
Transcriptome Analysis Reveals the Involvement of Alternative Splicing in the Nitrogen Starvation Response of Chlamydomonas reinhardtii
 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Transcriptome Data
2.2. Transcriptome Analysis
2.3. Gene Expression Correlation and Principal Component Analysis
2.4. Identification and Quantification of AS Events
2.5. Time-Series Cluster Analysis
2.6. Gene Functional Annotation and Enrichment Analysis
2.7. Statistical Analysis
3. Results
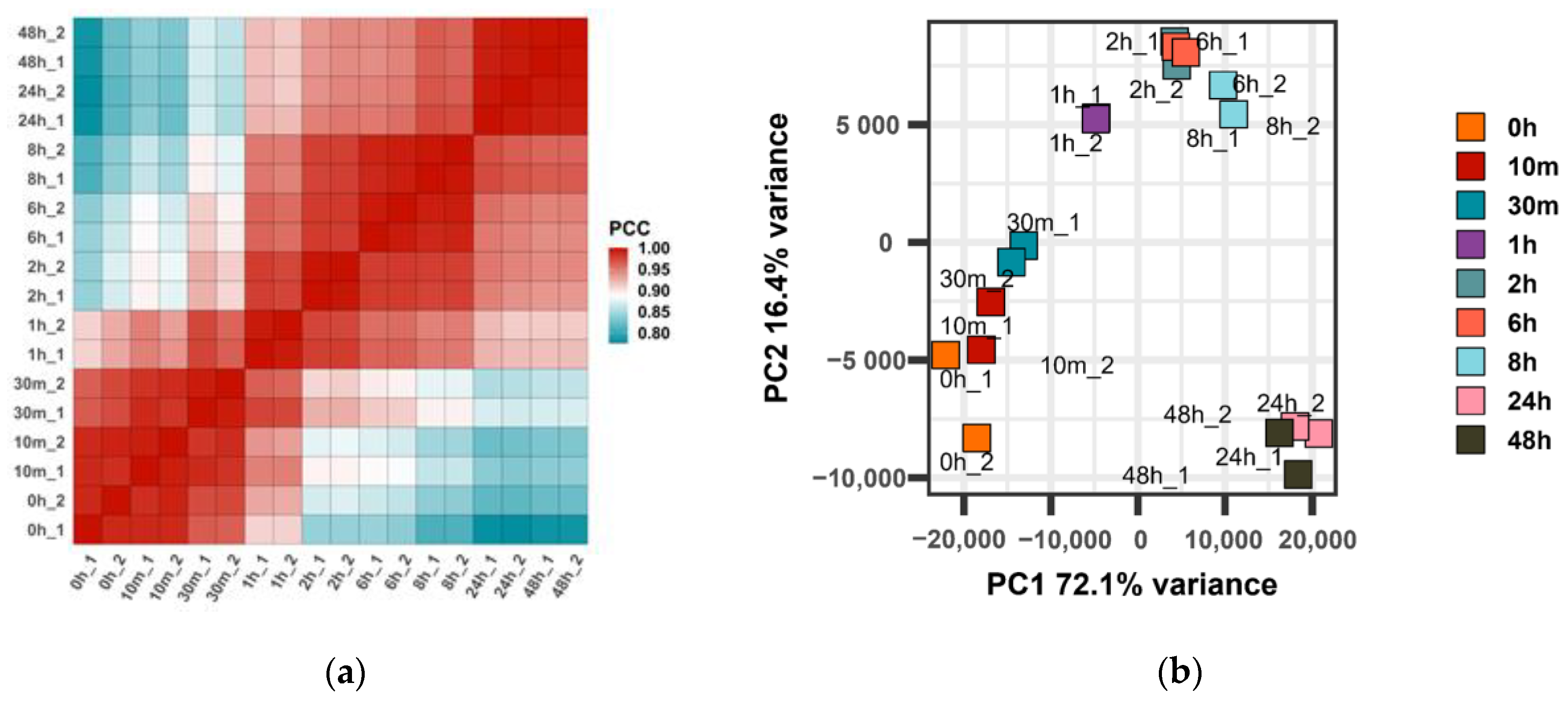
3.1. Gene Expression Correlation and Principal Component Analysis
3.2. Identification of AS Events
3.3. Quantification of AS Events and Time-Series Cluster Analysis
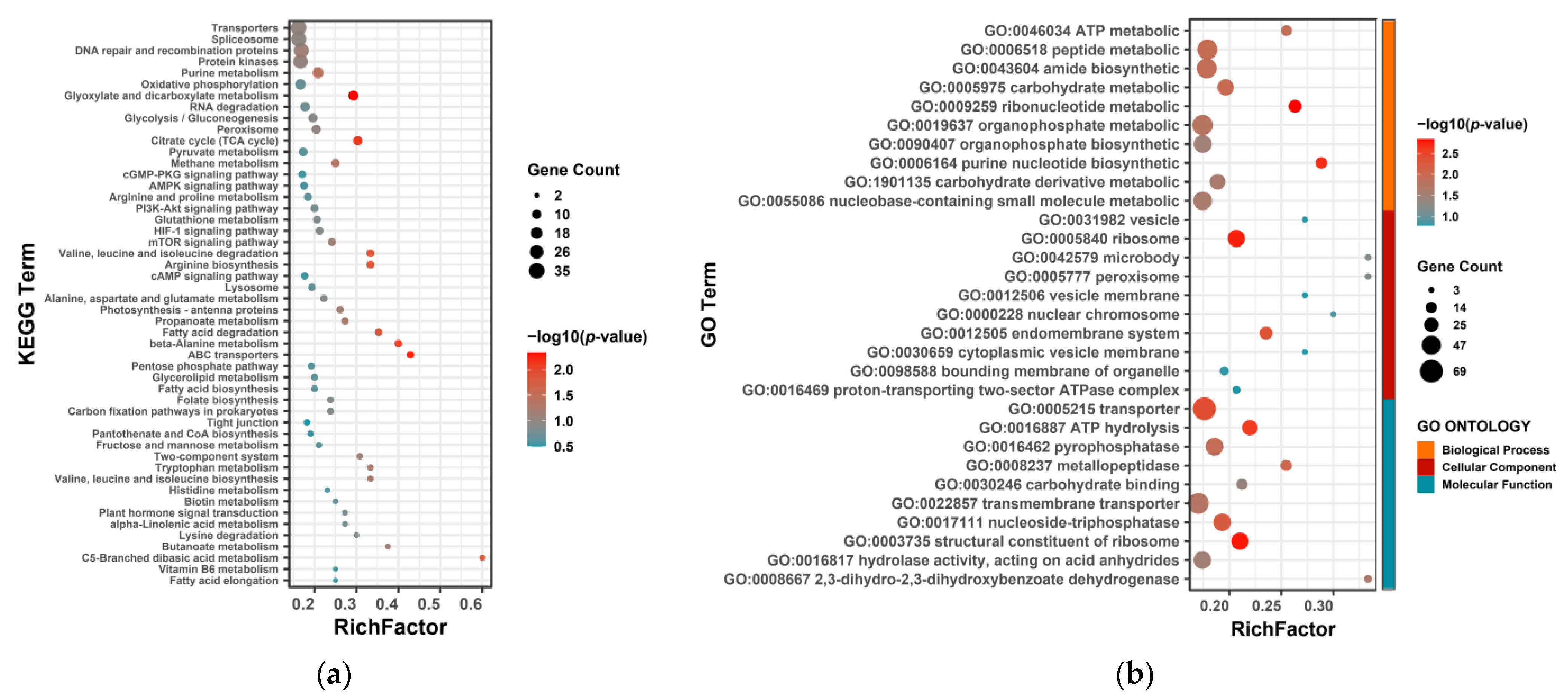
3.4. Functional Analysis of AS-Related Genes in Response to Nitrogen Starvation
3.5. Transcript Analysis of Genes Potentially Associated with Nitrogen Starvation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| AS | Alternative Splicing |
| TAP | Tris-Acetate-Phosphate |
| STAR | Spliced Transcripts Alignment to a Reference, the software used for genome alignment. (https://github.com/alexdobin/STAR) |
| StringTie | The software for Transcript assembly and quantification. |
| rMATS | Robust and Flexible Detection of Differential Alternative Splicing, the software used to identify alternative splicing events. (http://rnaseq-mats.sourceforge.net/, accessed on 14 November 2022) |
| mRNA | Messenger Ribonucleic Acid |
| SE | Skipped Exon |
| A3SS | the Alternative 3′ Splice Site |
| A5SS | the Alternative 5′ Splice Site |
| MXE | Mutually Exclusive Exons |
| RI | Retained Intron |
| EST | Expressed Sequence Tag |
| TAG | Triacylglycerol |
| SRA | Sequence Read Archive |
| NCBI | National Center for Biotechnology Information |
| SRAToolkit | The tools that download, manipulate and validate next-generation sequencing data stored in the NCBI SRA archive (https://github.com/ncbi/sra-tools, accessed on 14 November 2022) |
| Fastp | A tool designed to provide fast all-in-one preprocessing for FastQ files. (https://github.com/OpenGene/fastp, accessed on 14 November 2022) |
| TPM | Transcripts Per Kilobase per Million mapped reads |
| Mfuzz | the R package used to clustering |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| GO | Gene Ontology |
| ClusterProfile | the R package used for gene functional enrichment analysis |
| Phytozome v13 | a comparative platform for green plant genomics (https://phytozome-next.jgi.doe.gov/, accessed on 14 November 2022) |
| PC1 | Principal Component 1 |
| PC2 | Principal Component 2 |
| PCC | Pearson Correlation Coefficient |
| Mvalue | Membership value |
| ABC | ATP binding cassette |
| TCA | Tricarboxylic Acid Cycle |
| SR | Serine/arginine-Rich |
| SRS1 | serine/arginine-rich pre-mRNA splicing factor |
| SRE1 | SR-related pre-mRNA splicing factor |
| PGI1 | phosphoglucose isomerase |
| PKS3 | type III polyketide synthase |
| CDS | Coding sequence |
| NAD | Nicotinamide Adenine Dinucleotide |
| OsGS | glutamine synthetase of Oryza sativa |
| GLN | glutamine synthetase |
References
- Lee, Y.; Rio, D.C. Mechanisms and Regulation of Alternative Pre-mRNA Splicing. Annu. Rev. Biochem. 2015, 84, 291–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breitbart, R.E.; Andreadis, A.; Nadal-Ginard, B. Alternative Splicing: A Ubiquitous Mechanism For The Generation Of Multiple Protein Isoforms From Single Genes. Annu. Rev. Biochem. 1987, 56, 467–495. [Google Scholar] [CrossRef] [PubMed]
- Shen, S.; Park, J.W.; Lu, Z.-X.; Lin, L.; Henry, M.D.; Wu, Y.N.; Zhou, Q.; Xing, Y. rMATS: Robust and flexible detection of differential alternative splicing from replicate RNA-Seq data. Proc. Natl. Acad. Sci. USA 2014, 111, E5593–E5601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.W.; Tokheim, C.; Shen, S.; Xing, Y. Identifying Differential Alternative Splicing Events from RNA Sequencing Data Using RNASeq-MATS. In Deep Sequencing Data Analysis; Shomron, N., Ed.; Humana Press Inc.: Totowa, NJ, USA, 2013; Volume 1038, pp. 171–179. [Google Scholar]
- Labadorf, A.; Link, A.; Rogers, M.F.; Thomas, J.; Reddy, A.S.; Ben-Hur, A. Genome-wide analysis of alternative splicing in Chlamydomonas reinhardtii. BMC Genom. 2010, 11, 114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, Q.; Shai, O.; Lee, L.J.; Frey, B.J.; Blencowe, B.J. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat. Genet. 2008, 40, 1413–1415. [Google Scholar] [CrossRef] [PubMed]
- Marquez, Y.; Brown, J.W.; Simpson, C.; Barta, A.; Kalyna, M. Transcriptome survey reveals increased complexity of the alternative splicing landscape in Arabidopsis. Genome Res. 2012, 22, 1184–1195. [Google Scholar] [CrossRef] [Green Version]
- Ramani, A.K.; Calarco, J.A.; Pan, Q.; Mavandadi, S.; Wang, Y.; Nelson, A.C.; Lee, L.J.; Morris, Q.; Blencowe, B.J.; Zhen, M.; et al. Genome-wide analysis of alternative splicing in Caenorhabditis elegans. Genome Res. 2010, 21, 342–348. [Google Scholar] [CrossRef] [Green Version]
- Petrillo, E.; Kalyna, M.; Mandadi, K.K.; Tu, S.-L.; Simpson, C.G. Editorial: Alternative Splicing Regulation in Plants. Front. Plant Sci. 2020, 11, 913. [Google Scholar] [CrossRef]
- Punzo, P.; Grillo, S.; Batelli, G. Alternative splicing in plant abiotic stress responses. Biochem. Soc. Trans. 2020, 48, 2117–2126. [Google Scholar] [CrossRef]
- Thatcher, S.R.; Danilevskaya, O.N.; Meng, X.; Beatty, M.; Zastrow-Hayes, G.; Harris, C.; Van Allen, B.; Habben, J.; Li, B. Genome-Wide Analysis of Alternative Splicing during Development and Drought Stress in Maize. Plant Physiol. 2015, 170, 586–599. [Google Scholar] [CrossRef]
- Li-Beisson, Y.; Beisson, F.; Riekhof, W. Metabolism of acyl-lipids in Chlamydomonas reinhardtii. Plant J. 2015, 82, 504–522. [Google Scholar] [CrossRef] [PubMed]
- Raj-Kumar, P.-K.; Vallon, O.; Liang, C. In silico analysis of the sequence features responsible for alternatively spliced introns in the model green alga Chlamydomonas reinhardtii. Plant Mol. Biol. 2017, 94, 253–265. [Google Scholar] [CrossRef] [PubMed]
- Pandey, M.; Stormo, G.D.; Dutcher, S.K. Alternative Splicing During the Chlamydomonas reinhardtii Cell Cycle. G3 Genes Genomes Genet. 2020, 10, 3797–3810. [Google Scholar] [CrossRef] [PubMed]
- Goncalves, E.C.; Wilkie, A.C.; Kirst, M.; Rathinasabapathi, B. Metabolic regulation of triacylglycerol accumulation in the green algae: Identification of potential targets for engineering to improve oil yield. Plant Biotechnol. J. 2016, 14, 1649–1660. [Google Scholar] [CrossRef]
- Takeuchi, T.; Benning, C. Nitrogen-dependent coordination of cell cycle, quiescence and TAG accumulation in Chlamydomonas. Biotechnol. Biofuels 2019, 12, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Li-Beisson, Y.; Thelen, J.J.; Fedosejevs, E.; Harwood, J.L. The lipid biochemistry of eukaryotic algae. Prog. Lipid Res. 2019, 74, 31–68. [Google Scholar] [CrossRef]
- Yang, M.; Kong, F.; Xie, X.; Wu, P.; Chu, Y.; Cao, X.; Xue, S. Galactolipid DGDG and Betaine Lipid DGTS Direct De Novo Synthesized Linolenate into Triacylglycerol in a Stress-Induced Starchless Mutant of Chlamydomonas reinhardtii. Plant Cell Physiol. 2020, 61, 851–862. [Google Scholar] [CrossRef]
- Arora, N.; Pienkos, P.T.; Pruthi, V.; Poluri, K.M.; Guarnieri, M.T. Leveraging algal omics to reveal potential targets for augmenting TAG accumulation. Biotechnol. Adv. 2018, 36, 1274–1292. [Google Scholar] [CrossRef]
- Zou, S.M.; Huang, Z.; Wu, X.M.; Yu, X.K. Physiological and Genetic Regulation for High Lipid Accumulation by Chlorella sorokiniana Strains from Different Environments of an Arctic Glacier, Desert, and Temperate Lake under Nitrogen Deprivation Conditions. Microbiol. Spectr. 2022, 10, e00394-22. [Google Scholar] [CrossRef]
- Ngan, C.Y.; Wong, C.H.; Choi, C.; Yoshinaga, Y.; Louie, K.; Jia, J.; Chen, C.; Bowen, B.; Cheng, H.Y.; Leonelli, L.; et al. Lineage-specific chromatin signatures reveal a regulator of lipid metabolism in microalgae. Nat. Plants 2015, 1, 15107. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Craig, R.J.; Gallaher, S.D.; Shu, S.; Salomé, P.; Jenkins, J.W.; Blaby-Haas, C.E.; Purvine, S.O.; O’Donnell, S.; Barry, K.; Grimwood, J.; et al. The Chlamydomonas Genome Project, version 6: Reference assemblies for mating type plus and minus strains reveal extensive structural mutation in the laboratory. bioRxiv 2022. [Google Scholar] [CrossRef]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.-C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Futschik, M.E.; Carlisle, B. NOISE-ROBUST SOFT CLUSTERING OF GENE EXPRESSION TIME-COURSE DATA. J. Bioinform. Comput. Biol. 2005, 3, 965–988. [Google Scholar] [CrossRef] [PubMed]
- Cardoso-Moreira, M.; Halbert, J.; Valloton, D.; Velten, B.; Chen, C.; Shao, Y.; Liechti, A.; Ascenção, K.; Rummel, C.; Ovchinnikova, S.; et al. Gene expression across mammalian organ development. Nature 2019, 571, 505–509. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [Green Version]
- The Gene Ontology Consortium. The Gene Ontology resource: Enriching a GOld mine. Nucleic Acids Res. 2021, 49, D325–D334. [Google Scholar] [CrossRef]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef]
- Goodstein, D.M.; Shu, S.; Howson, R.; Neupane, R.; Hayes, R.D.; Fazo, J.; Mitros, T.; Dirks, W.; Hellsten, U.; Putnam, N.; et al. Phytozome: A comparative platform for green plant genomics. Nucleic Acids Res. 2012, 40, D1178–D1186. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.Y.; Shim, D.; Kong, F.; Auroy, P.; Lee, Y.; Li-Beisson, Y.; Lee, Y.; Yamaoka, Y. The Chlamydomonas transcription factor MYB1 mediates lipid accumulation under nitrogen depletion. New Phytol. 2022, 235, 595–610. [Google Scholar] [CrossRef] [PubMed]
- Merchant, S.S.; Prochnik, S.E.; Vallon, O.; Harris, E.H.; Karpowicz, S.J.; Witman, G.B.; Terry, A.; Salamov, A.; Fritz-Laylin, L.K.; Maréchal-Drouard, L.; et al. The Chlamydomonas Genome Reveals the Evolution of Key Animal and Plant Functions. Science 2007, 318, 245–250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, J.; Wang, X.; Xu, Q.; Zhao, S.; Tai, Y.; Wei, C. Global dissection of alternative splicing uncovers transcriptional diversity in tissues and associates with the flavonoid pathway in tea plant (Camellia sinensis). BMC Plant Biol. 2018, 18, 266. [Google Scholar] [CrossRef]
- Iñiguez, L.P.; Ramírez, M.; Barbazuk, W.B.; Hernández, G. Identification and analysis of alternative splicing events in Phaseolus vulgaris and Glycine max. BMC Genom. 2017, 18, 650. [Google Scholar] [CrossRef] [Green Version]
- Filichkin, S.A.; Priest, H.D.; Givan, S.A.; Shen, R.; Bryant, D.W.; Fox, S.E.; Wong, W.-K.; Mockler, T.C. Genome-wide mapping of alternative splicing in Arabidopsis thaliana. Genome Res. 2009, 20, 45–58. [Google Scholar] [CrossRef] [Green Version]
- Li, G.; Xu, D.; Huang, G.; Bi, Q.; Yang, M.; Shen, H.; Liu, H. Analysis of Whole-Transcriptome RNA-Seq Data Reveals the Involvement of Alternative Splicing in the Drought Response of Glycyrrhiza uralensis. Front. Genet. 2022, 13, 885651. [Google Scholar] [CrossRef]
- Wahl, M.C.; Will, C.L.; Lührmann, R. The Spliceosome: Design Principles of a Dynamic RNP Machine. Cell 2009, 136, 701–718. [Google Scholar] [CrossRef] [Green Version]
- Staiger, D.; Brown, J.W. Alternative Splicing at the Intersection of Biological Timing, Development, and Stress Responses. Plant Cell 2013, 25, 3640–3656. [Google Scholar] [CrossRef] [Green Version]
- Tian, C.; Lin, X.; Saetan, W.; Huang, Y.; Shi, H.; Jiang, D.; Chen, H.; Deng, S.; Wu, T.; Zhang, Y.; et al. Transcriptome analysis of liver provides insight into metabolic and translation changes under hypoxia and reoxygenation stress in silver sillago (Sillago sihama). Comp. Biochem. Physiol. Part D Genom. Proteom. 2020, 36, 100715. [Google Scholar] [CrossRef]
- ter Beek, J.; Guskov, A.; Slotboom, D.J. Structural diversity of ABC transporters. J. Gen. Physiol. 2014, 143, 419–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.E.; Ryoo, G.; Lee, W. Alternative Splicing: Expanding Diversity in Major ABC and SLC Drug Transporters. AAPS J. 2017, 19, 1643–1655. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Zheng, Y.; Zhan, J.; He, C.; Wang, Q. Comparative metabolic profiling of the lipid-producing green microalga Chlorella reveals that nitrogen and carbon metabolic pathways contribute to lipid metabolism. Biotechnol. Biofuels 2017, 10, 153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hounslow, E.; Evans, C.A.; Pandhal, J.; Sydney, T.; Couto, N.; Pham, T.K.; Gilmour, D.J.; Wright, P.C. Quantitative proteomic comparison of salt stress in Chlamydomonas reinhardtii and the snow alga Chlamydomonas nivalis reveals mechanisms for salt-triggered fatty acid accumulation via reallocation of carbon resources. Biotechnol. Biofuels 2021, 14, 121. [Google Scholar] [CrossRef]
- Cai, Y.; Chen, H.; Tang, X.; Zhao, J.; Zhang, H.; Chen, Y.Q.; Chen, W. The relationship between amino acid and lipid metabolism in oleaginous eukaryotic microorganism. Appl. Microbiol. Biotechnol. 2022, 106, 3405–3417. [Google Scholar] [CrossRef]
- Lin, H.; Goodenough, U.W. Gametogenesis in the Chlamydomonas reinhardtii minus Mating Type Is Controlled by Two Genes, MID and MTD1. Genetics 2007, 176, 913–925. [Google Scholar] [CrossRef] [Green Version]
- Morton, M.; AlTamimi, N.; Butt, H.; Reddy, A.S.; Mahfouz, M. Serine/Arginine-rich protein family of splicing regulators: New approaches to study splice isoform functions. Plant Sci. 2019, 283, 127–134. [Google Scholar] [CrossRef]
- Mukherjee, N.; Corcoran, D.L.; Nusbaum, J.D.; Reid, D.W.; Georgiev, S.; Hafner, M.; Ascano, M.; Tuschl, T.; Ohler, U.; Keene, J.D. Integrative Regulatory Mapping Indicates that the RNA-Binding Protein HuR Couples Pre-mRNA Processing and mRNA Stability. Mol. Cell 2011, 43, 327–339. [Google Scholar] [CrossRef] [Green Version]
- Koo, K.M.; Jung, S.; Lee, B.S.; Kim, J.-B.; Jo, Y.D.; Choi, H.-I.; Kang, S.-Y.; Chung, G.-H.; Jeong, W.-J.; Ahn, J.-W. The Mechanism of Starch Over-Accumulation in Chlamydomonas reinhardtii High-Starch Mutants Identified by Comparative Transcriptome Analysis. Front. Microbiol. 2017, 8, 858. [Google Scholar] [CrossRef]
- Msanne, J.; Vu, H.S.; Cahoon, E.B. Acyl-acyl carrier protein pool dynamics with oil accumulation in nitrogen-deprived Chlamydomonas reinhardtii microalgal cells. J. Am. Oil Chem. Soc. 2021, 98, 1107–1112. [Google Scholar] [CrossRef]
- Liu, X.; Tian, Y.; Chi, W.; Zhang, H.; Yu, J.; Chen, G.; Wu, W.; Jiang, X.; Wang, S.; Lin, Z.; et al. Alternative splicing of OsGS1;1 affects nitrogen-use efficiency, grain development, and amylose content in rice. Plant J. 2022, 110, 1751–1762. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene_ID | Symbol | AS | Pfam_ID | Description | KEGG Pathway |
|---|---|---|---|---|---|
| Cre01.g005150_4532 | SGA1 | RI | PF00266 | Serine glyoxylate aminotransferase | Glyoxylate and dicarboxylate metabolism, Peroxisome |
| Cre01.g006950_4532 | FBA1 | A5SS | PF00274 | Fructose-1,6-bisphosphate aldolase | Glycolysis |
| Cre01.g042750_4532 | ACH1 | SE | PF00330, PF00694 | Aconitate hydratase | Glyoxylate and dicarboxylate metabolism, Citrate cycle |
| Cre02.g088250_4532 | MCT2 | A3SS | PF00698, PF12796, PF13857 | Malonyl-CoA:acyl-carrier-protein transacylase | Fatty acid biosynthesis |
| Cre02.g119000_4532 | PEX19 | RI | PF04614 | Putative peroxisome biogenesis protein | Peroxisome |
| Cre02.g141400_4532 | PCK1 | SE | PF01293 | Phosphoenolpyruvate carboxykinase | Citrate cycle, Glycolysis, Pyruvate metabolism |
| Cre02.g146050_4532 | ATO2 | SE | PF00108, PF00109, PF02803 | Acetyl-CoA acyltransferase | Glyoxylate and dicarboxylate metabolism, Pyruvate metabolism, Fatty acid degradation, Valine, leucine, and isoleucine degradation |
| Cre03.g144807_4532 | MAS1 | SE, MXE | PF01274 | Malate synthase | Glyoxylate and dicarboxylate metabolism, Pyruvate metabolism |
| Cre03.g149100_4532 | CIS2 | SE, MXE | PF00285 | Citrate synthase, glyoxysomal/microbody form | Glyoxylate and dicarboxylate metabolism, Citrate cycle |
| Cre03.g168700_4532 | NA | SE | PF13242 | Phosphoglycolate phosphatase/4-nitrophenylphosphatase | Glyoxylate and dicarboxylate metabolism |
| Cre03.g175400_4532 | PGI1 | A3SS,SE | PF00342 | Phosphoglucose isomerase 1 | Glycolysis |
| Cre03.g193850_4532 | SCLA1 | SE | PF00549, PF02629 | Succinyl-CoA ligase alpha chain | Citrate cycle |
| Cre03.g194850_4532 | MDH1 | A5SS | PF00056, PF02866 | NAD-dependent malate dehydrogenase 1, chloroplastic | Glyoxylate and dicarboxylate metabolism, Citrate cycle, Pyruvate metabolism |
| Cre04.g214500_4532 | IDH3 | SE | PF00180 | Isocitrate dehydrogenase, NADP-dependent | Citrate cycle, Peroxisome |
| Cre06.g254400_4532 | FUM1 | SE | PF05681, PF05683 | Fumarate hydratase 1 | Citrate cycle, Pyruvate metabolism |
| Cre06.g262900_4532 | PFK1 | SE | PF00365 | Phosphofructokinase | Glycolysis |
| Cre06.g263602_4532 | PEX12 | MXE | PF04757 | Peroxin 12 | Peroxisome |
| Cre06.g272050_4532 | GPM1 | A3SS | PF01676, PF06415 | Phosphoglycerate mutase 1 | Glycolysis |
| Cre07.g343700_4532 | OGD2 | SE, MXE | PF00198, PF00364 | Dihydrolipoamide succinyltransferase, oxoglutarate dehydrogenase E2 component | Citrate cycle |
| Cre07.g347100_4532 | NA | SE | PF01263 | Putative sugar epimerase | Glycolysis |
| Cre07.g353450_4532 | ACS3 | A3SS, SE, MXE | PF00501, PF13193, PF16177 | Acetyl-CoA synthetase/ligase | Glyoxylate and dicarboxylate metabolism, Glycolysis, Pyruvate metabolism |
| Cre11.g467723_4532 | KAS1 | SE | PF00109, PF02801 | 3-ketoacyl-ACP-synthase | Fatty acid biosynthesis |
| Cre12.g483950_4532 | MDH4 | SE | PF00056, PF02866 | Malate dehydrogenase 4 | Glyoxylate and dicarboxylate metabolism, Citrate cycle, Pyruvate metabolism |
| Cre12.g500150_4532 | ALDH2 | SE | PF00171 | Aldehyde dehydrogenase | Glycolysis, Pyruvate metabolism, Fatty acid degradation, Valine, leucine, and isoleucine degradation |
| Cre12.g507400_4532 | LCS3 | SE | PF00501 | Long-chain acyl-CoA synthetase | Peroxisome, Fatty acid degradation, Fatty acid biosynthesis |
| Cre12.g510650_4532 | FBP2 | RI | PF00316 | Fructose-1,6-bisphosphatase, chloroplastic | Glycolysis |
| Cre12.g513200_4532 | ENO1 | A5SS, SE | PF00113, PF03952 | Enolase | Glycolysis |
| Cre12.g530600_4532 | GLN3 | A5SS | PF00120, PF03951 | Glutamine synthetase | Glyoxylate and dicarboxylate metabolism, Arginine biosynthesis |
| Cre12.g537200_4532 | OGD1 | SE | PF00676, PF02779, PF16078, PF16870 | 2-oxoglutarate dehydrogenase, E1 subunit | Citrate cycle |
| Cre15.g637761_4532 | NA | MXE | PF00005, PF06472 | Peroxisomal long-chain acyl-CoA transporter, ABC superfamily | Peroxisome |
| Cre16.g664550_4532 | SHMT1 | SE | PF00464 | Serine hydroxymethyltransferase | Glyoxylate and dicarboxylate metabolism |
| Cre16.g679200_4532 | PEX1 | SE | PF00004 | Peroxisome biogenesis protein | Peroxisome |
| Cre16.g689050_4532 | ACX1 | SE | PF01756, PF02770, PF14749 | Acyl-CoA oxidase/dehydrogenase | Peroxisome, Fatty acid degradation |
| Cre16.g695050_4532 | ECH3 | SE | PF00725, PF02737, PF16113, PF00378 | Enoyl-CoA hydratase 1 | Fatty acid degradation |
| Cre16.g695100_4532 | NA | SE | PF00441, PF02770, PF02771 | Putative Acyl-CoA oxidase | Peroxisome, Fatty acid degradation |
| Cre17.g722150_4532 | PKS3 | SE | PF02797, PF08392 | Type III polyketide synthase | Fatty acid elongation |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, X.; Li, X.; Zhao, J.; Xie, M.; Li, X.; Jia, B.; Huang, Y. Transcriptome Analysis Reveals the Involvement of Alternative Splicing in the Nitrogen Starvation Response of Chlamydomonas reinhardtii. Processes 2022, 10, 2719. https://doi.org/10.3390/pr10122719
Yang X, Li X, Zhao J, Xie M, Li X, Jia B, Huang Y. Transcriptome Analysis Reveals the Involvement of Alternative Splicing in the Nitrogen Starvation Response of Chlamydomonas reinhardtii. Processes. 2022; 10(12):2719. https://doi.org/10.3390/pr10122719
Chicago/Turabian StyleYang, Xingcai, Xiangyu Li, Jialin Zhao, Mingshi Xie, Xinyi Li, Bin Jia, and Ying Huang. 2022. "Transcriptome Analysis Reveals the Involvement of Alternative Splicing in the Nitrogen Starvation Response of Chlamydomonas reinhardtii" Processes 10, no. 12: 2719. https://doi.org/10.3390/pr10122719