
Developing and Regenerating Cofactors for Sustainable Enzymatic CO2 Conversion


Abstract
:1. Introduction
2. Natural Cofactor Regeneration
2.1. Enzymatic Regeneration
2.2. Chemical Regeneration
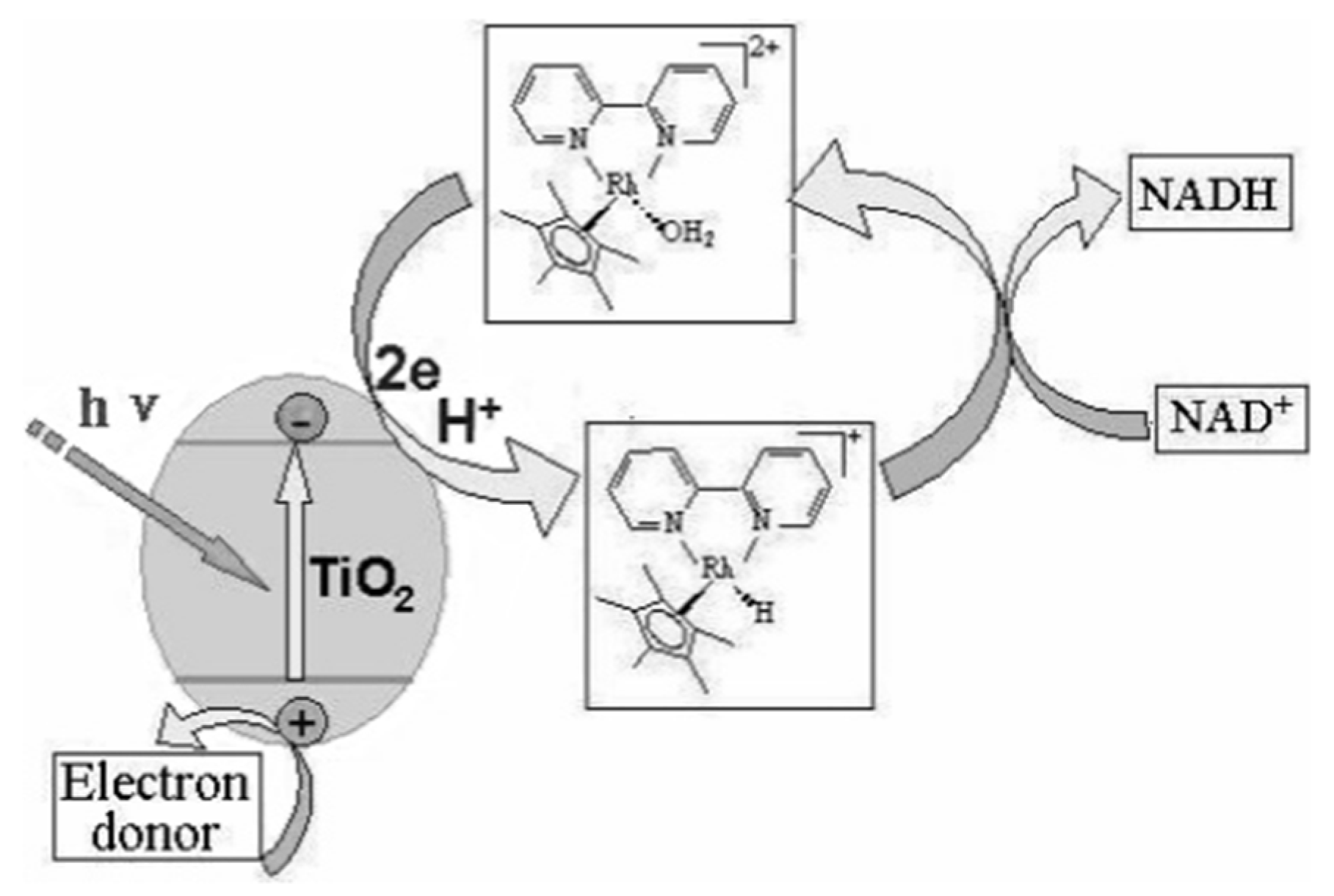
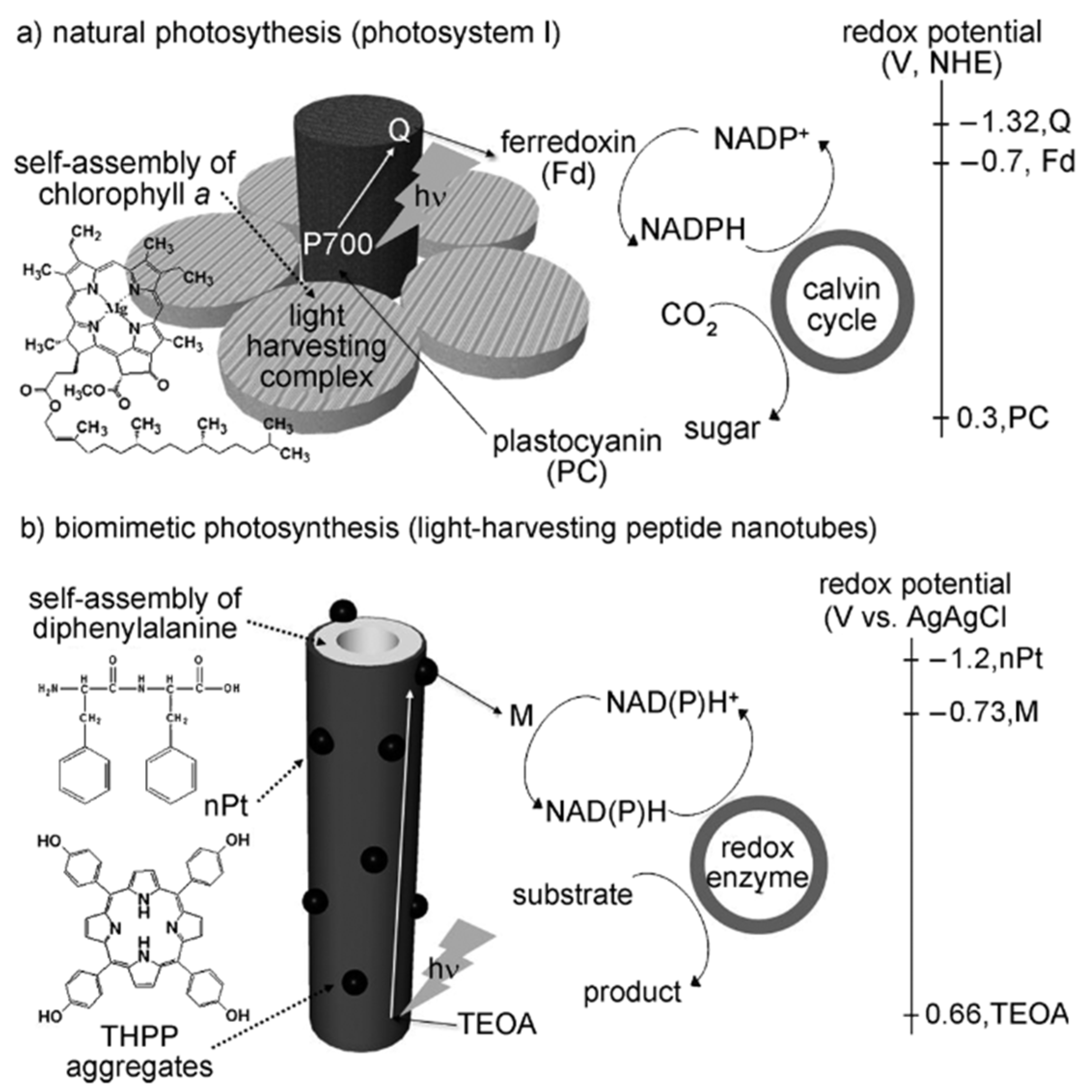
2.3. Photocatalytic Regeneration
2.3.1. Inorganic Photosensitizer
2.3.2. Organic Photosensitizer
2.4. Electrochemical Regeneration
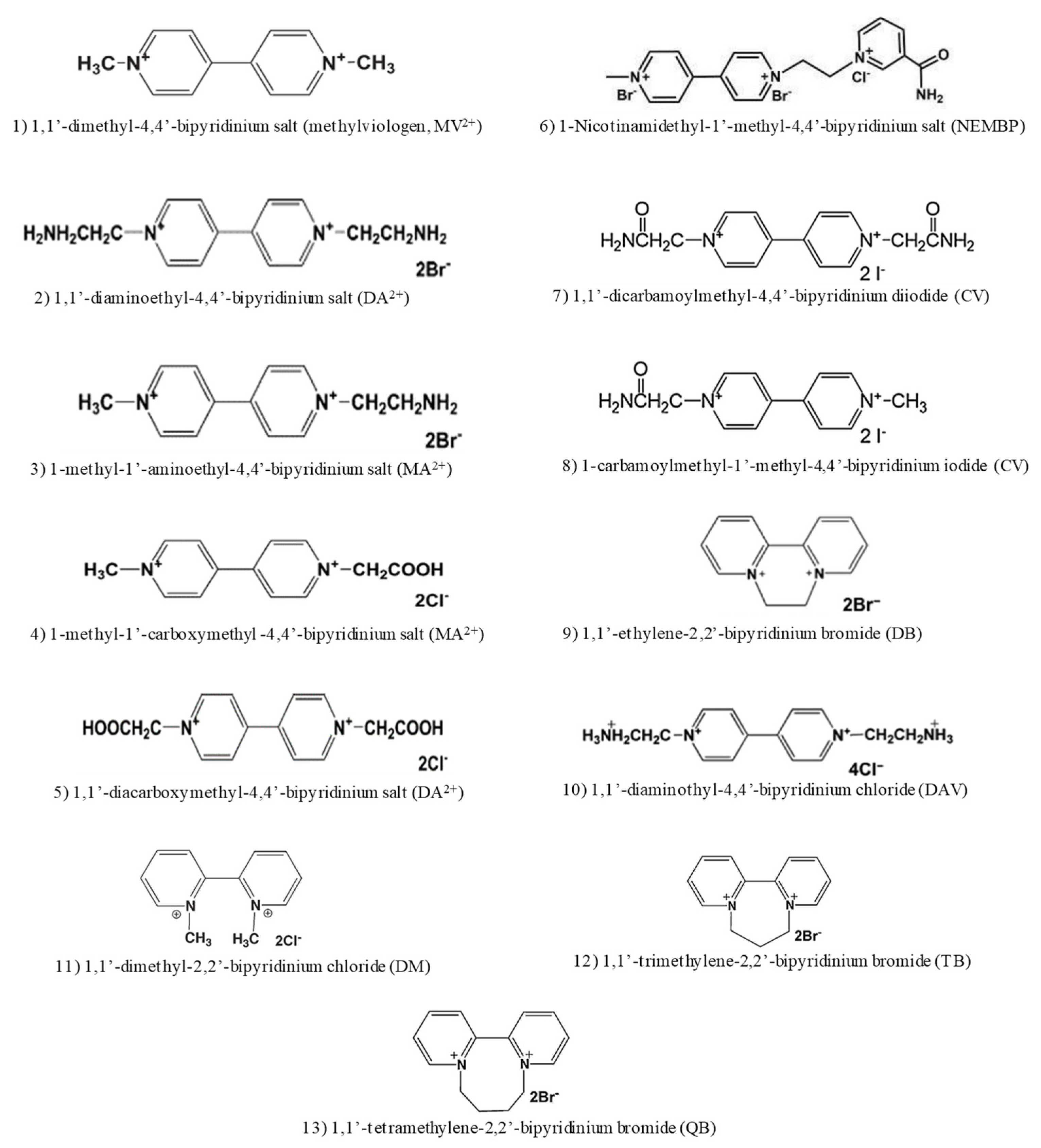
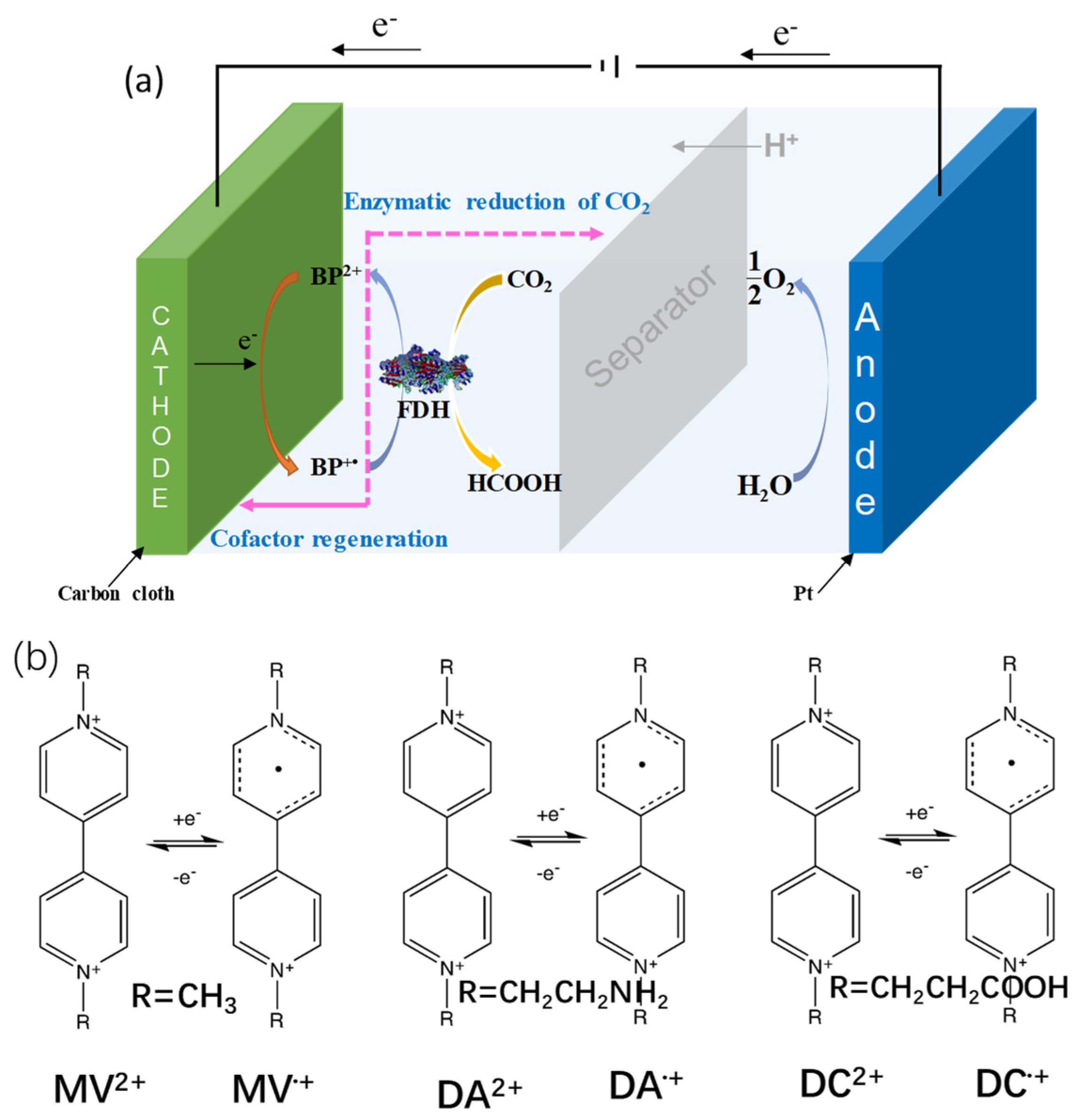
3. Artificial Cofactor Development and Regeneration
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wang, W.; Wang, S.P.; Ma, X.B.; Gong, J.L. Recent advances in catalytic hydrogenation of carbon dioxide. Chem. Soc. Rev. 2011, 40, 3703–3727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kondratenko, E.V.; Mul, G.; Baltrusaitis, J.; Larrazabal, G.O.; Perez-Ramirez, J. Status and perspectives of CO2 conversion into fuels and chemicals by catalytic, photocatalytic and electrocatalytic processes. Energy Environ. Sci. 2013, 6, 3112–3135. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.B.; Li, F.F.; Nie, Y.; Zhang, X.P.; Zhang, S.J.; Ji, X.Y. Zinc-based deep eutectic solvent—An efficient carbonic anhydrase mimic for CO2 hydration and conversion. Sep. Purif. Technol. 2021, 276, 119446. [Google Scholar] [CrossRef]
- Zhang, Z.; Li, F.; Bai, Y.; Nie, Y.; Ji, X. Application and Challenge of Ionic Liquids as Co-solvent for Electro-enzymatic Conversion of CO2 to Methanol. Proc. Chin. Soc. Electr. Eng. 2021, 41, 3657–3665. [Google Scholar]
- Li, H.; Opgenorth, P.H.; Wernick, D.G.; Rogers, S.; Wu, T.Y.; Higashide, W.; Malati, P.; Huo, Y.X.; Cho, K.M.; Liao, J.C. Integrated Electromicrobial Conversion of CO2 to Higher Alcohols. Science 2012, 335, 1596. [Google Scholar] [CrossRef]
- Zhang, Z.B.; Xu, B.H.; Luo, J.Q.; Von Solms, N.; He, H.Y.; Zhang, Y.Q.; Pinelo, M.; Zhang, S.J. Ionic Liquids as Bifunctional Cosolvents Enhanced CO2 Conversion Catalysed by NADH-Dependent Formate Dehydrogenase. Catalysts 2018, 8, 304. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Lin, W.B. Metal-organic frameworks for artificial photosynthesis and photocatalysis. Chem. Soc. Rev. 2014, 43, 5982–5993. [Google Scholar] [CrossRef]
- Benson, E.E.; Kubiak, C.P.; Sathrum, A.J.; Smieja, J.M. Electrocatalytic and homogeneous approaches to conversion of CO2 to liquid fuels. Chem. Soc. Rev. 2009, 38, 89–99. [Google Scholar] [CrossRef]
- Shi, J.F.; Jiang, Y.J.; Jiang, Z.Y.; Wang, X.Y.; Wang, X.L.; Zhang, S.H.; Han, P.P.; Yang, C. Enzymatic conversion of carbon dioxide. Chem. Soc. Rev. 2015, 44, 5981–6000. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.L.; Li, Z.; Shi, J.F.; Wu, H.; Jiang, Z.Y.; Zhang, W.Y.; Song, X.K.; Ai, Q.H. Bioinspired Approach to Multienzyme Cascade System Construction for Efficient Carbon Dioxide Reduction. Acs Catal. 2014, 4, 962–972. [Google Scholar] [CrossRef]
- Zhang, Z.B.; Muschiol, J.; Huang, Y.H.; Sigurdardottir, S.B.; von Solms, N.; Daugaard, A.E.; Wei, J.; Luo, J.Q.; Xu, B.H.; Zhang, S.J.; et al. Efficient ionic liquid-based platform for multi-enzymatic conversion of carbon dioxide to methanol. Green Chem. 2018, 20, 4339–4348. [Google Scholar] [CrossRef] [Green Version]
- Ji, X.Y.; Su, Z.G.; Wang, P.; Ma, G.H.; Zhang, S.P. Tethering of Nicotinamide Adenine Dinucleotide Inside Hollow Nanofibers for High-Yield Synthesis of Methanol from Carbon Dioxide Catalyzed by Coencapsulated Multienzymes. Acs Nano 2015, 9, 4600–4610. [Google Scholar] [CrossRef] [PubMed]
- Jahns, T. Unusually stable NAD-specific glutamate dehydrogenase from the alkaliphile Amphibacillus xylanus. Antonie Van Leeuwenhoek Int. J. Gen. 1996, 70, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Ren, S.Z.; Wang, Z.Y.; Bilal, M.; Feng, Y.X.; Jiang, Y.H.; Jia, S.R.; Cui, J.D. Co-immobilization multienzyme nanoreactor with co-factor regeneration for conversion of CO2. Int. J. Biol. Macromol. 2020, 155, 110–118. [Google Scholar] [CrossRef] [PubMed]
- Netto, C.G.C.M.; Andrade, L.H.; Toma, H.E. Carbon dioxide/methanol conversion cycle based on cascade enzymatic reactions supported on superparamagnetic nanoparticles. An. Da Acad. Bras. De Cienc. 2018, 90, 593–606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Zahab, B.; Donnelly, D.; Wang, P. Particle-tethered NADH for production of methanol from CO2 catalyzed by coimmobilized enzymes. Biotechnol. Bioeng. 2008, 99, 508–514. [Google Scholar] [CrossRef]
- Yu, X.J.; Niks, D.; Ge, X.; Liu, H.Z.; Hille, R.; Mulchandani, A. Synthesis of Formate from CO2 Gas Catalyzed by an O-2-Tolerant NAD-Dependent Formate Dehydrogenase and Glucose Dehydrogenase. Biochemistry 2019, 58, 1861–1868. [Google Scholar] [CrossRef]
- Marpani, F.; Sarossy, Z.; Pinelo, M.; Meyer, A.S. Kinetics based reaction optimization of enzyme catalyzed reduction of formaldehyde to methanol with synchronous cofactor regeneration. Biotechnol. Bioeng. 2017, 114, 2762–2770. [Google Scholar] [CrossRef]
- Scriven, E.F.V. 2.05—Pyridines and their Benzo Derivatives: (ii) Reactivity at Ring Atoms. In Comprehensive Heterocyclic Chemistry; Katritzky, A.R., Rees, C.W., Eds.; Pergamon: Oxford, UK, 1984; pp. 165–314. [Google Scholar]
- Schopfer, L.M.; Massey, V.; Nishino, T. Rapid Reaction Studies on the Reduction and Oxidation of Chicken Liver Xanthine Dehydrogenase by the Xanthine Urate and Nad Nadh Couples. J. Biol. Chem 1988, 263, 13528–13538. [Google Scholar] [CrossRef]
- Saba, T.; Burnett, J.W.H.; Li, J.W.; Kechagiopoulos, P.N.; Wang, X.D. A facile analytical method for reliable selectivity examination in cofactor NADH regeneration. Chem. Commun. 2020, 56, 1231–1234. [Google Scholar] [CrossRef] [Green Version]
- Jones, J.B.; Lewis, A.J.; Sneddon, D.W.; Higgins, W. Preparative-scale reductions of cyclic ketone and aldehyde substrates of horse liver alcohol-dehydrogenase with in-situ sodium dithionite bicycling of catalytic amounts of nad. J. Chem. Soc. Chem. Commun. 1972, 15, 856–857. [Google Scholar] [CrossRef]
- Stincone, A.; Prigione, A.; Cramer, T.; Wamelink, M.M.C.; Campbell, K.; Cheung, E.; Olin-Sandoval, V.; Gruning, N.M.; Kruger, A.; Alam, M.T.; et al. The return of metabolism: Biochemistry and physiology of the pentose phosphate pathway. Biol. Rev. 2015, 90, 927–963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hofler, G.T.; Fernandez-Fueyo, E.; Pesic, M.; Younes, S.H.; Choi, E.G.; Kim, Y.H.; Urlacher, V.B.; Arends, I.; Hollmann, F. A Photoenzymatic NADH Regeneration System. Chembiochem 2018, 19, 2344–2347. [Google Scholar] [CrossRef] [PubMed]
- Schneider, J.; Matsuoka, M.; Takeuchi, M.; Zhang, J.L.; Horiuchi, Y.; Anpo, M.; Bahnemann, D.W. Understanding TiO2 Photocatalysis: Mechanisms and Materials. Chem. Rev. 2014, 114, 9919–9986. [Google Scholar] [CrossRef]
- Wang, X.; Saba, T.; Yiu, H.H.P.; Howe, R.F.; Anderson, J.A.; Shi, J. Cofactor NAD(P)H Regeneration Inspired by Heterogeneous Pathways. Chem 2017, 2, 621–654. [Google Scholar] [CrossRef] [Green Version]
- Ryu, J.; Lee, S.H.; Nam, D.H.; Park, C.B. Rational Design and Engineering of Quantum-Dot-Sensitized TiO2 Nanotube Arrays for Artificial Photosynthesis. Adv. Mater. 2011, 23, 1883–1888. [Google Scholar] [CrossRef]
- Jiang, Z.Y.; Lu, C.Q.; Wu, H. Photoregeneration of NADH using carbon-containing TiO2. Ind. Eng. Chem. Res. 2005, 44, 4165–4170. [Google Scholar] [CrossRef]
- Shi, Q.; Yang, D.; Jiang, Z.Y.; Li, J. Visible-light photocatalytic regeneration of NADH using P-doped TiO2 nanoparticles. J. Mol. Catal. B-Enzym. 2006, 43, 44–48. [Google Scholar] [CrossRef]
- Geng, J.Q.; Yang, D.; Zhu, J.H.; Chen, D.M.; Jiang, Z.Y. Nitrogen-doped TiO2 nanotubes with enhanced photocatalytic activity synthesized by a facile wet chemistry method. Mater. Res. Bull. 2009, 44, 146–150. [Google Scholar] [CrossRef]
- Chen, D.; Yang, D.; Wang, Q.; Jiang, Z.Y. Effects of boron doping on photocatalytic activity and microstructure of titanium dioxide nanoparticles. Ind. Eng. Chem. Res. 2006, 45, 4110–4116. [Google Scholar] [CrossRef]
- Nam, D.H.; Lee, S.H.; Park, C.B. CdTe, CdSe, and CdS Nanocrystals for Highly Efficient Regeneration of Nicotinamide Cofactor Under Visible Light. Small 2010, 6, 922–926. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Lee, M.; Lee, J.S.; Park, C.B. Self-Assembled Light-Harvesting Peptide Nanotubes for Mimicking Natural Photosynthesis. Angew. Chem.-Int. Ed. 2012, 51, 517–520. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Lee, S.H.; Lee, J.S.; Lee, M.; Park, C.B. Zn-containing porphyrin as a biomimetic light-harvesting molecule for biocatalyzed artificial photosynthesis. Chem. Commun. 2011, 47, 10227–10229. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.B.; Tong, J.H.; Meng, X.L.; Cai, Y.J.; Ma, S.S.; Huo, F.; Luo, J.Q.; Xu, B.H.; Zhang, S.J.; Pinelo, M. Development of an Ionic Porphyrin-Based Platform as a Biomimetic Light-Harvesting Agent for High-Performance Photoenzymatic Synthesis of Methanol from CO2. ACS Sustain. Chem. Eng. 2021, 9, 11503–11511. [Google Scholar] [CrossRef]
- Yadav, R.K.; Baeg, J.O.; Oh, G.H.; Park, N.J.; Kong, K.J.; Kim, J.; Hwang, D.W.; Biswas, S.K. A Photocatalyst-Enzyme Coupled Artificial Photosynthesis System for Solar Energy in Production of Formic Acid from CO2. J. Am. Chem. Soc. 2012, 134, 11455–11461. [Google Scholar] [CrossRef]
- Park, C.B.; Lee, S.H.; Subramanian, E.; Kale, B.B.; Lee, S.M.; Baeg, J.O. Solar energy in production of L-glutamate through visible light active photocatalyst-redox enzyme coupled bioreactor. Chem. Commun. 2008, 42, 5423–5425. [Google Scholar] [CrossRef]
- Yadav, R.K.; Oh, G.H.; Park, N.J.; Kumar, A.; Kong, K.J.; Baeg, J.O. Highly Selective Solar-Driven Methanol from CO2 by a Photocatalyst/Biocatalyst Integrated System. J. Am. Chem. Soc. 2014, 136, 16728–16731. [Google Scholar] [CrossRef]
- Schmakel, C.O.; Santhanam, K.S.V.; Elving, P.J. Nicotinamide Adenine-Dinucleotide (Nad+) and Related Compounds—Electrochemical Redox Pattern and Allied Chemical Behavior. J. Am. Chem. Soc. 1975, 97, 5083–5092. [Google Scholar] [CrossRef]
- Kohlmann, C.; Markle, W.; Lutz, S. Electroenzymatic synthesis. J. Mol. Catal. B-Enzym. 2008, 51, 57–72. [Google Scholar] [CrossRef]
- Zhang, Z.B.; Li, J.J.; Ji, M.B.; Liu, Y.R.; Wang, N.; Zhang, X.P.; Zhang, S.J.; Ji, X.Y. Encapsulation of multiple enzymes in a metal-organic framework with enhanced electro-enzymatic reduction of CO2 to methanol. Green Chem. 2021, 23, 2362–2371. [Google Scholar] [CrossRef]
- Damian, A.; Maloo, K.; Omanovic, S. Direct electrochemical regeneration of NADH on Au, Cu and Pt-Au electrodes. Chem. Biochem. Eng. Q 2007, 21, 21–32. [Google Scholar]
- Ali, I.; Gill, A.; Omanovic, S. Direct electrochemical regeneration of the enzymatic cofactor 1,4-NADH employing nano-patterned glassy carbon/Pt and glassy carbon/Ni electrodes. Chem. Eng. J. 2012, 188, 173–180. [Google Scholar] [CrossRef]
- Ali, I.; Khan, T.; Omanovic, S. Direct electrochemical regeneration of the cofactor NADH on bare Ti, Ni, Co and Cd electrodes: The influence of electrode potential and electrode material. J. Mol. Catal. A-Chem. 2014, 387, 86–91. [Google Scholar] [CrossRef]
- Yuan, M.W.; Kummer, M.J.; Milton, R.D.; Quah, T.; Minteer, S.D. Efficient NADH Regeneration by a Redox Polymer-Immobilized Enzymatic System. ACS Catal. 2019, 9, 5486–5495. [Google Scholar] [CrossRef]
- Siu, E.; Won, K.; Park, C.B. Electrochemical regeneration of NADH using conductive vanadia-silica xerogels. Biotechnol. Prog. 2007, 23, 293–296. [Google Scholar] [CrossRef] [PubMed]
- Ali, I.; Ullah, N.; McArthur, M.A.; Coulombe, S.; Omanovic, S. Direct electrochemical regeneration of enzymatic cofactor 1,4-NADH on a cathode composed of multi-walled carbon nanotubes decorated with nickel nanoparticles. Can. J. Chem. Eng. 2018, 96, 68–73. [Google Scholar] [CrossRef]
- Kim, S.; Kim, M.K.; Lee, S.H.; Yoon, S.; Jung, K.-D. Conversion of CO2 to formate in an electroenzymatic cell using Candida boidinii formate dehydrogenase. J. Mol. Catal. B Enzym. 2014, 102, 9–15. [Google Scholar] [CrossRef]
- Zhang, L.; Vila, N.; Kohring, G.W.; Walcarius, A.; Etienne, M. Covalent Immobilization of (2,2′-Bipyridyl) (Pentamethylcyclopentadienyl)-Rhodium Complex on a Porous Carbon Electrode for Efficient Electrocatalytic NADH Regeneration. ACS Catal. 2017, 7, 4386–4394. [Google Scholar] [CrossRef]
- Ruschig, U.; Muller, U.; Willnow, P.; Hopner, T. CO2 Reduction to Formate by Nadh Catalyzed by Formate Dehydrogenase from Pseudomonas-Oxalaticus. Eur. J. Biochem. 1976, 70, 325–330. [Google Scholar] [CrossRef]
- Ikeyama, S.; Amao, Y. An Artificial Co-enzyme Based on the Viologen Skeleton for Highly Efficient CO2 Reduction to Formic Acid with Formate Dehydrogenase. Chemcatchem 2017, 9, 833–838. [Google Scholar] [CrossRef]
- Miyaji, A.; Amao, Y. Visible-Light Driven CO2 Reduction to Formate with Electron Mediated Nicotinamide-Modified Viologen in the System of Water-Soluble Zinc Porphyrin and Formate Dehydrogenase. Chemnanomat 2021, 7, 626–634. [Google Scholar] [CrossRef]
- Ikeyama, S.; Katagiri, T.; Amao, Y. The improvement of formic acid production from CO2 with visible-light energy and formate dehydrogenase by the function of the viologen derivative with carbamoylmethyl group as an electron carrier. J. Photoch. Photobio. A 2018, 358, 362–367. [Google Scholar] [CrossRef]
- Ikeyama, S.; Amao, Y. A novel electron carrier molecule based on a viologen derivative for visible light-driven CO2 reduction to formic acid with the system of zinc porphyrin and formate dehydrogenase. Sustain. Energy Fuels 2017, 1, 1730–1733. [Google Scholar] [CrossRef]
- Amao, Y.; Abe, R.; Shiotani, S. Effect of chemical structure of bipyridinium salts as electron carrier on the visible-light induced conversion of CO2 to formic acid with the system consisting of water-soluble zinc porphyrin and formate dehydrogenase. J. Photochem. Photobiol. A Chem. 2015, 313, 149–153. [Google Scholar] [CrossRef] [Green Version]
- Amao, Y.; Ikeyama, S. Discovery of the Reduced Form of Methylviologen Activating Formate Dehydrogenase in the Catalytic Conversion of Carbon Dioxide to Formic Acid. Chem. Lett. 2015, 44, 1182–1184. [Google Scholar] [CrossRef]
- Ishibashi, T.; Ikeyama, S.; Ito, M.; Ikeda, S.; Amao, Y. Light-driven CO2 Reduction to Formic Acid with a Hybrid System of Biocatalyst and Semiconductor Based Photocatalyst. Chem. Lett. 2018, 47, 1505–1508. [Google Scholar] [CrossRef] [Green Version]
- Toyodome, T.; Amao, Y.; Higashi, M. Photoelectrochemical reduction of CO2 to formate over a hybrid system of CuInS2 photocathode and formate dehydrogenase under visible-light irradiation. New J. Chem. 2021, 45, 14803–14807. [Google Scholar] [CrossRef]
- Zhang, Z.B.; Vasiliu, T.; Li, F.F.; Laaksonen, A.; Mocci, F.; Ji, X.Y. Electrochemically driven efficient enzymatic conversion of CO2 to formic acid with artificial cofactors. J. CO2 Util. 2021, 52, 101679. [Google Scholar] [CrossRef]
- Jayathilake, B.S.; Bhattacharya, S.; Vaidehi, N.; Narayanan, S.R. Efficient and Selective Electrochemically Driven Enzyme-Catalyzed Reduction of Carbon Dioxide to Formate using Formate Dehydrogenase and an Artificial Cofactor. Acc. Chem. Res. 2019, 52, 676–685. [Google Scholar] [CrossRef] [PubMed]

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
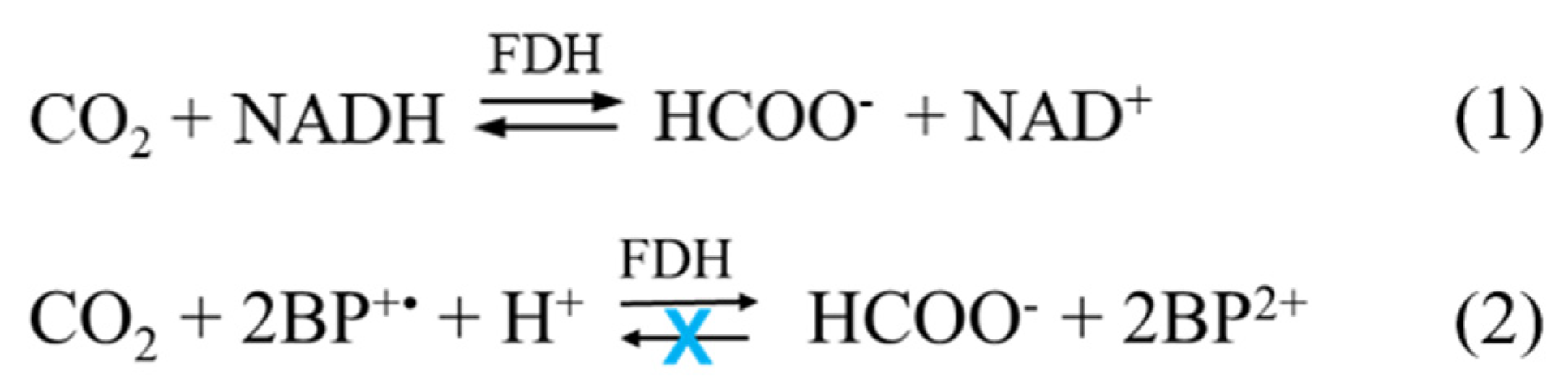{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NADH Regeneration Solution | Advantages | Disadvantages |
|---|---|---|
| Enzymatic method | Environmentally friendly, high efficiency, high selectivity | High cost of enzymes and coenzymes. By-product separation, instability of enzyme |
| Chemical method | Low cost | By-products separation, unfriendly environment, unfriendly to enzyme, low efficiency, low selectivity, unsustainable. |
| Photochemical method | Low cost, environmentally friendly, the efficiency of energy utilization | Unstable photocurrent, unstable photosensitizer, byproduct separation |
| Electrochemical method | Low cost, environmentally friendly, high efficiency | Instability of electrocatalyst, low selectivity. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Z.; Zhang, X.; Ji, X. Developing and Regenerating Cofactors for Sustainable Enzymatic CO2 Conversion. Processes 2022, 10, 230. https://doi.org/10.3390/pr10020230
Zhang Z, Zhang X, Ji X. Developing and Regenerating Cofactors for Sustainable Enzymatic CO2 Conversion. Processes. 2022; 10(2):230. https://doi.org/10.3390/pr10020230
Chicago/Turabian StyleZhang, Zhibo, Xiangping Zhang, and Xiaoyan Ji. 2022. "Developing and Regenerating Cofactors for Sustainable Enzymatic CO2 Conversion" Processes 10, no. 2: 230. https://doi.org/10.3390/pr10020230