learning Models for Molecular Design of Ty-rosine Kinase Inhibitors Using Variational Autoencoders and Perturbation-Based Approach of Chemical Space Exploration. Int. J. Mol. Sci. 2022, 23, 11262. [Google Scholar] [CrossRef]
- Hatamleh, M.; Chong, J.W.; Tan, R.R.; Aviso, K.B.; Janairo, J.I.B.; Chemmangattuvalappil, N.G. Design of mosquito repellent molecules via the integration of hyperbox machine learning and computer aided molecular design. Digit. Chem. Eng. 2022, 3, 100018. [Google Scholar] [CrossRef]
- Ooi, Y.J.; Aung, K.N.G.; Chong, J.W.; Tan, R.R.; Aviso, K.B.; Chemmangattuvalappil, N.G. Design of fragrance molecules using computer-aided molecular design with machine learning. Comput. Chem. Eng. 2021, 157, 107585. [Google Scholar] [CrossRef]
- Radhakrishnapany, K.T.; Wong, C.Y.; Tan, F.K.; Chong, J.W.; Tan, R.R.; Aviso, K.B.; Janairo, J.I.B.; Chemmangattuvalappil, N.G. Design of fragrant molecules through the incorporation of rough sets into computer-aided molecular design. Mol. Syst. Des. Eng. 2020, 5, 1391–1416. [Google Scholar] [CrossRef]
- Helmy, M.; Smith, D.; Selvarajoo, K. Systems biology approaches integrated with artificial intelligence for optimized food-focused metabolic engineering. Metab. Eng. Commun. 2020, 11, e00149. [Google Scholar] [CrossRef]
- Ji, Z.; Su, J.; Liu, C.; Wang, H.; Huang, D.; Zhou, X. Integrating Genomics and Proteomics Data to Predict Drug Effects Using Binary Linear Programming. PLoS ONE 2014, 9, e102798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, Z.; Wu, D.; Zhao, W.; Peng, H.; Zhao, S.; Huang, D.; Zhou, X. Systemic modeling myeloma-osteoclast interactions under normoxic/hypoxic condition using a novel computational approach. Sci. Rep. 2015, 5, 13291. [Google Scholar] [CrossRef] [Green Version]
- Peng, H.; Zhao, W.; Tan, H.; Ji, Z.; Li, J.; Li, K.; Zhou, X. Prediction of treatment efficacy for prostate cancer using a mathematical model. Sci. Rep. 2016, 6, 21599. [Google Scholar] [CrossRef]
- Corey, E.J.; Wipke, W.T. Computer-Assisted Design of Complex Organic Syntheses. Science 1969, 166, 178–192. [Google Scholar] [CrossRef] [PubMed]
- Cook, A.; Johnson, A.P.; Law, J.; Mirzazadeh, M.; Ravitz, O.; Simon, A. Computer-aided synthesis design: 40 years on. WIREs Comput. Mol. Sci. 2011, 2, 79–107. [Google Scholar] [CrossRef]
- Ihlenfeldt, W.-D.; Gasteiger, J. Computer-Assisted Planning of Organic Syntheses: The Second Generation of Programs. Angew. Chem. Int. Ed. 1996, 34, 2613–2633. [Google Scholar] [CrossRef]
- Todd, M.H. Computer-aided organic synthesis. Chem. Soc. Rev. 2005, 34, 247–266. [Google Scholar] [CrossRef] [PubMed]
- Ruddigkeit, L.; van Deursen, R.; Blum, L.C.; Reymond, J.-L. Enumeration of 166 Billion Organic Small Molecules in the Chemical Universe Database GDB-17. J. Chem. Inf. Model. 2012, 52, 2864–2875. [Google Scholar] [CrossRef]
- Davies, I.W. The digitization of organic synthesis. Nature 2019, 570, 175–181. [Google Scholar] [CrossRef] [Green Version]
- Coley, C.W.; Eyke, N.S.; Jensen, K.F. Autonomous Discovery in the Chemical Sciences Part I: Progress. Angew. Chem. Int. Ed. 2020, 59, 22858–22893. [Google Scholar] [CrossRef]
- Coley, C.W.; Eyke, N.S.; Jensen, K.F. Autonomous Discovery in the Chemical Sciences Part II: Outlook. Angew. Chem. Int. Ed. 2019, 59, 23414–23436. [Google Scholar] [CrossRef] [Green Version]
- Shen, Y.; Borowski, J.E.; Hardy, M.A.; Sarpong, R.; Doyle, A.G.; Cernak, T. Automation and computer-assisted planning for chemical synthesis. Nat. Rev. Methods Prim. 2021, 1, 23. [Google Scholar] [CrossRef]
- Klucznik, T.; Mikulak-Klucznik, B.; McCormack, M.P.; Lima, H.; Szymkuć, S.; Bhowmick, M.; Molga, K.; Zhou, Y.; Rickershauser, L.; Gajewska, E.P.; et al. Efficient Syntheses of Diverse, Medicinally Relevant Targets Planned by Computer and Executed in the Laboratory. Chem 2018, 4, 522–532. [Google Scholar] [CrossRef] [Green Version]
- Ravitz, O. Data-driven computer aided synthesis design. Drug Discov. Today: Technol. 2013, 10, e443–e449. [Google Scholar] [CrossRef]
- Law, J.; Zsoldos, Z.; Simon, A.; Reid, D.; Liu, Y.; Khew, S.Y.; Johnson, A.P.; Major, S.; Wade, R.A.; Ando, H.Y. Route Designer: A Retrosynthetic Analysis Tool Utilizing Au-tomated Retrosynthetic Rule Generation. J. Chem. Inf. Model. 2009, 49, 593–602. [Google Scholar] [CrossRef]
- Christ, C.D.; Zentgraf, M.; Kriegl, J.M. Mining Electronic Laboratory Notebooks: Analysis, Retrosynthesis, and Reaction Based Enumeration. J. Chem. Inf. Model. 2012, 52, 1745–1756. [Google Scholar] [CrossRef]
- Segler, M.H.S.; Waller, M.P. Neural-Symbolic Machine Learning for Retrosynthesis and Reaction Prediction. Chem. A Eur. J. 2017, 23, 5966–5971. [Google Scholar] [CrossRef]
- Coley, C.W.; Rogers, L.; Green, W.H.; Jensen, K.F. Computer-Assisted Retrosynthesis Based on Molecular Similarity. ACS Central Sci. 2017, 3, 1237–1245. [Google Scholar] [CrossRef]
- Segler, M.H.S.; Waller, M.P. Modelling Chemical Reasoning to Predict and Invent Reactions. Chem. A Eur. J. 2017, 23, 6118–6128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baylon, J.L.; Cilfone, N.A.; Gulcher, J.R.; Chittenden, T.W. Enhancing retro synthetic reaction prediction with deep learning using multiscale reaction classification. J. Chem. Inf. Model. 2019, 59, 673–688. [Google Scholar] [CrossRef] [PubMed]
- Thakkar, A.; Kogej, T.; Reymond, J.-L.; Engkvist, O.; Bjerrum, E.J. Datasets and their influence on the development of computer assisted synthesis planning tools in the pharmaceutical domain. Chem. Sci. 2019, 11, 154–168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Genheden, S.; Thakkar, A.; Chadimová, V.; Reymond, J.-L.; Engkvist, O.; Bjerrum, E. AiZynthFinder: A fast, robust and flexible open-source software for retrosynthetic planning. J. Cheminformatics 2020, 12, 70. [Google Scholar] [CrossRef]
- Tu, Z.; Coley, C.W. Permutation Invariant Graph-to-Sequence Model for Template-Free Retrosynthesis and Reaction Prediction. J. Chem. Inf. Model. 2022, 62, 3503–3513. [Google Scholar] [CrossRef]
- Cadeddu, A.; Wylie, E.K.; Jurczak, J.; Wampler-Doty, M.; Grzybowski, B.A. Organic Chemistry as a language and the implications of chemical linguistics for structural and retrosynthetic analyses. Angew. Chem. Int. Ed. 2014, 53, 8108–8112. [Google Scholar] [CrossRef]
- Schwaller, P.; Laino, T.; Gaudin, T.; Bolgar, P.; Hunter, C.A.; Bekas, C.; Lee, A.A. Molecular Transformer: A Model for Uncertainty-Calibrated Chemical Reaction Prediction. ACS Central Sci. 2019, 5, 1572–1583. [Google Scholar] [CrossRef] [Green Version]
- Lin, K.; Xu, Y.; Pei, J.; Lai, L. Automatic retrosynthetic route planning using template-free models. Chem. Sci. 2020, 11, 3355–3364. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Li, P.; Song, S. Decomposing Retrosynthesis into Reactive Center Prediction and Molecule Generation. bioRxiv 2019, 677849. [Google Scholar] [CrossRef]
- Schwaller, P.; Petraglia, R.; Zullo, V.; Nair, V.H.; Haeuselmann, R.A.; Pisoni, R.; Bekas, C.; Iuliano, A.; Laino, T. Predicting retrosynthetic pathways using trans-former-based models and a hyper-graph exploration strategy. Chem. Sci. 2020, 11, 3316–3325. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Li, Y.; Qiu, J.; Chen, G.; Liu, H.; Liao, B.; Hsieh, C.-Y.; Yao, X. RetroPrime: A Diverse, plausible and Transformer-based method for Single-Step retrosynthesis predictions. Chem. Eng. J. 2021, 420, 129845. [Google Scholar] [CrossRef]
- Tetko, I.V.; Karpov, P.; Van Deursen, R.; Godin, G. State-of-the-art augmented NLP transformer models for direct and single-step retrosynthesis. Nat. Commun. 2020, 11, 5575. [Google Scholar] [CrossRef]
- Ucak, U.V.; Ashyrmamatov, I.; Ko, J.; Lee, J. Retrosynthetic reaction pathway prediction through neural machine translation of atomic environments. Nat. Commun. 2022, 1, 1186. [Google Scholar] [CrossRef]
- Duan, H.; Wang, L.; Zhang, C.; Guo, L.; Li, J. Retrosynthesis with attention-based NMT model and chemical analysis of “wrong” predictions. RSC Adv. 2020, 10, 1371–1378. [Google Scholar] [CrossRef] [Green Version]
- Mao, K.; Xiao, X.; Xu, T.; Rong, Y.; Huang, J.; Zhao, P. Molecular graph enhanced transformer for retrosynthesis prediction. Neurocomputing 2021, 457, 193–202. [Google Scholar] [CrossRef]
- Chen, B.; Shen, T.; Jaakkola, T.S.; Barzilay, R. Learning to Make Generalizable and Diverse Predictions for Retrosynthesis. arXiv 2019, arXiv:1910.09688. [Google Scholar] [CrossRef]
- Pesciullesi, G.; Schwaller, P.; Laino, T.; Reymond, J.-L. Transfer learning enables the molecular transformer to predict regio- and stereoselective reactions on carbohydrates. Nat. Commun. 2020, 11, 4874. [Google Scholar] [CrossRef]
- Coley, C.W.; Barzilay, R.; Jaakkola, T.S.; Green, W.H.; Jensen, K.F. Prediction of Organic Reaction Outcomes Using Machine Learning. ACS Central Sci. 2017, 3, 434–443. [Google Scholar] [CrossRef] [Green Version]
- Wei, J.N.; Duvenaud, D.; Aspuru-Guzik, A. Neural Networks for the Prediction of Organic Chemistry Reactions. ACS Central Sci. 2016, 2, 725–732. [Google Scholar] [CrossRef]
- Jin, W.; Coley, C.; Barzilay, R.; Jaakkola, T. Predicting Organic Reaction Outcomes with Weisfeiler-Lehman Network. In Advances in Neural Information Processing Systems 30 (NIPS 2017); Curran Associates, Inc.: Red Hook, NY, USA, 2017; pp. 2607–2616. [Google Scholar]
- Schwaller, P.; Gaudin, T.; Lányi, D.; Bekas, C.; Laino, T. “Found in Translation”: Predicting outcomes of complex organic chemistry reactions using neural sequence-to-sequence models. Chem. Sci. 2018, 9, 6091–6098. [Google Scholar] [CrossRef] [Green Version]
- Perera, D.; Tucker, J.W.; Brahmbhatt, S.; Helal, C.J.; Chong, A.; Farrell, W.; Richardson, P.; Sach, N.W. A platform for automated nanomole-scale reaction screening and micromole-scale synthesis in flow. Science 2018, 359, 429–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahneman, D.T.; Estrada, J.G.; Lin, S.; Dreher, S.D.; Doyle, A.G. Predicting reaction performance in C–N cross-coupling using machine learning. Science 2018, 360, 186–190. [Google Scholar] [CrossRef]
- Schwaller, P. Learning the Language of Chemical Reactions-Atom by Atom. Linguistics-Inspired Machine Learning Methods for Chemical Reaction Tasks. Ph.D. Thesis, Universität Bern, Bern, Switzerland, 2021. Available online: https://boristheses.unibe.ch/id/eprint/2736 (accessed on 27 May 2021).
- Żurański, A.M.; Alvarado, J.I.M.; Shields, B.J.; Doyle, A.G. Predicting Reaction Yields via Supervised Learning. Acc. Chem. Res. 2021, 54, 1856–1865. [Google Scholar] [CrossRef]
- Haywood, A.L.; Redshaw, J.; Hanson-Heine, M.W.D.; Taylor, A.; Brown, A.; Mason, A.M.; Gärtner, T.; Hirst, J.D. Kernel Methods for Predicting Yields of Chemical Reactions. J. Chem. Inf. Model. 2021, 62, 2077–2092. [Google Scholar] [CrossRef] [PubMed]
- Sandfort, F.; Strieth-Kalthoff, F.; Kühnemund, M.; Beecks, C.; Glorius, F. A Structure-Based Platform for Predicting Chemical Reactivity. Chem 2020, 6, 1379–1390. [Google Scholar] [CrossRef]
- Sato, A.; Miyao, T.; Funatsu, K. Prediction of Reaction Yield for Buchwald-Hartwig Cross-coupling Reactions Using Deep Learning. Mol. Inform. 2021, 41, 2100156. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, M.K.; Ahneman, D.T.; Riera, O.; Doyle, A.G. Deoxyfluorination with Sulfonyl Fluorides: Navigating Reaction Space with Machine Learning. J. Am. Chem. Soc. 2018, 140, 5004–5008. [Google Scholar] [CrossRef]
- Li, J.; Eastgate, M.D. Making Better Decisions during Synthetic Route Design: Leveraging Prediction to Achieve Green-ness-by-Design. React. Chem. Eng. 2019, 4, 1595–1607. [Google Scholar] [CrossRef]
- Christensen, M.; Yunker, L.P.E.; Adedeji, F.; Häse, F.; Roch, L.M.; Gensch, T.; Gomes, G.D.P.; Zepel, T.; Sigman, M.S.; Aspuru-Guzik, A.; et al. Data-science driven autonomous process optimization. Commun. Chem. 2021, 4, 112. [Google Scholar] [CrossRef]
- Angello, N.H.; Rathore, V.; Beker, W.; Wołos, A.; Jira, E.R.; Roszak, R.; Wu, T.C.; Schroeder, C.M.; Aspuru-Guzik, A.; Grzybowski, B.A.; et al. Closed-loop optimization of general reaction conditions for heteroaryl Suzuki-Miyaura coupling. Science 2022, 378, 399–405. [Google Scholar] [CrossRef]
- Ley, S.V.; Fitzpatrick, D.E.; Ingham, R.J.; Myers, R.M. Organic Synthesis: March of the Machines. Angew. Chem. Int. Ed. 2015, 54, 3449–3464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ley, S.V.; Fitzpatrick, D.E.; Myers, R.M.; Battilocchio, C.; Ingham, R.J. Machine-Assisted Organic Synthesis. Angew. Chem. Int. Ed. 2015, 54, 10122–10136. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Ballmer, S.G.; Gillis, E.P.; Fujii, S.; Schmidt, M.J.; Palazzolo, A.M.E.; Lehmann, J.W.; Morehouse, G.F.; Burke, M.D. Synthesis of many different types of organic small molecules using one automated process. Science 2015, 347, 1221–1226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chatterjee, S.; Guidi, M.; Seeberger, P.H.; Gilmore, K. Automated radial synthesis of organic molecules. Nature 2020, 579, 379–384. [Google Scholar] [CrossRef]
- Mehr, S.H.M.; Craven, M.; Leonov, A.I.; Keenan, G.; Cronin, L. A universal system for digitization and automatic execution of the chemical synthesis literature. Science 2020, 370, 101–108. [Google Scholar] [CrossRef]
- Steiner, S.; Wolf, J.; Glatzel, S.; Andreou, A.; Granda, J.M.; Keenan, G.; Hinkley, T.; Aragon-Camarasa, G.; Kitson, P.J.; Angelone, D.; et al. Organic synthesis in a modular robotic system driven by a chemical programming language. Science 2019, 363, eaav2211. [Google Scholar] [CrossRef] [Green Version]
- Gao, W.; Raghavan, P.; Coley, C.W. Autonomous platforms for data-driven organic synthesis. Nat. Commun. 2022, 13, 1075. [Google Scholar] [CrossRef]
- Burger, B.; Maffettone, P.M.; Gusev, V.V.; Aitchison, C.M.; Bai, Y.; Wang, X.; Li, X.; Alston, B.M.; Li, B.; Clowes, R.; et al. A mobile robotic chemist. Nature 2020, 583, 237–241. [Google Scholar] [CrossRef]
- Xu, H.; Lin, J.; Liu, Q.; Chen, Y.; Zhang, J.; Yang, Y.; Young, M.C.; Xu, Y.; Zhang, D.; Mo, F. High-throughput discovery of chemical structure-polarity relationships combining automation and machine-learning techniques. Chem 2022, 8, 3202–3214. [Google Scholar] [CrossRef]
- Angelone, D.; Hammer, A.J.S.; Rohrbach, S.; Krambeck, S.; Granda, J.M.; Wolf, J.; Zalesskiy, S.; Chisholm, G.; Cronin, L. Convergence of multiple synthetic paradigms in a universally programmable chemical synthesis machine. Nat. Chem. 2020, 13, 63–69. [Google Scholar] [CrossRef]
- Bédard, A.-C.; Adamo, A.; Aroh, K.C.; Russell, M.G.; Bedermann, A.A.; Torosian, J.; Yue, B.; Jensen, K.F.; Jamison, T.F. Re-configurable system for automated optimization of diverse chemical reactions. Science 2018, 361, 1220–1225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coley, C.W.; Thomas, D.A.; Lummiss, J.A.M.; Jaworski, J.N.; Breen, C.P.; Schultz, V.; Hart, T.; Fishman, J.S.; Rogers, L.; Gao, H.; et al. A robotic platform for flow synthesis of organic compounds informed by AI planning. Science 2019, 365, 557–565. [Google Scholar] [CrossRef]
- Nambiar, A.M.K.; Breen, C.P.; Hart, T.; Kulesza, T.; Jamison, T.F.; Jensen, K.F. Bayesian Optimization of Computer-Proposed Multistep Synthetic Routes on an Automated Robotic Flow Platform. ACS Central Sci. 2022, 8, 825–836. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Callahan, A.J.; Phadke, K.S.; Bellaire, B.; Farquhar, C.E.; Zhang, G.; Schissel, C.K.; Mijalis, A.J.; Hartrampf, N.; Loas, A.; et al. Automated Flow Synthesis of Peptide–PNA Conjugates. ACS Central Sci. 2021, 8, 205–213. [Google Scholar] [CrossRef]
- MacLeod, B.P.; Parlane, F.G.L.; Morrissey, T.D.; Häse, F.; Roch, L.M.; Dettelbach, K.E.; Moreira, R.; Yunker, L.P.E.; Rooney, M.B.; Deeth, J.R.; et al. Self-driving laboratory for accelerated discovery of thin-film materials. Sci. Adv. 2020, 6, eaaz8867. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Salley, D.; Sharma, A.; Keenan, G.; Mullin, M.; Cronin, L. An artificial intelligence enabled chemical synthesis robot for exploration and optimization of nanomaterials. Sci. Adv. 2022, 8, eabo2626. [Google Scholar] [CrossRef]
- Soto, F.; Wang, J.; Ahmed, R.; Demirci, U. Medical Micro/Nanorobots in Precision Medicine. Adv. Sci. 2020, 7, 2002203. [Google Scholar] [CrossRef]
- Plante, O.J.; Palmacci, E.R.; Seeberger, P.H. Automated Solid-Phase Synthesis of Oligosaccharides. Science 2001, 291, 1523–1527. [Google Scholar] [CrossRef] [Green Version]
- Caruthers, M.H. Gene Synthesis Machines: DNA Chemistry and Its Uses. Science 1985, 230, 281–285. [Google Scholar] [CrossRef] [Green Version]
- Ghislieri, D.; Gilmore, K.; Seeberger, P.H. Chemical Assembly Systems: Layered Control for Divergent, Continuous, Multi-step Syntheses of Active Pharmaceutical Ingredients. Angew. Chem. Int. Ed. 2014, 54, 678–682. [Google Scholar] [CrossRef]
- Britton, J.; Jamison, T.F. A Unified Continuous Flow Assembly Line Synthesis of Highly Substituted Pyrazoles and Pyrazolines. Angew. Chem. Int. Ed. 2017, 54, 678–682. [Google Scholar]
- Weissman, S.A.; Anderson, N.G. Design of Experiments (DoE) and Process Optimization. A Review of Recent Publications. Org. Process. Res. Dev. 2014, 19, 1605–1633. [Google Scholar] [CrossRef]
- Skilton, R.A.; Bourne, R.; Amara, Z.; Horvath, R.; Jin, J.; Scully, M.J.; Streng, E.S.; Tang, S.L.Y.; Summers, P.A.; Wang, J.; et al. Remote-controlled experiments with cloud chemistry. Nat. Chem. 2014, 7, 1–5. [Google Scholar] [CrossRef] [PubMed]
- McMullen, J.P.; Stone, M.T.; Buchwald, S.L.; Jensen, K.F. An Integrated Microreactor System for Self-Optimization of a Heck Reaction: From Micro- to Mesoscale Flow Systems. Angew. Chem. Int. Ed. 2010, 49, 7076–7080. [Google Scholar] [CrossRef]
- Aworinde, S.M.; Schweidtmann, A.M.; Lapkin, A.A. The concept of selectivity control by simultaneous distribution of the oxygen feed and wall temperature in a microstructured reactor. Chem. Eng. J. 2018, 331, 765–776. [Google Scholar] [CrossRef]
- Bhaskar, V.; Gupta, S.K.; Ray, A.K. Applications of Multiobjective Optimization in Chemical Engineering. Rev. Chem. Eng. 2000, 16, 1–54. [Google Scholar] [CrossRef]
- Xu, M.; Bhat, S.; Smith, R.; Stephens, G.; Sadhukhan, J. Multi-objective optimisation of metabolic productivity and thermo-dynamic performance. Comput. Chem. Eng. 2009, 33, 1438–1450. [Google Scholar] [CrossRef]
- Shields, B.J.; Stevens, J.; Li, J.; Parasram, M.; Damani, F.; Alvarado, J.I.M.; Janey, J.M.; Adams, R.P.; Doyle, A.G. Bayesian reaction optimization as a tool for chemical synthesis. Nature 2021, 590, 89–96. [Google Scholar] [CrossRef]
- Shambhawi, S.; Csányi, G.; Lapkin, A.A. Active Learning Training Strategy for Predicting O Adsorption Free Energy on Perovskite Catalysts using Inexpensive Catalyst Features. Chem. Methods 2021, 1, 444–450. [Google Scholar] [CrossRef]
- Nandiwale, K.Y.; Hart, T.; Zahrt, A.F.; Nambiar, A.M.K.; Mahesh, P.T.; Mo, Y.; Nieves-Remacha, M.J.; Johnson, M.D.; García-Losada, P.; Mateos, C.; et al. Continuous stirred-tank reactor cascade platform for self-optimization of reactions involving solids. React. Chem. Eng. 2022, 7, 1315–1327. [Google Scholar] [CrossRef]
- Felton, K.C.; Rittig, J.G.; Lapkin, A.A. Summit: Benchmarking Machine Learning Methods for Reaction Optimisation. Chem 2021, 1, 116–122. [Google Scholar] [CrossRef]
- Bradford, E.; Schweidtmann, A.M.; Lapkin, A. Efficient multiobjective optimization employing Gaussian processes, spectral sampling and a genetic algorithm. J. Glob. Optim. 2018, 71, 407–438. [Google Scholar] [CrossRef] [Green Version]
- Häse, F.; Roch, L.M.; Aspuru-Guzik, A. Chimera: Enabling hierarchy based multi-objective optimization for self-driving laboratories. Chem. Sci. 2018, 9, 7642–7655. [Google Scholar] [CrossRef] [PubMed]
- Häse, F.; Aldeghi, M.; Hickman, R.J.; Roch, L.M.; Aspuru-Guzik, A. Gryffin: An algorithm for Bayesian optimization of categorical variables informed by expert knowledge. Appl. Phys. Rev. 2021, 8, 031406. [Google Scholar] [CrossRef]
- Häse, F.; Roch, L.M.; Kreisbeck, C.; Aspuru-Guzik, A. Phoenics: A Bayesian Optimizer for Chemistry. ACS Central Sci. 2018, 4, 1134–1145. [Google Scholar] [CrossRef]
- Schweidtmann, A.M.; Clayton, A.D.; Holmes, N.; Bradford, E.; Bourne, R.A.; Lapkin, A.A. Machine learning meets continuous flow chemistry: Automated optimization towards the Pareto front of multiple objectives. Chem. Eng. J. 2018, 352, 277–282. [Google Scholar] [CrossRef]
- Felton, K.C.; Wigh, D.S.; Lapkin, A.A. Multi-task Bayesian Optimization of Chemical Reactions. ChemRxiv 2020. [Google Scholar] [CrossRef]
- Snoek, J.; Larochelle, H.; Adams, R.P. Practical Bayesian optimization of machine learning algorithms. Adv. Neural Inf. Process. Syst. 2012, 25, 2951–2959. [Google Scholar]
- Clayton, A.D.; Schweidtmann, A.M.; Clemens, G.; Manson, J.A.; Taylor, C.; Niño, C.G.; Chamberlain, T.W.; Kapur, N.; Blacker, A.J.; Lapkin, A.A.; et al. Automated self-optimisation of multi-step reaction and separation processes using machine learning. Chem. Eng. J. 2019, 384, 123340. [Google Scholar] [CrossRef]
- Jeraal, M.I.; Sung, S.; Lapkin, A.A. A Machine Learning- Enabled Autonomous Flow Chemistry Platform for Process Opti-mization of Multiple Reaction Metrics. Chem. Methods 2021, 1, 71–77. [Google Scholar] [CrossRef]
- Murray, P.M.; Tyler, S.N.G.; Moseley, J.D. Beyond the Numbers: Charting Chemical Reaction Space. Org. Process. Res. Dev. 2013, 17, 40–46. [Google Scholar] [CrossRef]
- Amar, Y.; Schweidtmann, A.M.; Deutsch, P.; Cao, L.; Lapkin, A. Machine learning and molecular descriptors enable rational solvent selection in asymmetric catalysis. Chem. Sci. 2019, 10, 6697–6706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; Amar, Y.; Cao, L.; Lapkin, A.A. Solvent Selection for Mitsunobu Reaction Driven by an Active Learning Surrogate Model. Org. Process. Res. Dev. 2020, 24, 2864–2873. [Google Scholar] [CrossRef]
- Jorayev, P.; Russo, D.; Tibbetts, J.D.; Schweidtmann, A.M.; Deutsch, P.; Bull, S.D.; Lapkin, A.A. Multi-objective Bayesian op-timisation of a two-step synthesis of p-cymene from crude sulphate turpentine. Chem. Eng. Sci. 2022, 247, 116938. [Google Scholar] [CrossRef]
- Langner, S.; Häse, F.; Perea, J.D.; Stubhan, T.; Hauch, J.; Roch, L.M.; Heumueller, T.; Aspuru-Guzik, A.; Brabec, C.J. Beyond Ternary OPV: High-Throughput Experimentation and Self-Driving Laboratories Optimize Multicomponent Systems. Adv. Mater. 2020, 32, e1907801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pollice, R.; Gomes, G.D.P.; Aldeghi, M.; Hickman, R.J.; Krenn, M.; Lavigne, C.; Lindner-D’Addario, M.; Nigam, A.; Ser, C.T.; Yao, Z.; et al. Data-Driven Strategies for Accelerated Materials Design. Accounts Chem. Res. 2021, 54, 849–860. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, T.-Y.; Vlachos, D. NEXTorch: A Design and Bayesian Optimization Toolkit for Chemical Sciences and Engineering. J. Chem. Inf. Model. 2021, 61, 5312–5319. [Google Scholar] [CrossRef]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
He, C.; Zhang, C.; Bian, T.; Jiao, K.; Su, W.; Wu, K.-J.; Su, A. A Review on Artificial Intelligence Enabled Design, Synthesis, and Process Optimization of Chemical Products for Industry 4.0. Processes 2023, 11, 330. https://doi.org/10.3390/pr11020330
He C, Zhang C, Bian T, Jiao K, Su W, Wu K-J, Su A. A Review on Artificial Intelligence Enabled Design, Synthesis, and Process Optimization of Chemical Products for Industry 4.0. Processes. 2023; 11(2):330. https://doi.org/10.3390/pr11020330
Chicago/Turabian StyleHe, Chasheng, Chengwei Zhang, Tengfei Bian, Kaixuan Jiao, Weike Su, Ke-Jun Wu, and An Su. 2023. "A Review on Artificial Intelligence Enabled Design, Synthesis, and Process Optimization of Chemical Products for Industry 4.0" Processes 11, no. 2: 330. https://doi.org/10.3390/pr11020330