
Mathematical Models for Cholesterol Metabolism and Transport

Abstract
:1. Introduction
2. Overview of Cholesterol Metabolism and Transport
3. Mathematical Models of Cholesterol Synthesis and Metabolisms in Liver
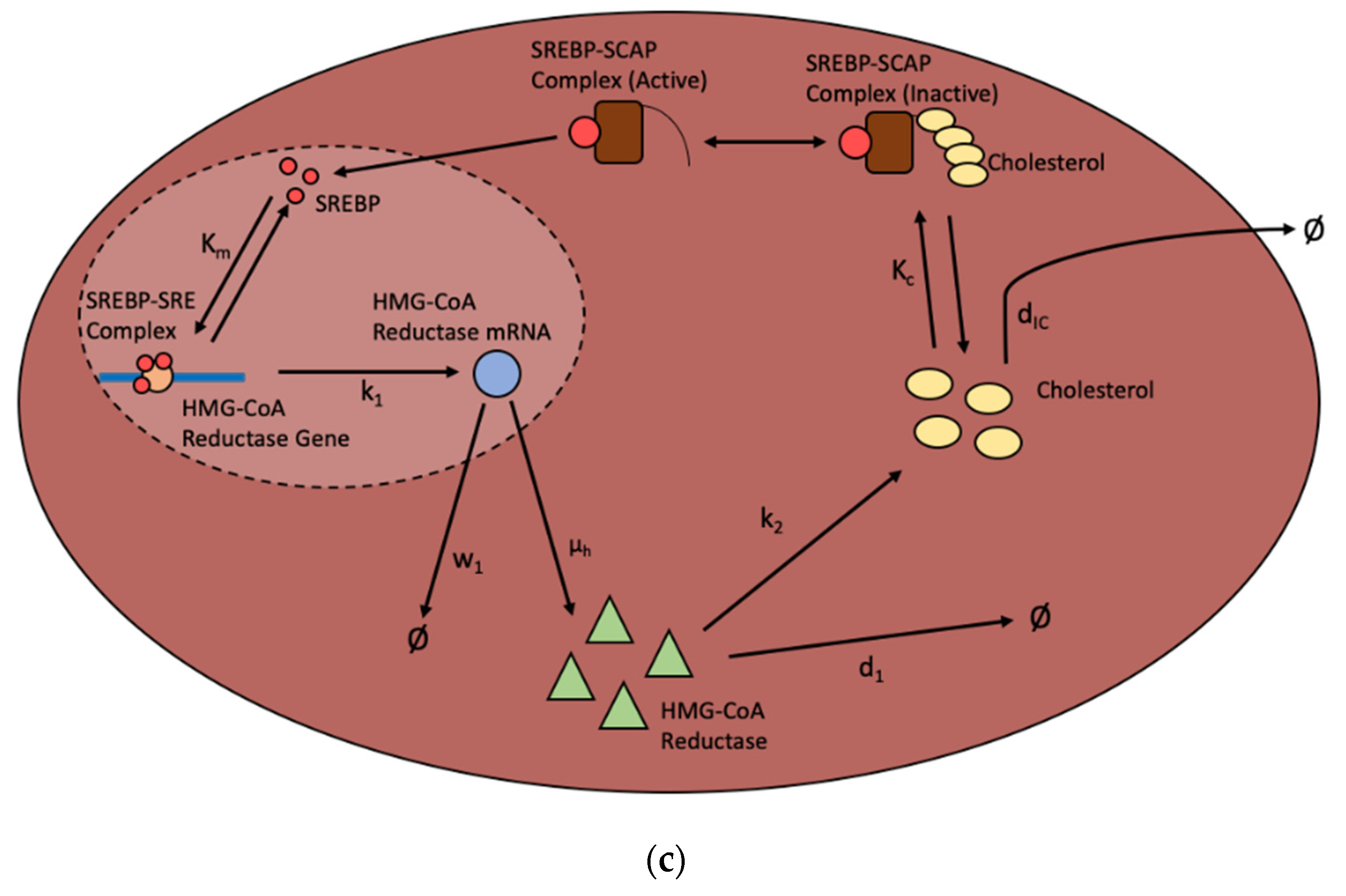
3.1. Cholesterol Metabolism with SREBP Negative Feedback
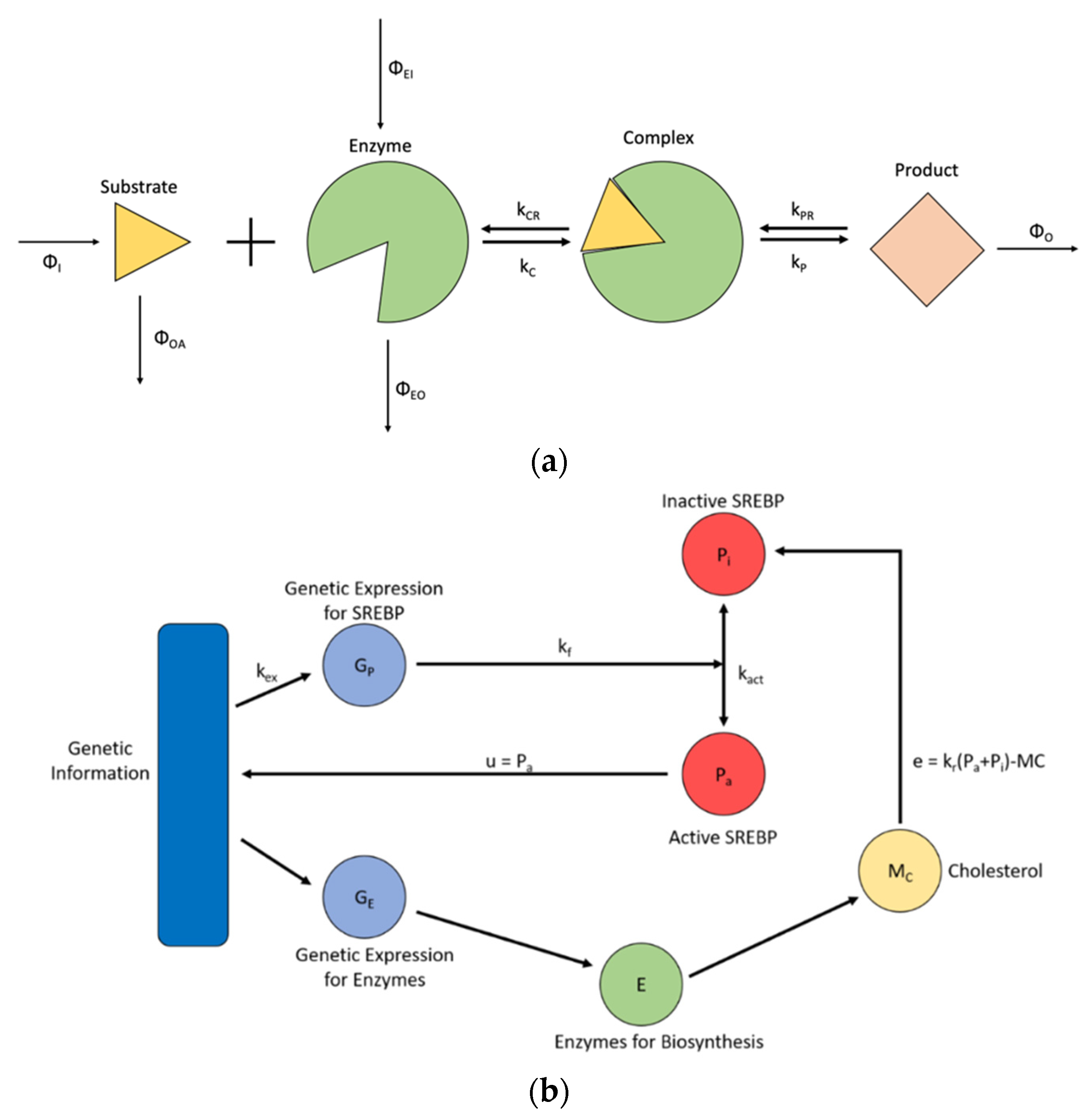
3.2. Steady State Cholesterol Biosynthesis
3.3. De Novo Cholesterol Biosynthesis
3.4. Hepatocyte Cellular Cholesterol Biosynthesis and Lipoprotein Metabolism
3.5. Boolean Modeling of Cholesterol Biosynthesis Regulation
3.6. Bile Acid Biosynthesis
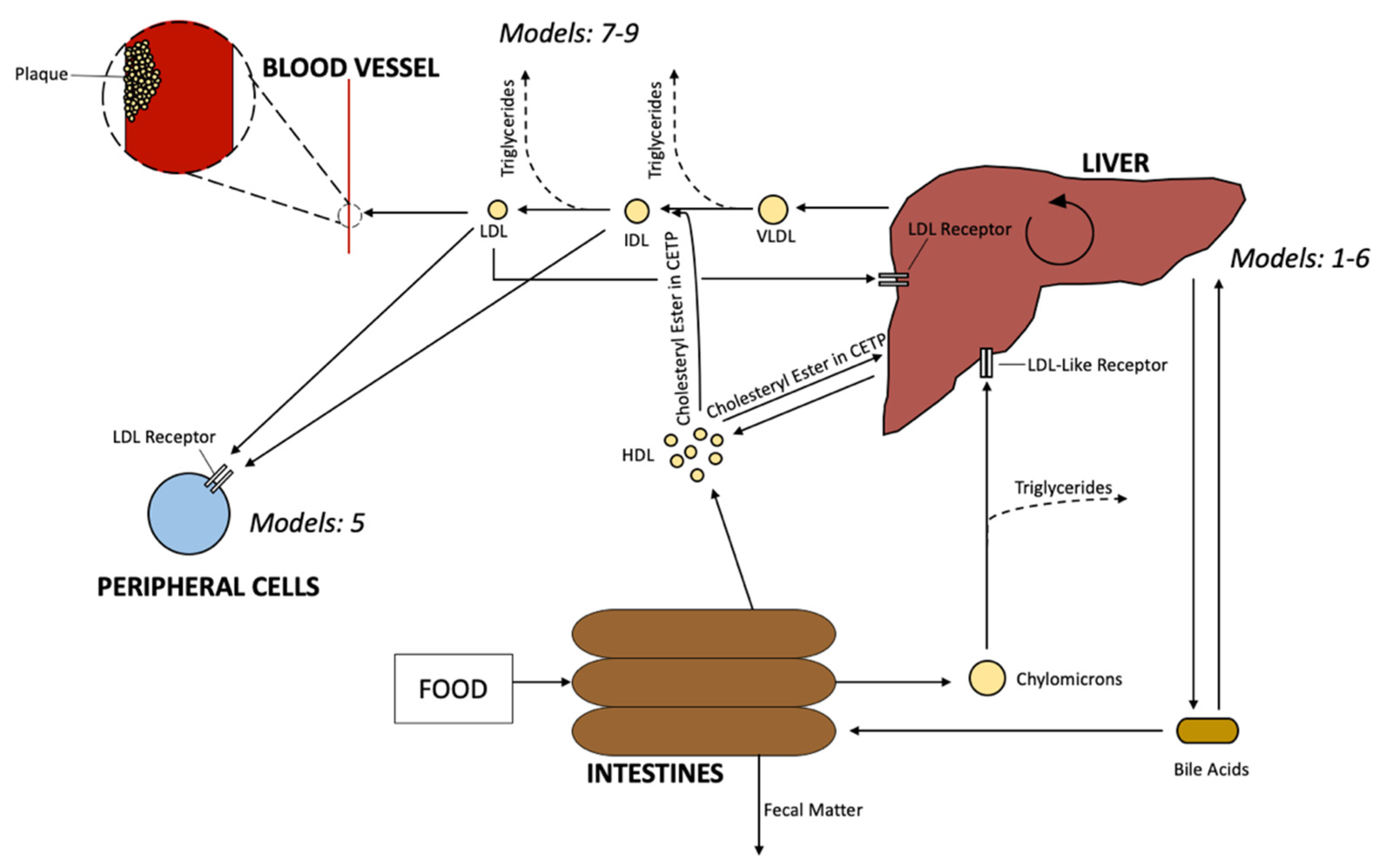
4. Mathematical Models of Cholesterol Transport and Metabolisms between Compartments in Human Body
4.1. Lipoprotein Transport and Metabolism
4.2. Blood Plasma Model
4.3. Two-Pool Model of Cholesterol Transport
4.4. Drug Delivery Model
5. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A
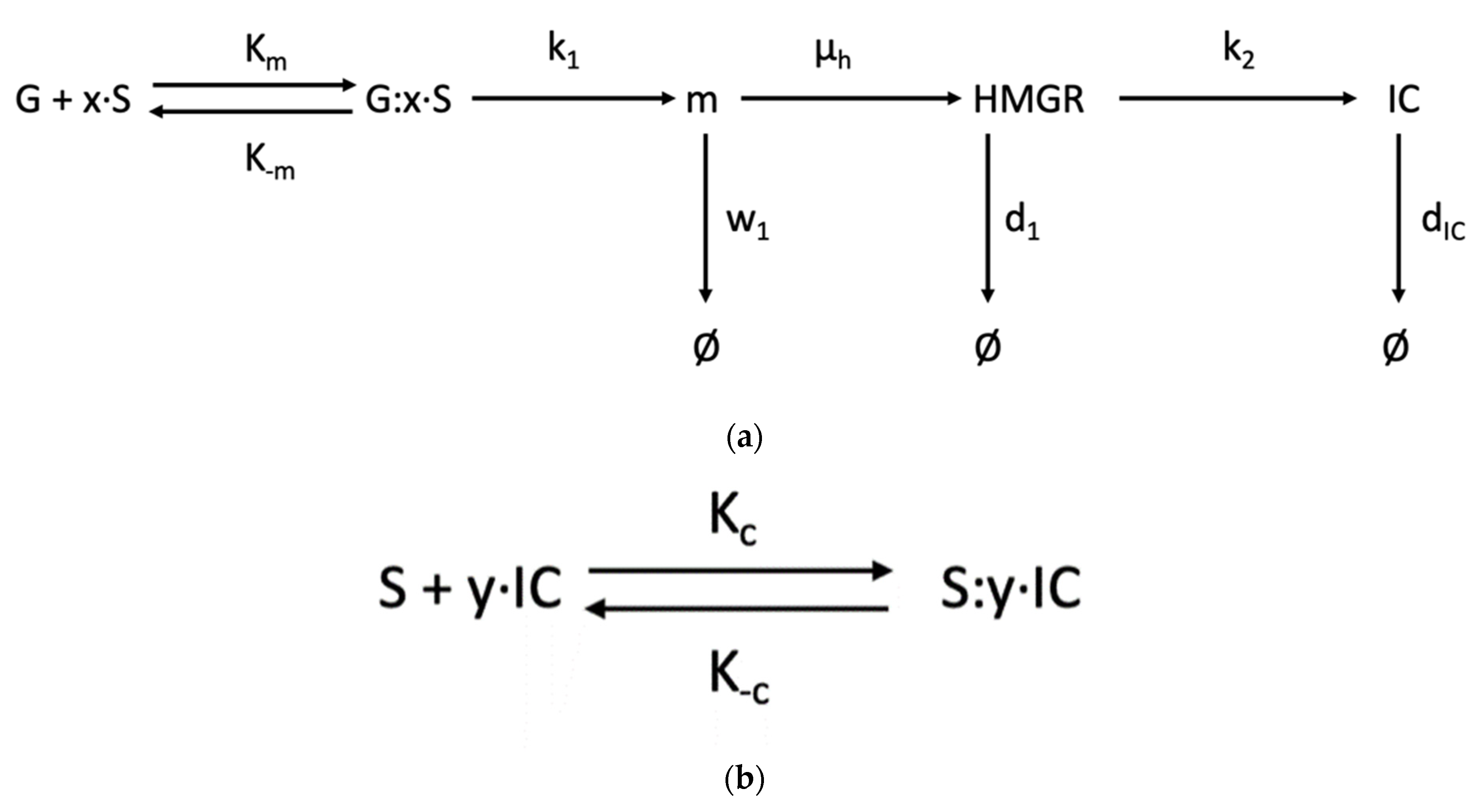
- Model 1. SREBP Regulation [35].
- d[m]/dt = change in mRNA concentration over time
- k1 = rate of HMG-CoA reductase gene transcription
- Km = equilibrium rate of SREBP-gene binding
- [IC] = concentration of intracellular cholesterol
- KC = equilibrium rate of SREBP-cholesterol binding
- S0 = initial concentration of SREBP
- y = required molecules of cholesterol to bind to SREBP
- x = required molecules of SREBP to bind to gene
- w1 = degradation rate of HMG-CoA reductase mRNA
- [m] = concentration of HMG-CoA reductase mRNA
- d[HMGR]/dt = change in HMG-CoA reductase concentration over time
- μh = rate of HMG-CoA reductase mRNA translation
- d1 = degradation rate of HMG-CoA reductase
- [HMGR] = concentration of HMG-CoA reductase
- d[IC]/dt = change in intracellular cholesterol concentration over time
- k2 = rate of HMG-CoA reductase converting to intracellular cholesterol
- dIC = degradation rate of intracellular cholesterol
- css = concentration of intracellular cholesterol at steady state
- Model 2. Steady State Cholesterol Biosynthesis [38].
- d[S]/dt = change in substrate concentration over time
- ΦI = influx of substrate
- ΦOA = outflux of substrate
- kC = forward binding rate of substrate to enzyme
- [E] = concentration of enzyme
- [S] = concentration of substrate
- kCR = reverse binding rate of substrate to enzyme
- [C] = concentration of substrate-enzyme complex
- dE/dt = change in enzyme concentration over time
- ΦEI = influx of enzyme
- kP = forward reaction and release rate of substrate to product
- kPR = reverse release rate of product and enzyme
- [P] = concentration of product
- ΦEO = outflux of enzyme
- d[IC]/dt = change in intracellular cholesterol concentration over time
- d[P]/dt = change in product concentration over time
- ΦO = outflux of product
- r = ratio of reaction rates
- EN* = new normalized steady state concentration of enzyme
- f* = ΦOAss*/ΦIss*
- f = ΦOAss/ΦIss
- SN* = new normalized steady state concentration of substrate
- PN* = new normalized steady state concentration of product
- u(s) = control signal as a function of signal frequency
- kact = rate SREBP becomes active
- s = frequency
- keG = elimination rate of SREBP mRNA
- keP = elimination rate of SREBP
- kf = rate of SREBP formation from gene expression
- kex = rate of SREBP gene expression
- Model 3. De Novo Cholesterol Biosynthesis [22].
- d[HMGR]/dt = change in HMG-CoA reductase over time
- k1 = rate of HMG-CoA reductase gene transcription
- b1 = attenuation coefficient for cholesterol control of HMG-CoA reductase
- [IC] = concentration intracellular cholesterol
- d1 = rate of HMG-CoA reductase degradation
- [HMGR] = concentration of HMG-CoA reductase
- d[IC]/dt = change in intracellular cholesterol over time
- k2 = rate of HMG-CoA conversion by HMG-CoA reductase
- [HMG-CoA] = concentration of HMG-CoA
- km1 = Michaelis–Menten constant
- dIC = rate of cholesterol use and degradation
- [m]* = steady state concentration of mRNA
- g1 = rate of transcription
- gm = rate of DNA degradation
- w1 = rate of mRNA degradation
- n = Hill cooperativity
- [HMGR]* = steady state concentration of HMG-CoA reductase
- Model 4. Cellular cholesterol biosynthesis and lipoprotein metabolism [48].
- J = scaling parameter
- μmh = transcription rate of HMGCR
- κmh = transcription rate of HMGCR by SREBP-2
- c = intracellular cholesterol
- xc = binding sites number for cholesterol to bind to SREBP-2
- xh = the number of SREBP binding sites on the HMGCR gene
- xr = number of binding sites on the LDLR gene SREBP-2 binds to
- δmh = HMGCR mRNA degradation rate
- mh = concentration of HMGCR mRNA
- mr = concentration of LDLR mRNA
- μmr = transcription rate of LDLR mRNA
- κmr = transcription rate of LDLR mRNA by SREBP-2
- δmr = receptor mRNA degradation rate
- mr = LDLR mRNA
- μh = synthesis rate of HMGCR
- μr = synthesis rate of LDLR mRNA
- μc = synthesis rate of cholesterol
- δh = HMGCR degradation rate
- h = intracellular HMGCR
- κc = SREBP-2 negatively regulation rate
- W = scaling parameter
- xυ = VLDL dilapidation rate to LDL
- αυ = VLDL binding rate on the cell surface
- α-υ = VLDL unbinding rate on the cell surface
- αL = LDL binding rate on the cell surface
- α-L = LDL unbinding rate on the cell surface
- βυ = VLDL occupied receptor endocytosis rate
- βL = LDL occupied receptor endocytosis rate
- β0 = empty receptor endocytosis rate
- γv = rate of cholesterol extraction from VLDL
- γL = rate of cholesterol extraction from LDL
- ƒ = rate of cholesterol receptor recycled from endosome
- γr = rate of cholesterol receptor recycled from intracellular space to cell surface
- lE = LDL in the media surrounding cell
- lRB = the LDL bound receptors
- lI = internalized LDL particles
- δc = the rate of esterification of LDL particles
- υE = concentration of extracellular VLDL
- ω = constant serum supply of VLDL
- P = empty receptors
- φ = degradation
- Model 5. Boolean Modeling of Cholesterol Biosynthesis Regulation [37].
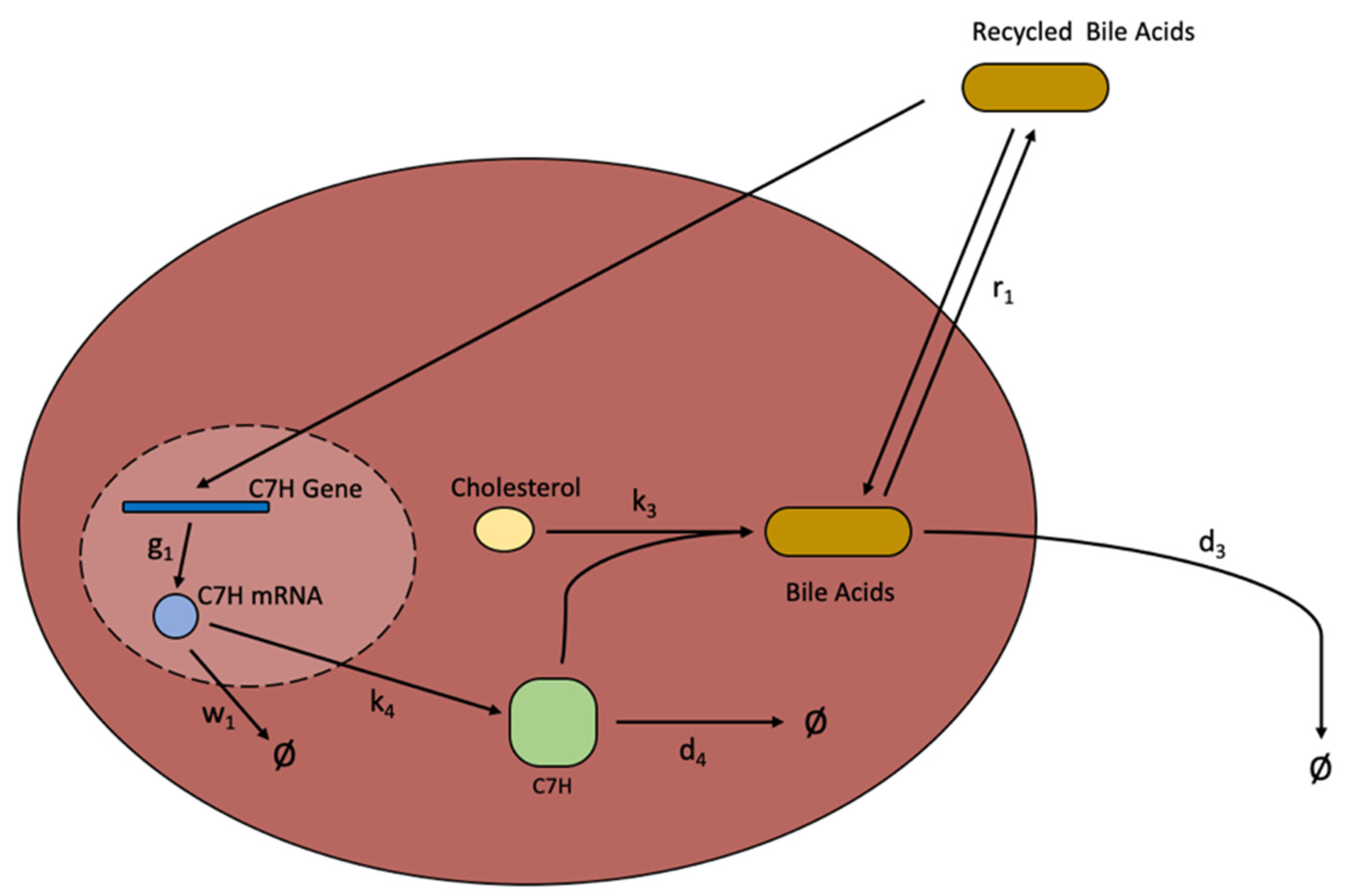
- Model 6. Bile Acid Biosynthesis [22]
- d[IC]/dt = change in intracellular cholesterol over time
- k3 = rate of cholesterol to bile acid conversion
- [C7H] = concentration of C7H
- [IC] = concentration of intracellular cholesterol
- km2 = C7H Michaelis–Menten constant
- d[BA]/dt = change in bile acid concentration over time
- d3 = rate of unrecycled bile acid degradation
- η = percent of bile acids that are not recycled
- [BA] = concentration of bile acids
- r1 = rate of bile acid recycling
- k5 = rate of recycled bile acids rejoining bile acid pool
- [RBA] = concentration of recycled bile acids
- d[C7H]/dt = change in C7H concentration over time
- k4 = rate of transcription
- b2 = regulation of C7H attenuation coefficient
- d4 = rate of C7H degradation
- [RBA]* = steady state concentration of recycled bile acids
- [C7H]* = steady state concentration of recycled bile acids
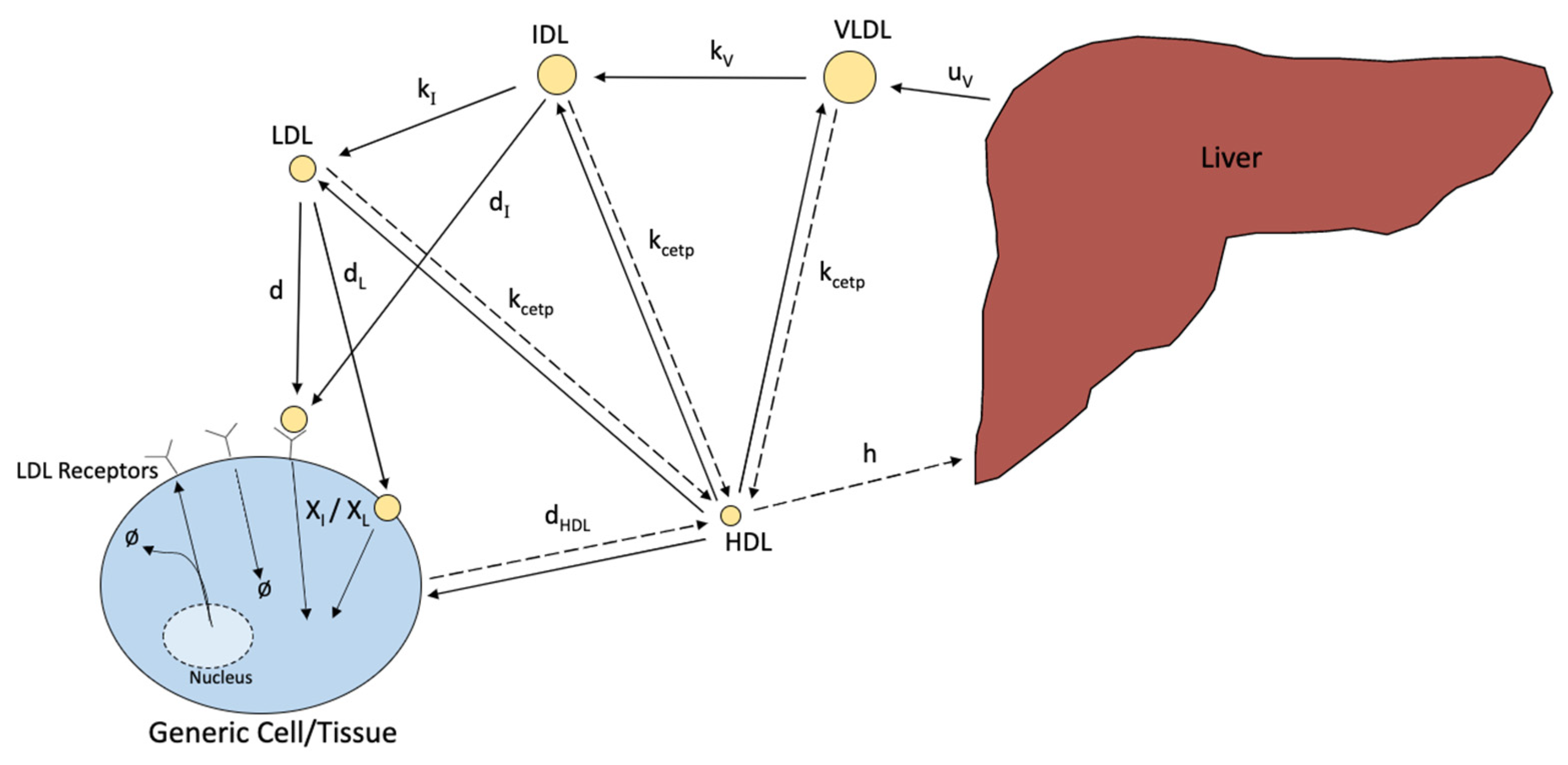
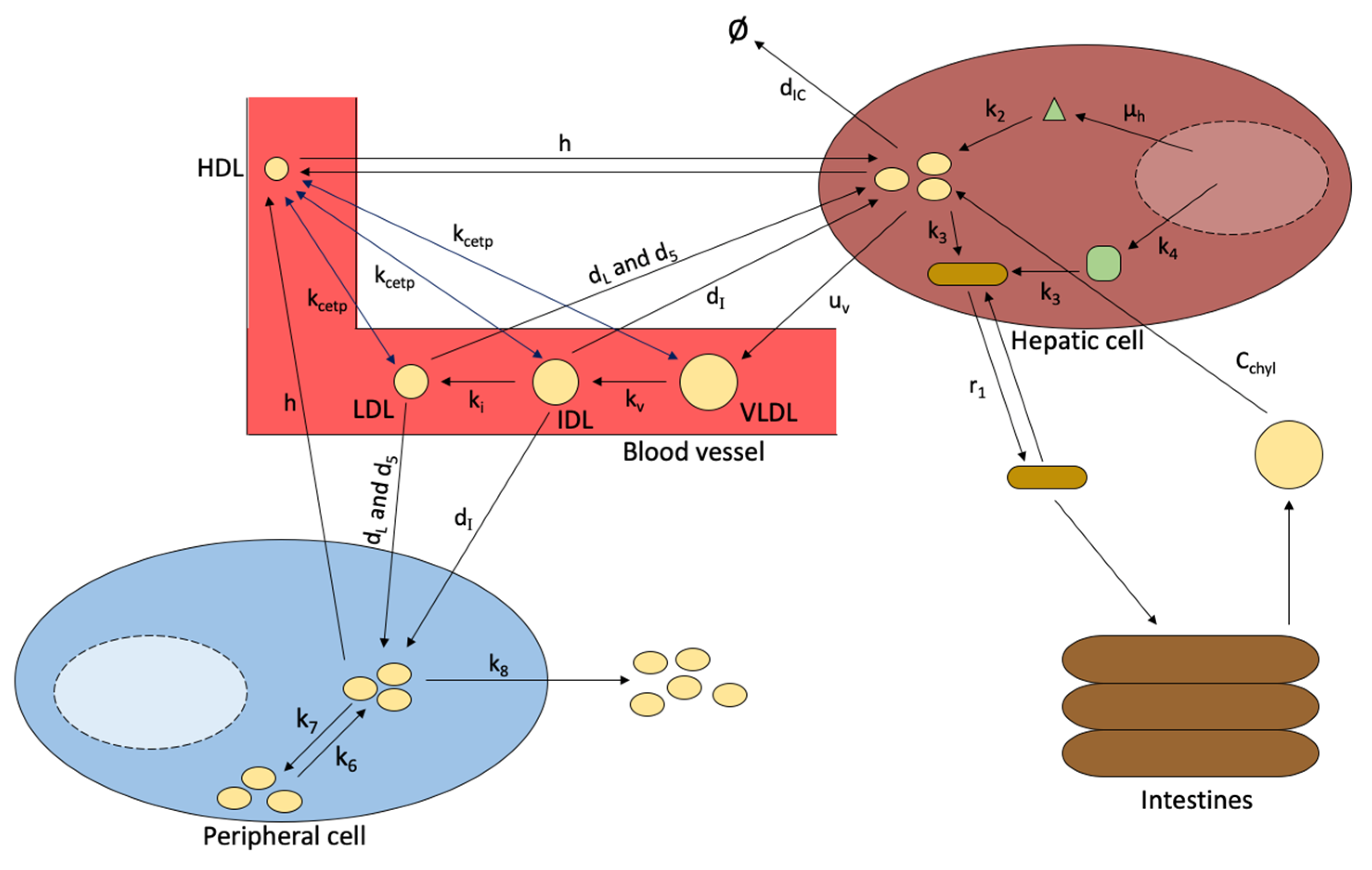
- Model 7. Lipoprotein transportation and Metabolism [22].
- d[V]/dt = change in VLDL cholesterol concentration over time
- uv = rate of VLDL production by the liver
- kv = conversion rate of VLDL to IDL
- [V] = concentration of VLDL cholesterol
- kcetp = rate of CETP transfer
- [H] = concentration of HDL cholesterol
- av = cholesterol gradient attenuation coefficient of HDL and VLDL equilibrium
- d[I]/dt = change in IDL cholesterol concentration over time
- ki = conversion rate of IDL to LDL
- [I] = concentration of IDL cholesterol
- ai = cholesterol gradient attenuation coefficient of HDL and IDL equilibrium
- dI = rate of internalization of IDL into a cell by receptors
- ΦLR = particles of cholesterol taken by receptors
- d[L]/dt = change in LDL cholesterol concentration over time
- dL = rate of internalization of LDL into a cell by receptors
- [L] = concentration of LDL cholesterol
- d5 = rate of internalization of LDL into a cell without the use of receptors
- al = cholesterol gradient attenuation coefficient of HDL and LDL equilibrium
- d[ΦLR]/dt = change in LDL receptors over time
- b = combined receptor internalization rate of LDL and IDL
- χI = fraction of cholesterol that makes up IDL
- χL = fraction of cholesterol that makes up LDL
- c = rate of receptor reuse
- [IC] = concentration of intracellular cholesterol
- d[IC]/dt = change in intracellular cholesterol concentration over time
- dHDL = rate cholesterol is transferred to HDL
- p = cholesterol transfer to HDL attenuation coefficient
- dIC = rate of cholesterol uses and degradation
- h = rate HDL is taken into the liver
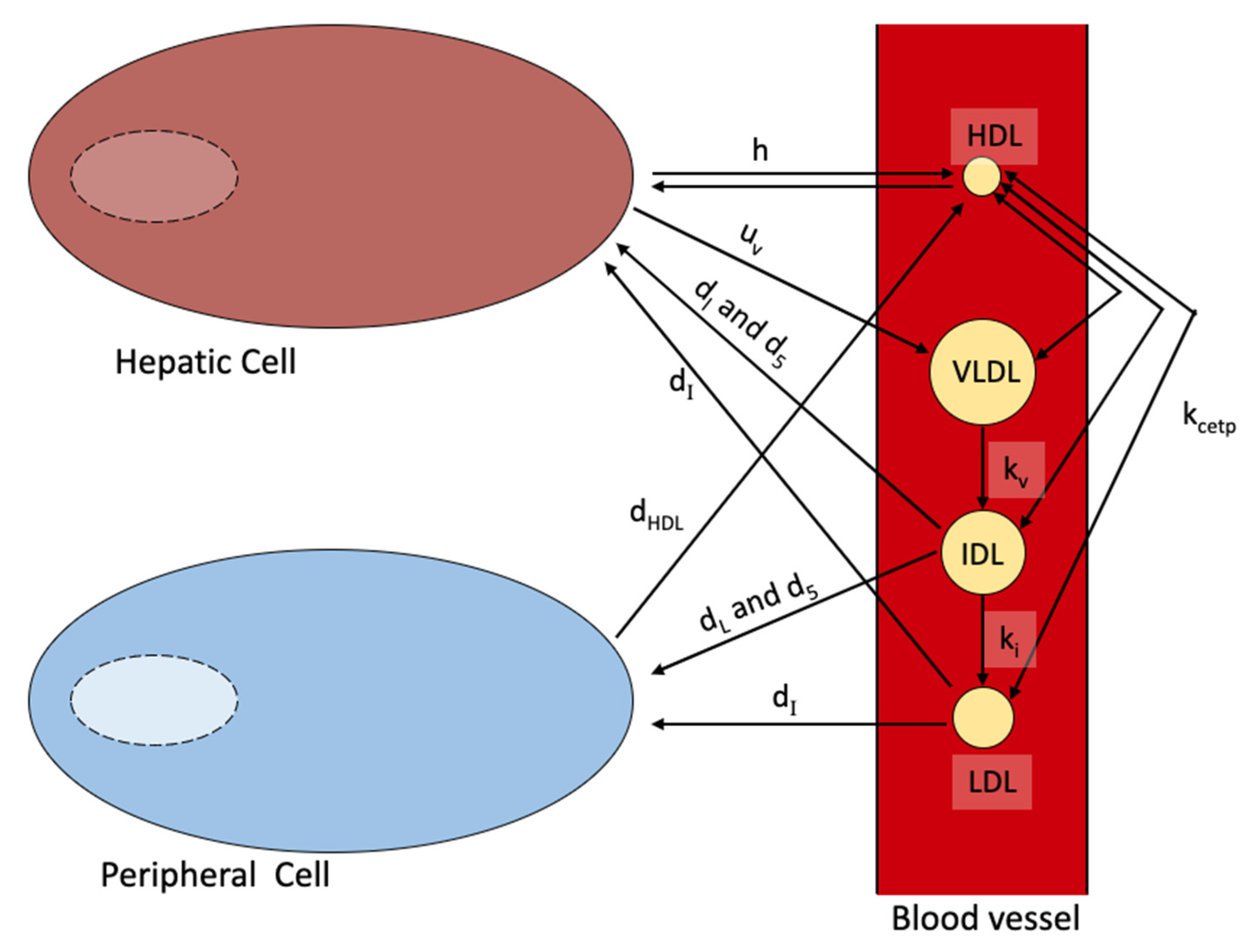
- Model 8. Blood Plasma Model [22].
- d[V]/dt = change in VLDL cholesterol concentration over time
- uv = rate VLDL is expelled by the liver
- [IC-H] = concentration of hepatic intracellular cholesterol
- kv = rate of conversion of VLDL to LDL
- [V] = concentration of VLDL cholesterol
- kcetp = rate of CETP transfer
- [H] = concentration of HDL cholesterol
- av = cholesterol gradient attenuation coefficient of HDL and VLDL equilibrium
- d[I]/dt = change in IDL cholesterol concentration over time
- ki = rate of conversion of IDL to LDL
- [I] = concentration of IDL cholesterol
- ai = cholesterol gradient attenuation coefficient of HDL and IDL equilibrium
- dI = rate of IDL internalization using receptors
- ΦHLR = particles of cholesterol taken by hepatic LDL receptors
- ΦPLR = particles of cholesterol taken by peripheral LDL receptors
- d[L]/dt = change in LDL cholesterol concentration over time
- d5 = rate of LDL internalization without the use of receptors
- [L] = concentration of LDL cholesterol
- dL = rate of LDL internalized using LDL receptors
- al = cholesterol gradient attenuation coefficient of HDL and LDL equilibrium
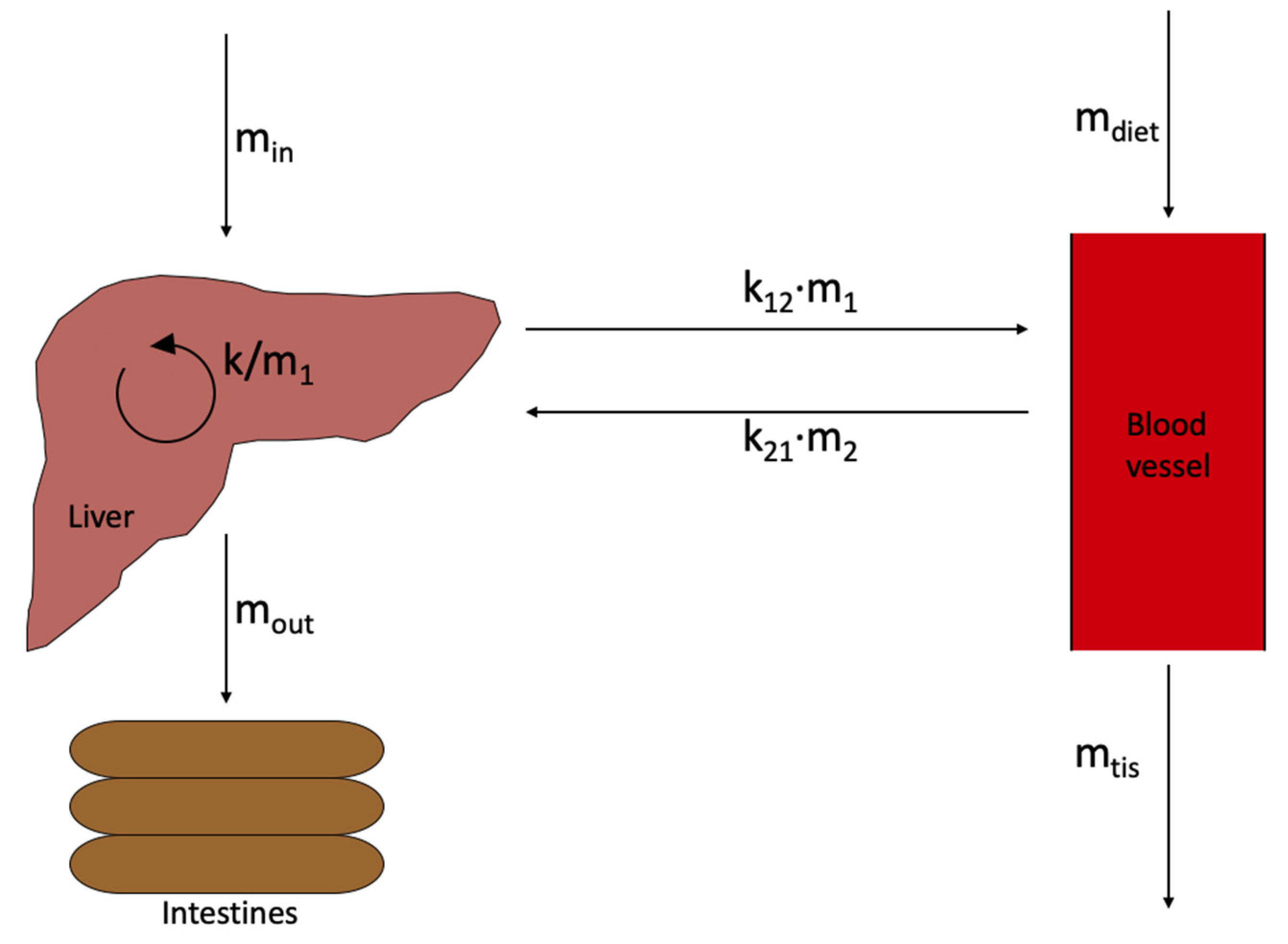
- dm1/dt = change in cholesterol amount in the liver over time
- k = rate of cholesterol synthesis in the liver
- k21 = rate of cholesterol moving from the bloodstream to the liver
- m2 = amount of cholesterol in the bloodstream
- k12 = rate of cholesterol moving from the liver to the bloodstream
- m1 = amount of cholesterol in the liver
- mout = amount of cholesterol used to make bile acids
- min = amount of cholesterol received from intestines
- dm2/dt = change in cholesterol amount in the bloodstream over time
- mtis = amount of cholesterol used for reactions and entering peripheral muscle/tissue
- mdiet = amount of cholesterol obtained from diet
- m1* = steady state cholesterol amount in the liver
- m2* = steady state cholesterol amount in the bloodstream
- C2* = steady state concentration of cholesterol in the bloodstream
- C1* = steady state concentration of cholesterol in the liver
- V1 = volume of blood in the liver
- V2 = volume of blood in the bloodstream
- Model 10. Modeling Drug Delivery [39].
- ji = flux of drug
- Dip = diffusion coefficient for drug through carrier
- dci/dx = concentration gradient
- ∂ci/∂t = change in concentration over time
- dMt/dt = amount of drug released over time
- K = partition coefficient of drug
- re = external radius of spherical carrier
- ri = internal radius of spherical carrier
- ci2 = drug concentration inside carrier
- ci2 = drug concentration outside of carrier
- Mt = amount of drug released at any given time
- A = surface area of carrier
- cs = drug solubility in carrier
- c0 = initial drug concentration in carrier
- t = time
- Mt/M∞ = amount of drug released at given time over drug released at infinite time
- k = structure/geometry constant
- n = 1 for zero order and 0.5 for Fickian diffusion
- ε = dimensionless parameter of erosion
- <x> = diffusion distance of water
- λ = rate of degradation
- Deff = diffusivity of water
- Mn = average molecular weight of polymer
- NA = Avogadro’s number
- N = degree of polymerization
- ρ = density of polymer
- A = cross sectional area of carrier
- δ = thickness of carrier
- Lp = coefficient of permeability
- σ = coefficient of reflection
- Δπs = pressure of water
- De = Deborah number
- λ = relaxation time
- θ = diffusion time
- vd,eq = equilibrium concentration of drug
- vd = concentration of drug
- L = half-thickness of carrier
- A = constant
- B = constant
References
- Jesch, E.D.; Carr, T.P. Food Ingredients That Inhibit Cholesterol Absorption. Prev. Nutr. Food Sci. 2017, 22, 67–80. [Google Scholar]
- Mouritsen, O.G.; Zuckermann, M.J. What’s so Special about Cholesterol? Lipids 2004, 39, 1101–1113. [Google Scholar] [CrossRef]
- Tabas, I. Cholesterol in Health and Disease. J. Clin. Investig. 2002, 110, 583–590. [Google Scholar] [CrossRef]
- Chimento, A.; Casaburi, I.; Avena, P.; Trotta, F.; De Luca, A.; Rago, V.; Pezzi, V.; Sirianni, R. Cholesterol and Its Metabolites in Tumor Growth: Therapeutic Potential of Statins in Cancer Treatment. Front. Endocrinol. 2019, 10, 807. [Google Scholar] [CrossRef] [Green Version]
- Schade, D.S.; Shey, L.; Eaton, R.P. Cholesterol Review: A Metabolically Important Molecule. Endocr. Pract. 2020, 26, 1514–1523. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.J.; Sharpe, L.J. Cholesterol Synthesis. In Biochemistry of Lipids, Lipoproteins and Membranes; Elsevier: Amsterdam, The Netherlands, 2016; pp. 327–358. [Google Scholar]
- de Boer, J.F.; Kuipers, F.; Groen, A.K. Cholesterol Transport Revisited: A New Turbo Mechanism to Drive Cholesterol Excretion. Trends Endocrinol. Metab. 2018, 29, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Page, I.H.; Allen, E.V.; Chamberlain, F.L.; Keys, A.; Stamler, J.; Stare, F.J. Dietary Fat and Its Relation to Heart Attacks and Strokes: Report by the Central Committee for Medical and Community Program of the American Heart Association. JAMA 1961, 175, 389–391. [Google Scholar]
- Virani, S.S.; Alonso, A.; Benjamin, E.J.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Delling, F.N.; et al. Heart Disease and Stroke Statistics—2020 Update: A Report From the American Heart Association. Circulation 2020, 141, E139–E596. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease Control and Prevention. High Cholesterol Facts. Available online: https://www.cdc.gov/cholesterol/facts.htm (accessed on 3 August 2021).
- Puglielli, L.; Tanzi, R.E.; Kovacs, D.M. Alzheimer’s Disease: The Cholesterol Connection. Nat. Neurosci. 2003, 6, 345–351. [Google Scholar] [CrossRef] [PubMed]
- Kuzu, O.F.; Noory, M.A.; Robertson, G.P. The Role of Cholesterol in Cancer. Cancer Res. 2016, 76, 2063–2070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, X.H.; Zhang, D.W.; Zheng, X.L.; Tang, C.K. Cholesterol Transport System: An Integrated Cholesterol Transport Model Involved in Atherosclerosis. Prog. Lipid Res. 2019, 73, 65–91. [Google Scholar] [CrossRef]
- Mc Auley, M.T.; Wilkinson, D.J.; Jones, J.J.L.; Kirkwood, T.B.L. A Whole-Body Mathematical Model of Cholesterol Metabolism and Its Age-Associated Dysregulation. BMC Syst. Biol. 2012, 6, 130. [Google Scholar] [CrossRef] [Green Version]
- Hurtado, P.J. Building New Models: Rethinking and Revising ODE Model Assumptions. In Foundations for Undergraduate Research in Mathematics; Callender Highlander, H., Capaldi, A., Diaz Eaton, C., Eds.; Springer International Publishing: Cham, Switzerland, 2020; pp. 1–86. [Google Scholar]
- Kuipers, B. Qualitative Reasoning: Modeling and Simulation with Incomplete Knowledge. Automatica 1989, 25, 571–585. [Google Scholar] [CrossRef]
- Parton, A.; McGilligan, V.; O’Kane, M.; Baldrick, F.R.; Watterson, S. Computational Modelling of Atherosclerosis. Brief. Bioinform. 2016, 17, 562–575. [Google Scholar] [CrossRef] [PubMed]
- Barnaba, C.; Rodríguez-Estrada, M.T.; Lercker, G.; García, H.S.; Medina-Meza, I.G. Cholesterol Photo-Oxidation: A Chemical Reaction Network for Kinetic Modeling. Steroids 2016, 116, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Weinrich, S.; Koch, S.; Bonk, F.; Popp, D.; Benndorf, D.; Klamt, S.; Centler, F. Augmenting Biogas Process Modeling by Resolving Intracellular Metabolic Activity. Front. Microbiol. 2019, 10, 1095. [Google Scholar] [CrossRef]
- Wang, E.; Casciano, C.N.; Clement, R.P.; Johnson, W. HMG-CoA Reductase Inhibitors (Statins) Characterized as Direct Inhibitors of P-Glycoprotein. Pharm. Res. 2001, 18, 800–806. [Google Scholar] [CrossRef] [PubMed]
- Young, S.G.; Fong, L.G. Lowering Plasma Cholesterol by Raising LDL—Revisited. N. Engl. J. Med. 2012, 366, 1154. [Google Scholar] [CrossRef] [Green Version]
- Sy, J.; Barahona, M. A Model of Cholesterol Metabolism and Transport. Bachelor’s Thesis, Imperial College, London, UK, 2008. [Google Scholar]
- Cartocci, V.; Servadio, M.; Trezza, V.; Pallottini, V. Can Cholesterol Metabolism Modulation Affect Brain Function and Behavior? J. Cell. Physiol. 2017, 232, 281–286. [Google Scholar] [CrossRef]
- Jin, U.; Park, S.J.; Park, S.M. Cholesterol Metabolism in the Brain and Its Association with Parkinson’s Disease. Exp. Neurobiol. 2019, 28, 554. [Google Scholar] [CrossRef] [Green Version]
- Huang, B.; Song, B.; Xu, C. Cholesterol Metabolism in Cancer: Mechanisms and Therapeutic Opportunities. Nat. Metab. 2020, 2, 132–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chambers, K.F.; Day, P.E.; Aboufarrag, H.T.; Kroon, P.A. Polyphenol Effects on Cholesterol Metabolism via Bile Acid Biosynthesis, CYP7A1: A Review. Nutrition 2019, 11, 2588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morgan, A.E.; Mooney, K.M.; Wilkinson, S.J.; Pickles, N.A.; Mc Auley, M.T. Cholesterol Metabolism: A Review of How Ageing Disrupts the Biological Mechanisms Responsible for Its Regulation. Ageing Res. Rev. 2016, 27, 108–124. [Google Scholar] [CrossRef]
- Millar, J.S.; Cuchel, M. Cholesterol Metabolism in Humans: A Review of Methods and Comparison of Results. Curr. Opin. Lipidol. 2018, 29, 1–9. [Google Scholar] [CrossRef]
- Avgerinos, N.A.; Neofytou, P. Mathematical Modelling and Simulation of Atherosclerosis Formation and Progress: A Review. Ann. Biomed. Eng. 2019, 47, 1764–1785. [Google Scholar] [CrossRef]
- Ren, L.; Yi, J.; Li, W.; Zheng, X.; Liu, J.; Wang, J.; Du, G. Apolipoproteins and Cancer. Cancer Med. 2019, 8, 7032–7043. [Google Scholar] [CrossRef] [Green Version]
- Morgan, A.E.; Auley, M.T.M. Cholesterol Homeostasis: An In Silico Investigation into How Aging Disrupts Its Key Hepatic Regulatory Mechanisms. Biology 2020, 9, 314. [Google Scholar] [CrossRef]
- Morgan, A.E.; Mooney, K.M.; Wilkinson, S.J.; Pickles, N.A.; Mc Auley, M.T. Mathematically Modelling the Dynamics of Cholesterol Metabolism and Ageing. Biosystems 2016, 145, 19–32. [Google Scholar]
- Sokolov, V.; Helmlinger, G.; Nilsson, C.; Zhudenkov, K.; Skrtic, S.; Hamrén, B.; Peskov, K.; Hurt-Camejo, E.; Jansson-Löfmark, R. Comparative Quantitative Systems Pharmacology Modeling of Anti-PCSK9 Therapeutic Modalities in Hypercholesterolemia. J. Lipid Res. 2019, 60, 1610–1621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toroghi, M.K.; Cluett, W.R.; Mahadevan, R. A Multi-Scale Model for Low-Density Lipoprotein Cholesterol (LDL-C) Regulation in the Human Body: Application to Quantitative Systems Pharmacology. Comput. Chem. Eng. 2019, 130, 106507. [Google Scholar] [CrossRef]
- Bhattacharya, B.S.; Sweby, P.K.; Minihane, A.M.; Jackson, K.G.; Tindall, M.J. A Mathematical Model of the Sterol Regulatory Element Binding Protein 2 Cholesterol Biosynthesis Pathway. J. Theor. Biol. 2014, 349, 150–162. [Google Scholar] [CrossRef]
- Hrydziuszko, O.; Balbus, J.; Zulpo, M.; Wrona, A.; Kubica, K. Mathematical Analyses of Two-Compartment Model of Human Cholesterol Circulatory Transport in Application to High Blood Cholesterol Prevention, Diagnosis and Treatment. Theor. Comput. Sci. 2015, 608, 98–107. [Google Scholar] [CrossRef]
- Kervizic, G.; Corcos, L. Dynamical Modeling of the Cholesterol Regulatory Pathway with Boolean Networks. BMC Syst. Biol. 2008, 2, 99. [Google Scholar] [CrossRef] [Green Version]
- Belič, A.; Ačimovič, J.; Naik, A.; Goličnik, M. Analysis of the Steady-State Relations and Control-Algorithm Characterisation in a Mathematical Model of Cholesterol Biosynthesis. Simul. Model. Pract. Theory 2013, 33, 18–27. [Google Scholar] [CrossRef]
- Peppas, N.A.; Narasimhan, B. Mathematical Models in Drug Delivery: How Modeling Has Shaped the Way We Design New Drug Delivery Systems. J. Control. Release 2014, 190, 75–81. [Google Scholar] [CrossRef]
- Wang, H.H.; Garruti, G.; Liu, M.; Portincasa, P.; Wang, D.Q.H. Cholesterol and Lipoprotein Metabolism and Atherosclerosis: Recent Advances in Reverse Cholesterol Transport. Ann. Hepatol. 2017, 16, S27–S42. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.-H.; Elvington, A.; Randolph, G.J. The Role of the Lymphatic System in Cholesterol Transport. Front. Pharmacol. 2015, 6, 182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janapala, U.S.; Reddivari, A.K.R. Low Cholesterol Diet. Med. Comput. J. 2021, 1, 21–23. [Google Scholar]
- Gray, C.; Stern, C. Treatment of Blood Cholesterol to Reduce Atheroscelerotic Cardiovascular Risk in Adults. J. Contemp. Pharm. Pract. 2019, 66, 54–63. [Google Scholar] [CrossRef]
- Mayes, P.A.; Bender, D.A. Overview of Metabolism. In Harper’s Illustrated Biochemistry; Murray, R.K., Granner, D.K., Davis, J.C., Mayes, P.A., Rodwell, V.W., Eds.; McGraw-Hill Companies: New York, NY, USA, 2003; pp. 123–126. [Google Scholar]
- von Eckardstein, A. LDL Contributes to Reverse Cholesterol Transport. Circ. Res. 2020, 793–795. [Google Scholar] [CrossRef]
- Lent-Schochet, D.; Jialal, I. Biochemistry, Lipoprotein Metabolism. StatPearls 2021. Available online: https://www.ncbi.nlm.nih.gov/books/NBK553193/ (accessed on 5 August 2021).
- Holmes, M.V.; Ala-Korpela, M. What Is ‘LDL Cholesterol’? Nat. Rev. Cardiol. 2019, 16, 197–198. [Google Scholar] [CrossRef]
- Pool, F.; Sweby, P.K.; Tindall, M.J. An Integrated Mathematical Model of Cellular Cholesterol Biosynthesis and Lipoprotein Metabolism. Processes 2018, 6, 134. [Google Scholar] [CrossRef] [Green Version]
- Hrydziuszko, O.; Wrona, A.; Balbus, J.; Kubica, K. Mathematical Two-Compartment Model of Human Cholesterol Transport in Application to High Blood Cholesterol Diagnosis and Treatment. Electron. Notes Theor. Comput. Sci. 2014, 306, 19–30. [Google Scholar] [CrossRef] [Green Version]
- Gluchowski, N.L.; Becuwe, M.; Walther, T.C.; Farese, R.V. Lipid Droplets and Liver Disease: From Basic Biology to Clinical Implications. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 343–355. [Google Scholar] [CrossRef]
- Horton, J.D.; Shimomura, I.; Brown, M.S.; Hammer, R.E.; Goldstein, J.L.; Shimano, H. Activation of Cholesterol Synthesis in Preference to Fatty Acid Synthesis in Liver and Adipose Tissue of Transgenic Mice Overproducing Sterol Regulatory Element-Binding Protein-2. J. Clin. Investig. 1998, 101, 2331–2339. [Google Scholar] [CrossRef]
- Wu, N.; Sarna, L.K.; Hwang, S.Y.; Zhu, Q.; Wang, P.; Siow, Y.L.; Karmin, O. Activation of 3-Hydroxy-3-Methylglutaryl Coenzyme A (HMG-CoA) Reductase during High Fat Diet Feeding. Biochim. Biophys. Acta-Mol. Basis Dis. 2013, 1832, 1560–1568. [Google Scholar] [CrossRef] [Green Version]
- Luo, J.; Yang, H.; Song, B.-L. Mechanisms and Regulation of Cholesterol Homeostasis. Nat. Rev. Mol. Cell Biol. 2019, 21, 225–245. [Google Scholar] [CrossRef]
- Turley, S.D.; Dietschy, J.M. The Intestinal Absorption of Biliary and Dietary Cholesterol as a Drug Target for Lowering the Plasma Cholesterol Level. Prev. Cardiol. 2003, 6, 29–33. [Google Scholar] [CrossRef]
- Petroglou, D.; Kanellos, I.; Savopoulos, C.; Kaiafa, G.; Chrysochoou, A.; Skantzis, P.; Daios, S.; Hatzitolios, A.I.; Giannoglou, G. The LDL-Receptor and Its Molecular Properties: From Theory to Novel Biochemical and Pharmacological Approaches in Reducing LDL-Cholesterol. Curr. Med. Chem. 2018, 27, 317–333. [Google Scholar] [CrossRef]
- Jakulj, L.; van Dijk, T.H.; de Boer, J.F.; Kootte, R.S.; Schonewille, M.; Paalvast, Y.; Boer, T.; Bloks, V.W.; Boverhof, R.; Nieuwdorp, M.; et al. Transintestinal Cholesterol Transport Is Active in Mice and Humans and Controls Ezetimibe-Induced Fecal Neutral Sterol Excretion. Cell Metab. 2016, 24, 783–794. [Google Scholar] [CrossRef] [Green Version]
- Bhatt, A.; Rohatgi, A. HDL Cholesterol Efflux Capacity: Cardiovascular Risk Factor and Potential Therapeutic Target. Curr. Atheroscler. Rep. 2016, 18, 2. [Google Scholar] [CrossRef]
- Chan, D.C.; Hoang, A.; Barrett, P.H.R.; Wong, A.T.Y.; Nestel, P.J.; Sviridov, D.; Watts, G.F. Apolipoprotein B-100 and ApoA-II Kinetics as Determinants of Cellular Cholesterol Efflux. J. Clin. Endocrinol. Metab. 2012, 97, E1658–E1666. [Google Scholar] [CrossRef] [Green Version]
- Sozen, E.; Ozer, N.K. Impact of High Cholesterol and Endoplasmic Reticulum Stress on Metabolic Diseases: An Updated Mini-Review. Redox Biol. 2017, 12, 456–461. [Google Scholar] [CrossRef]
- Brown, M.S.; Radhakrishnan, A.; Goldstein, J.L. Retrospective on Cholesterol Homeostasis: The Central Role of Scap. Annu. Rev. Biochem. 2018, 87, 783–807. [Google Scholar] [CrossRef] [Green Version]
- Tan, J.M.E.; Cook, E.C.L.; van den Berg, M.; Scheij, S.; Zelcer, N.; Loregger, A. Differential Use of E2 Ubiquitin Conjugating Enzymes for Regulated Degradation of the Rate-Limiting Enzymes HMGCR and SQLE in Cholesterol Biosynthesis. Atherosclerosis 2019, 281, 137–142. [Google Scholar] [CrossRef] [Green Version]
- Lu, X.-Y.; Shi, X.-J.; Hu, A.; Wang, J.-Q.; Ding, Y.; Jiang, W.; Sun, M.; Zhao, X.; Luo, J.; Qi, W.; et al. Feeding Induces Cholesterol Biosynthesis via the MTORC1–USP20–HMGCR Axis. Nature 2020, 588, 479–484. [Google Scholar] [CrossRef]
- Li, Q.; Zhang, H.; Zou, J.; Feng, X.; Feng, D. Bisphenol A Induces Cholesterol Biosynthesis in HepG2 Cells via SREBP-2/HMGCR Signaling Pathway. J. Toxicol. Sci. 2019, 44, 481–491. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Chen, S.; Cai, D.; Bian, D.; Wang, F. Long Noncoding RNA LncARSR Promotes Hepatic Cholesterol Biosynthesis via Modulating Akt/SREBP-2/HMGCR Pathway. Life Sci. 2018, 203, 48–53. [Google Scholar] [CrossRef]
- Ma, S.; Sun, W.; Gao, L.; Liu, S. Therapeutic Targets of Hypercholesterolemia: HMGCR and LDLR. Diabetes Metab. Syndr. Obes. Targets Ther. 2019, 12, 1543–1553. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.; Meng, M.; Feng, J.E. Observability of Boolean Networks via Matrix Equations. Automatica 2020, 111, 108621. [Google Scholar] [CrossRef]
- Brown, A.J.; Coates, H.W.; Sharpe, L.J. Cholesterol Synthesis. In Biochemistry of Lipids, Lipoproteins and Membranes; Ridgway, N.D., McLeod, R.S., Eds.; Elsevier: Amsterdam, The Netherlands, 2021; pp. 317–355. [Google Scholar]
- Eberlé, D.; Hegarty, B.; Bossard, P.; Ferré, P.; Foufelle, F. SREBP Transcription Factors: Master Regulators of Lipid Homeostasis. Biochimie 2004, 86, 839–848. [Google Scholar] [CrossRef]
- Bengoechea-Alonso, M.T.; Ericsson, J. SREBP in Signal Transduction: Cholesterol Metabolism and Beyond. Curr. Opin. Cell Biol. 2007, 19, 215–222. [Google Scholar] [CrossRef]
- Shefer, S.; Salen, G.; Batta, A.K.; Honda, A.; Tint, G.S.; Irons, M.; Elias, E.R.; Chen, T.C.; Holick, M.F. Markedly Inhibited 7-Dehydrocholesterol-Delta 7-Reductase Activity in Liver Microsomes from Smith-Lemli-Opitz Homozygotes. J. Clin. Investig. 1995, 96, 1779–1785. [Google Scholar] [CrossRef]
- Chiang, J.Y.L.; Li, T. Regulation of Bile Acid and Cholesterol Metabolism by PPARs. PPAR Res. 2009, 2009, 501739. [Google Scholar]
- Chiang, J.Y.L. Bile Acid Metabolism and Signaling in Liver Disease and Therapy. Liver Res. 2017, 1, 3–9. [Google Scholar] [CrossRef]
- Chiang, J.Y.L. Bile Acid Metabolism and Signaling. Compr. Physiol. 2013, 3, 1191. [Google Scholar]
- Xu, H.; Zhou, S.; Tang, Q.; Xia, H.; Bi, F. Cholesterol Metabolism: New Functions and Therapeutic Approaches in Cancer. Biochim. Biophys. Acta-Rev. Cancer 2020, 1874, 188394. [Google Scholar] [CrossRef]
- Ko, C.-W.; Qu, J.; Black, D.D.; Tso, P. Regulation of Intestinal Lipid Metabolism: Current Concepts and Relevance to Disease. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 169–183. [Google Scholar] [CrossRef]
- Iqbal, J.; Hussain, M.M. Intestinal Lipid Absorption. Am. J. Physiol. Endocrinol. Metab. 2009, 296, 1183–1194. [Google Scholar] [CrossRef] [Green Version]
- Mu, H.; Høy, C.E. The Digestion of Dietary Triacylglycerols. Prog. Lipid Res. 2004, 43, 105–133. [Google Scholar] [CrossRef]
- Trajkovska, K.T.; Topuzovska, S. High-Density Lipoprotein Metabolism and Reverse Cholesterol Transport: Strategies for Raising HDL Cholesterol. Anatol. J. Cardiol. 2017, 18, 149. [Google Scholar] [CrossRef]
- Jacobo-Albavera, L.; Domínguez-Pérez, M.; Medina-Leyte, D.J.; González-Garrido, A.; Villarreal-Molina, T. The Role of the ATP-Binding Cassette A1 (ABCA1) in Human Disease. Int. J. Mol. Sci. 2021, 22, 1593. [Google Scholar] [CrossRef]
- Wang, D.Q.-H.; Portincasa, P.; Tso, P. Transintestinal Cholesterol Excretion: A Secondary, Nonbiliary Pathway Contributing to Reverse Cholesterol Transport. Hepatology 2017, 66, 1337–1340. [Google Scholar] [CrossRef] [Green Version]
- Heeren, J.; Scheja, L. Metabolic-Associated Fatty Liver Disease and Lipoprotein Metabolism. Mol. Metab. 2021, 50, 101238. [Google Scholar] [CrossRef]
- Mittendorfer, B.; Patterson, B.W.; Klein, S. Effect of Sex and Obesity on Basal VLDL-Triacylglycerol Kinetics. Am. J. Clin. Nutr. 2003, 77, 573–579. [Google Scholar] [CrossRef]
- Bruinstroop, E.; Pei, L.; Ackermans, M.T.; Foppen, E.; Borgers, A.J.; Kwakkel, J.; Alkemade, A.; Fliers, E.; Kalsbeek, A. Hypothalamic Neuropeptide Y (NPY) Controls Hepatic VLDL-Triglyceride Secretion in Rats via the Sympathetic Nervous System. Diabetes 2012, 61, 1043–1050. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.H.; Yeo, Y. Controlled Drug Release from Pharmaceutical Nanocarriers. Chem. Eng. Sci. 2015, 125, 75–84. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Schnell, S.K.; Simon, J.-M.; Bedeaux, D.; Kjelstrup, S.; Bardow, A.; Vlugt, T.J.H. Fick Diffusion Coefficients of Liquid Mixtures Directly Obtained From Equilibrium Molecular Dynamics. J. Phys. Chem. B 2011, 115, 12921–12929. [Google Scholar] [CrossRef]
- Andrade, C.; Climent, M.A.; de Vera, G. Procedure for Calculating the Chloride Diffusion Coefficient and Surface Concentration from a Profile Having a Maximum beyond the Concrete Surface. Mater. Struct. 2015, 48, 863–869. [Google Scholar] [CrossRef]
- Maroni, A.; Zema, L.; Cerea, M.; Foppoli, A.; Palugan, L.; Gazzaniga, A. Erodible Drug Delivery Systems for Time-Controlled Release into the Gastrointestinal Tract. J. Drug Deliv. Sci. Technol. 2016, 32, 229–235. [Google Scholar] [CrossRef]
- Iñiguez-Franco, F.; Auras, R.; Ahmed, J.; Selke, S.; Rubino, M.; Dolan, K.; Soto-Valdez, H. Control of Hydrolytic Degradation of Poly(Lactic Acid) by Incorporation of Chain Extender: From Bulk to Surface Erosion. Polym. Test. 2018, 67, 190–196. [Google Scholar] [CrossRef]
- Kim, M.; Cha, C. Modulation of Functional Pendant Chains within Poly(ethylene glycol) Hydrogels for Refined Control of Protein Release. Sci. Rep. 2018, 8, 4315. [Google Scholar] [CrossRef] [Green Version]
- Laracuente, M.L.; Yu, M.H.; McHugh, K.J. Zero-Order Drug Delivery: State of the Art and Future Prospects. J. Control. Release 2020, 327, 834–856. [Google Scholar] [CrossRef]
- Carbinatto, F.M.; de Castro, A.D.; Evangelista, R.C.; Cury, B.S.F. Insights into the Swelling Process and Drug Release Mechanisms from Cross-Linked Pectin/High Amylose Starch Matrices. Asian J. Pharm. Sci. 2014, 9, 27–34. [Google Scholar] [CrossRef] [Green Version]
- Strachota, B.; Strachota, A.; Šlouf, M.; Brus, J.; Cimrová, V. Monolithic Intercalated PNIPAm/Starch Hydrogels with Very Fast and Extensive One-Way Volume and Swelling Responses to Temperature and PH: Prospective Actuators and Drug Release Systems. Soft Matter 2019, 15, 752–769. [Google Scholar] [CrossRef]
- Brazel, C.S.; Peppas, N.A. Modeling of Drug Release from Swellable Polymers. Eur. J. Pharm. Biopharm. 2000, 49, 47–58. [Google Scholar] [CrossRef]
- Salehi, A.; Zhao, J.; Cabelka, T.D.; Larson, R.G. A Unified Multicomponent Stress-Diffusion Model of Drug Release from Non-Biodegradable Polymeric Matrix Tablets. J. Control. Release 2016, 224, 43–58. [Google Scholar] [CrossRef]
- Ferrero, C.; Massuelle, D.; Doelker, E. Towards Elucidation of the Drug Release Mechanism from Compressed Hydrophilic Matrices Made of Cellulose Ethers. II. Evaluation of a Possible Swelling-Controlled Drug Release Mechanism Using Dimensionless Analysis. J. Control. Release 2010, 141, 223–233. [Google Scholar] [CrossRef]
- Liu, Z.; Wang, L.; Bao, C.; Li, X.; Cao, L.; Dai, K.; Zhu, L. Cross-Linked PEG via Degradable Phosphate Ester Bond: Synthesis, Water-Swelling, and Application as Drug Carrier. Biomacromolecules 2011, 12, 2389–2395. [Google Scholar] [CrossRef]
- Rahilly-Tierney, C.R.; Spiro, A.; Vokonas, P.; Gaziano, J.M. Relation Between High-Density Lipoprotein Cholesterol and Survival to Age 85 Years in Men (from the VA Normative Aging Study). Am. J. Cardiol. 2011, 107, 1173–1177. [Google Scholar] [CrossRef]
- Sahebkar, A. Effects of Resveratrol Supplementation on Plasma Lipids: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. Nutr. Rev. 2013, 71, 822–835. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model Number | Model | Inputs | Outputs | Known Parameters |
|---|---|---|---|---|
| 1 | SREBP Regulation [35] | Concentration of cholesterol, concentration of SREBP | Concentration of HMG-CoA reductase mRNA over time, concentration of HMG-CoA reductase over time, concentration of cholesterol over time, steady state concentration of cholesterol | Binding rates, rates of formation, rates of degradation, required molecules for successful binding |
| 2 | Steady State Cholesterol Biosynthesis [38] | Initial concentrations of enzyme and substrate | Changes in substrate, enzyme, complex, and product over time, normalized steady state enzyme and substrate concentration, control signal | In and out fluxes of enzyme and substrate, reaction and binding rates, frequency |
| 3 | De Novo Cholesterol Biosynthesis [22] | Initial intracellular cholesterol and HMG-CoA concentrations | Change in HMG-CoA reductase and intracellular cholesterol concentrations over time, steady state mRNA and HMG-CoA reductase concentrations | Rates of transcription, translation, conversion, and degradation, other constants |
| 4 | Cellular cholesterol biosynthesis and lipoprotein metabolism [48] | Initial cholesterol level | VLDL, LDL levels, VLDL, LDL that bound to receptors, and transcription rate of cholesterol receptors | Rates of transcription, translation, binding affinity, dissociation rate, rate of internalization, rate of cholesterol conversion, rate of receptor recycling |
| 5 | Boolean Modeling of Cholesterol Biosynthesis Regulation [37] | Acetoacetyl-CoA, acetyl-CoA, HMG-CoA synthase, desmosterol, 7-dehydrocholesterol, HMG-CoA reductase mRNA | HMG-CoA, HMG-CoA reductase, Cholesterol | None |
| 6 | Bile Acid Biosynthesis [22] | Initial C7H, intracellular cholesterol, bile acid, and returned bile acid concentrations | Change in intracellular cholesterol, C7H, and bile acids over time, steady state recycled bile acid and C7H concentrations | Rates of transcription, conversion, and degradation, other constants |
| 7 | Lipoprotein transportation and metabolism [22] | Inputs from lipoprotein metabolism, De Novo cholesterol biosynthesis, and bile acid biosynthesis. Initial lipoprotein cholesterol concentrations, amount of cholesterol particles taken by LDL receptors | Change in intracellular cholesterol, bile acids, and LDL receptors over time in liver cells, change in intracellular cholesterol and LDL receptors over time in peripheral cells. Change in VLDL, IDL, LDL, HDL, receptor, and intracellular cholesterol concentrations over time | Parameters from lipoprotein metabolism, De Novo cholesterol biosynthesis, and bile acid biosynthesis. Rates of transport, conversion, receptor reuse and internalization, attenuation coefficients, cholesterol fractions |
| 8 | Blood Plasma Model [22] | Initial concentrations of HDL, IDL, LDL, and VLDL cholesterol, particles of cholesterol taken by liver and peripheral cell LDL receptors | Change in VLDL, IDL, LDL, and HDL cholesterol over time | Rates of cholesterol transfer, rates of cholesterol internalization, attenuation coefficients |
| 9 | Two Pool Model of Cholesterol Transport [36,49] | Initial cholesterol concentrations in each pool, initial cholesterol amount in each pool, volume of blood in each pool | Change in cholesterol amount in each pool, steady state cholesterol amount in each pool, steady state cholesterol concentration in each pool | Rates of cholesterol transfer, rate of cholesterol synthesis, amounts of cholesterol in each pool, volume of blood in each pool, amount of cholesterol transferred from intestines, to bile, and to peripheral cells |
| 10 | Modeling Drug Delivery [39] | Dimensions of drug carrier, diffusion coefficients, initial concentrations and solubility, time | Drug release rates, cumulative amount of drug released, erosion coefficient | Equation coefficients and rates |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, F.; Macshane, B.; Searcy, R.; Huang, Z. Mathematical Models for Cholesterol Metabolism and Transport. Processes 2022, 10, 155. https://doi.org/10.3390/pr10010155
Zhang F, Macshane B, Searcy R, Huang Z. Mathematical Models for Cholesterol Metabolism and Transport. Processes. 2022; 10(1):155. https://doi.org/10.3390/pr10010155
Chicago/Turabian StyleZhang, Fangyuan, Brittany Macshane, Ryan Searcy, and Zuyi Huang. 2022. "Mathematical Models for Cholesterol Metabolism and Transport" Processes 10, no. 1: 155. https://doi.org/10.3390/pr10010155