
Gas Chromatography Multiresidue Method for Enantiomeric Fraction Determination of Psychoactive Substances in Effluents and River Surface Waters



Abstract
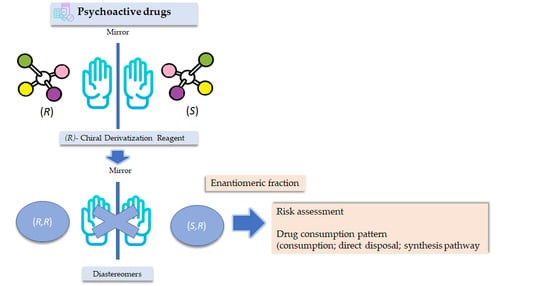
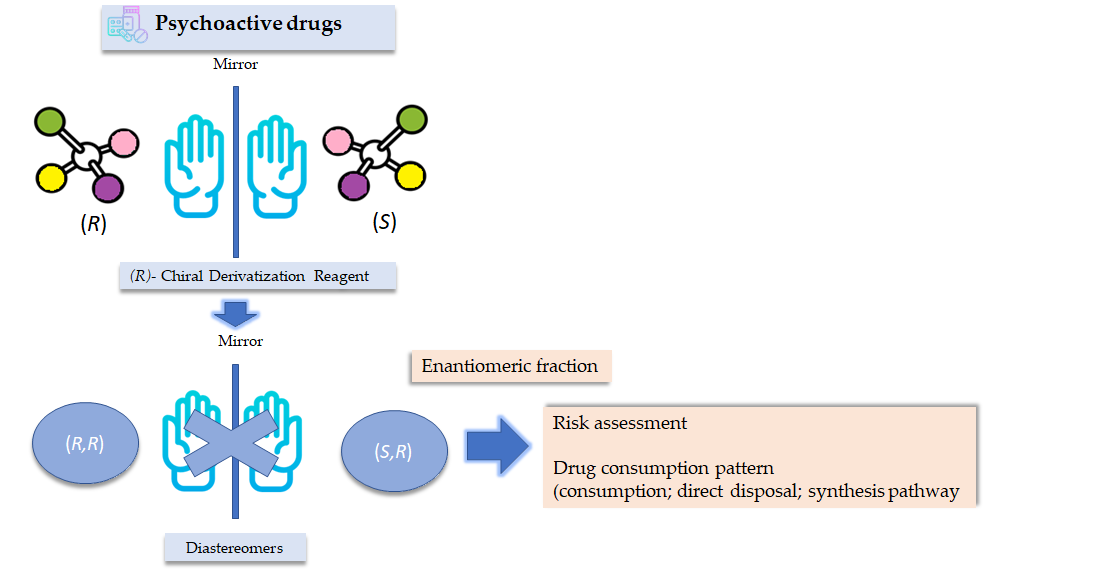
:
1. Introduction
2. Materials and Methods
2.1. Chemicals and Materials
2.2. Equipment
2.2.1. Chromatographic System
2.2.2. Other Equipment
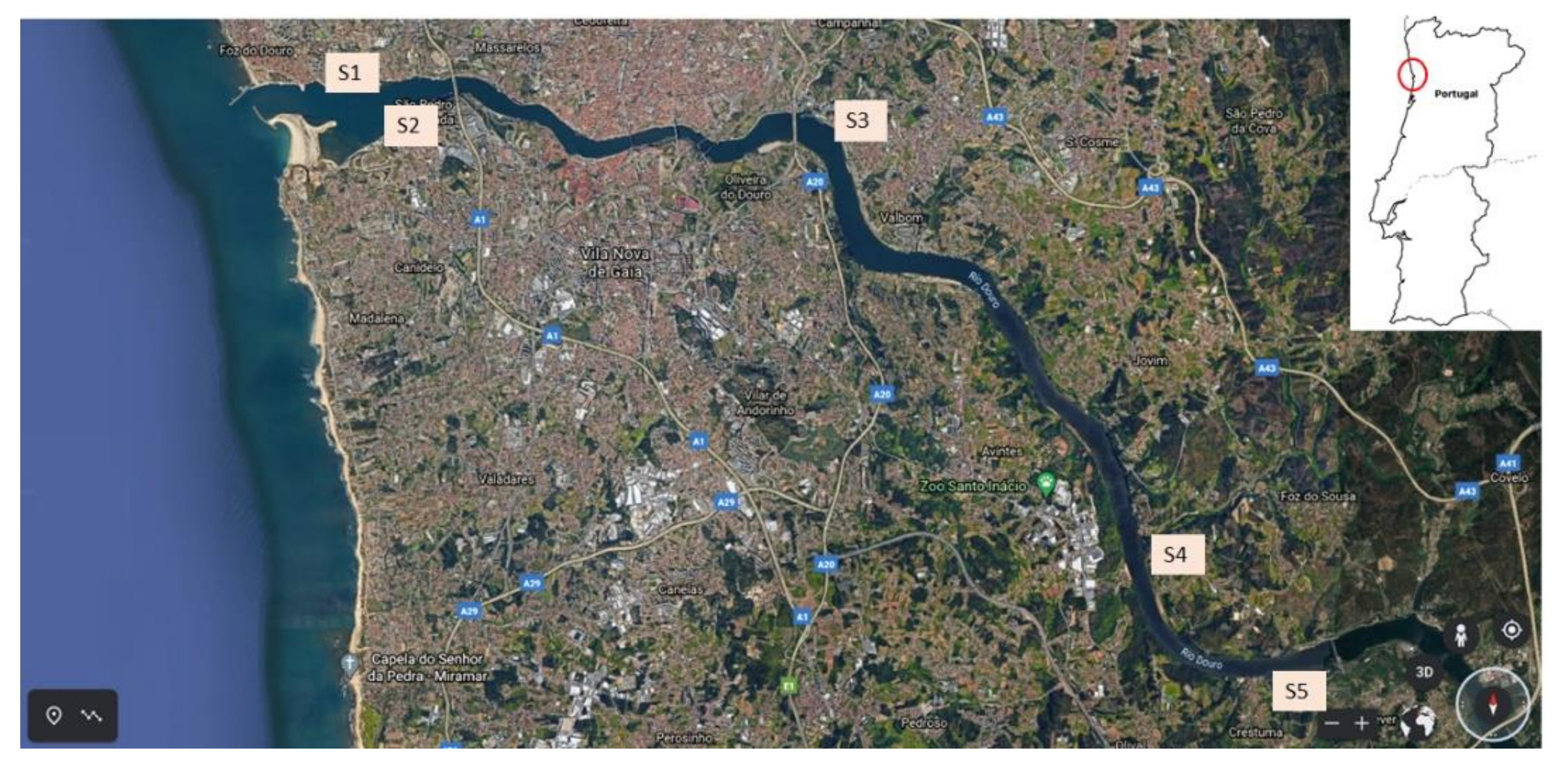
2.3. Sample Collection
2.4. Solid-Phase Extraction (SPE) Procedure
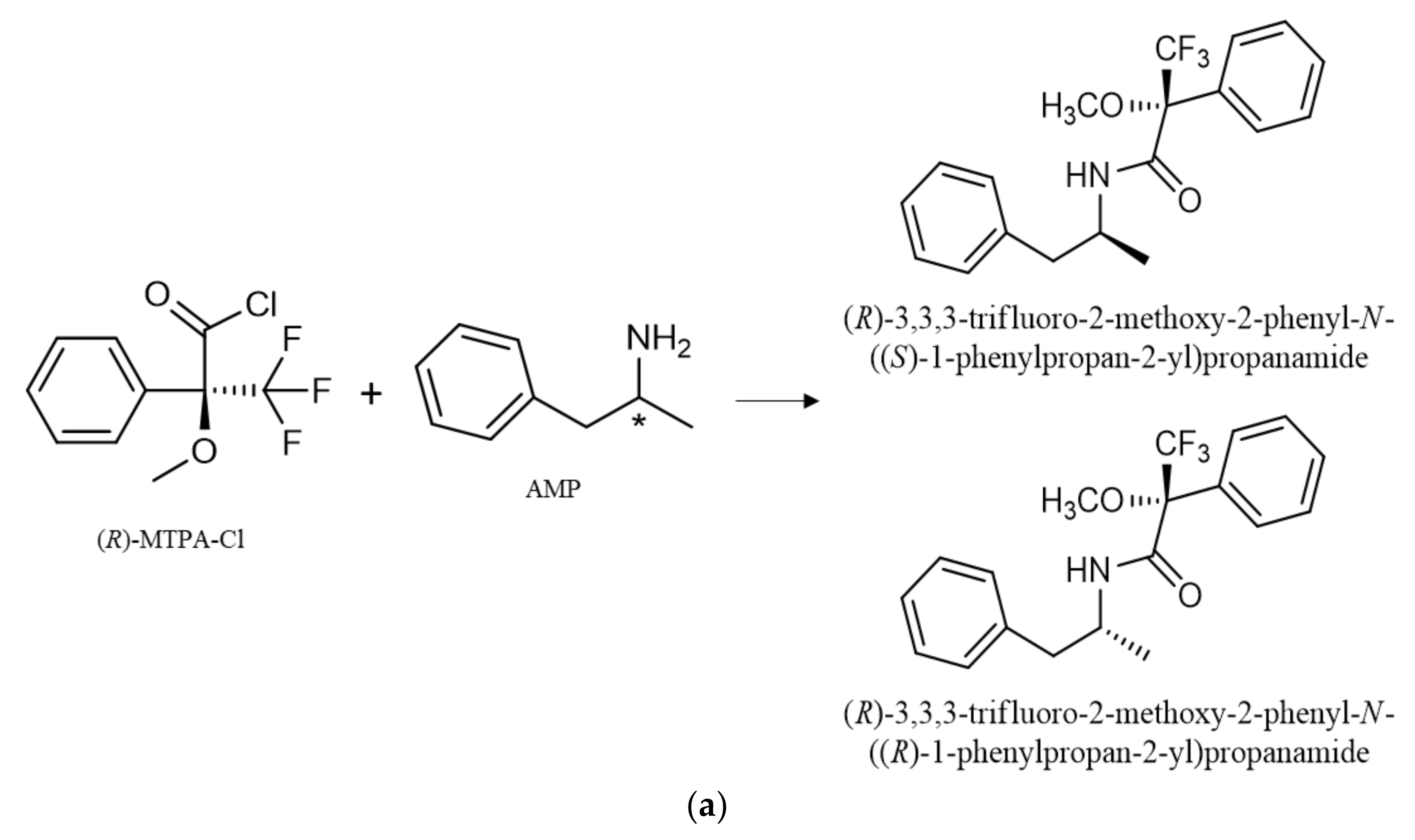
2.5. Derivatization with Chiral Derivatization Reagent
2.6. Chromatographic Conditions of the GC-MS
2.7. Method Parameters and Validation
3. Results and Discussion
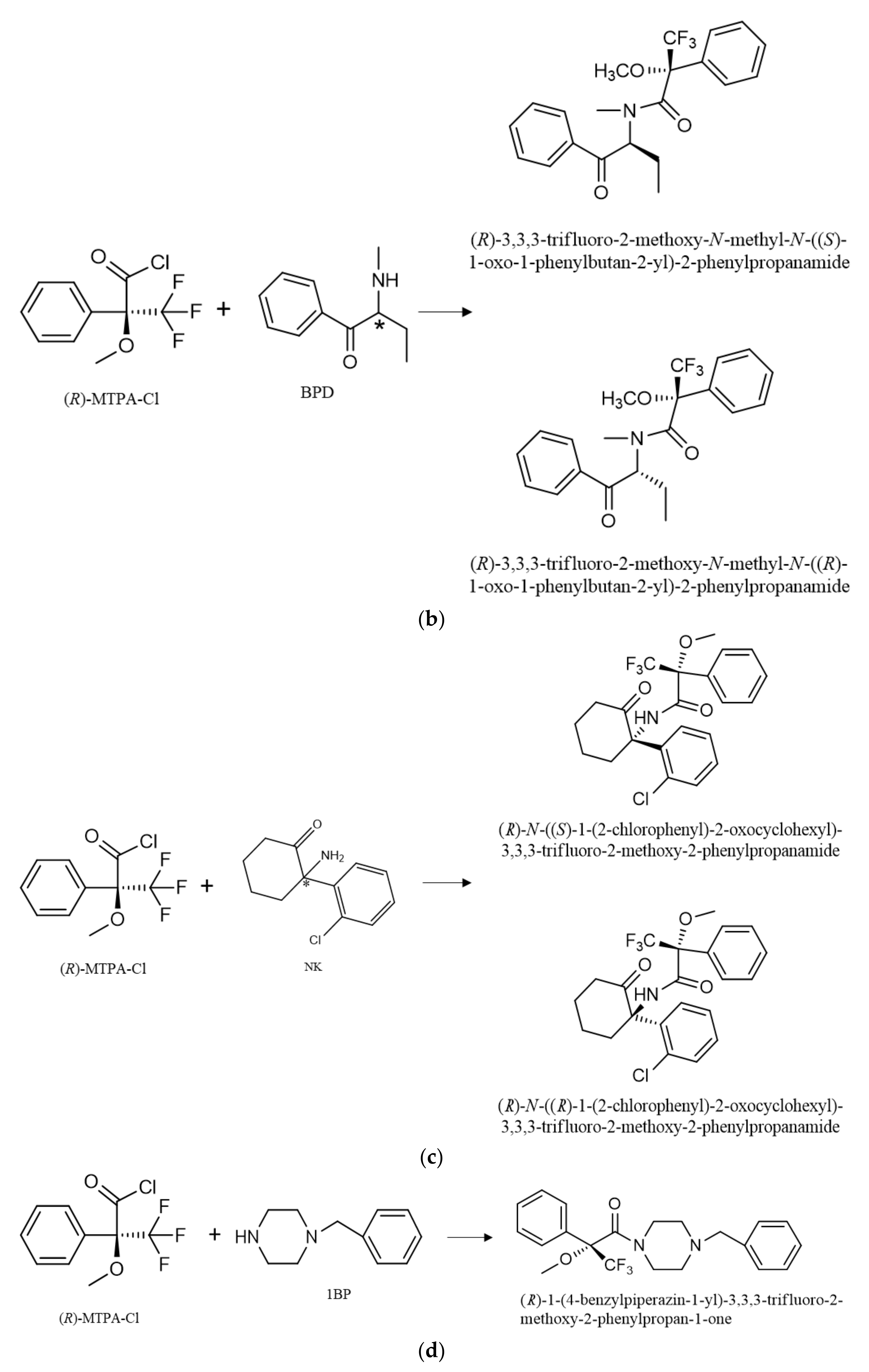
3.1. Derivatization with Chiral Derivatization Reagent
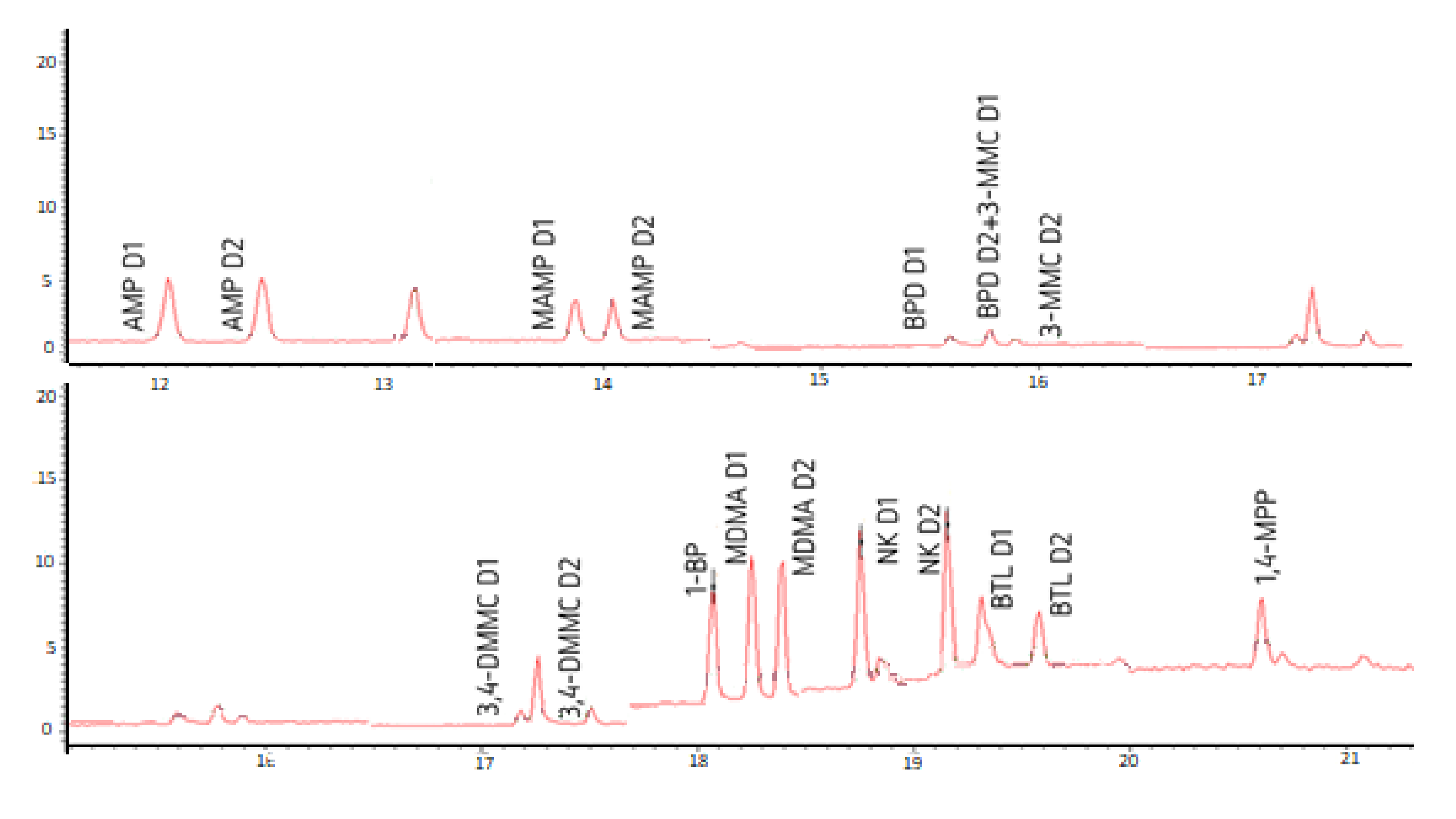
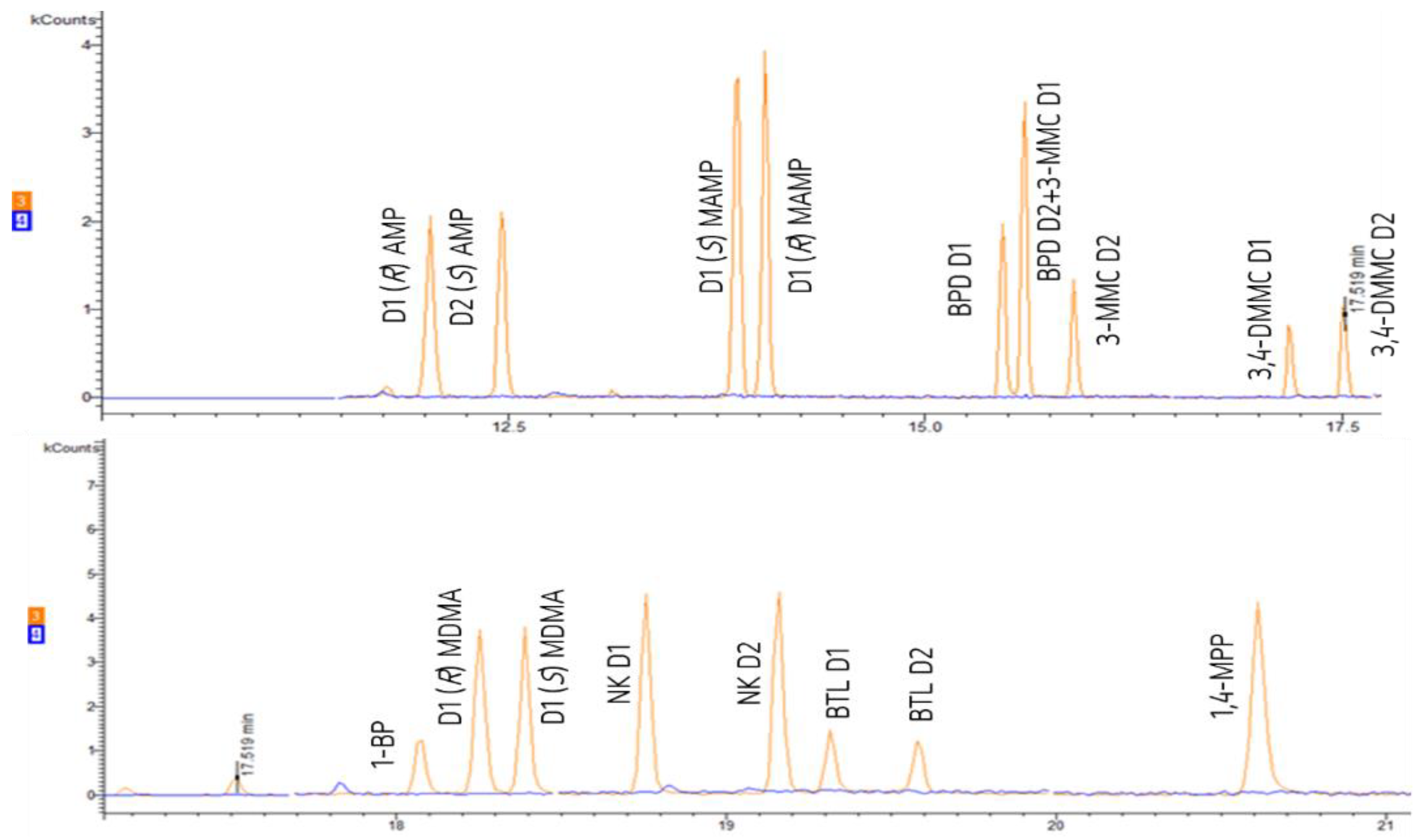
3.2. Optimization of the Chromatographic Separation of the Diastereomers and Piperazine Derivatives
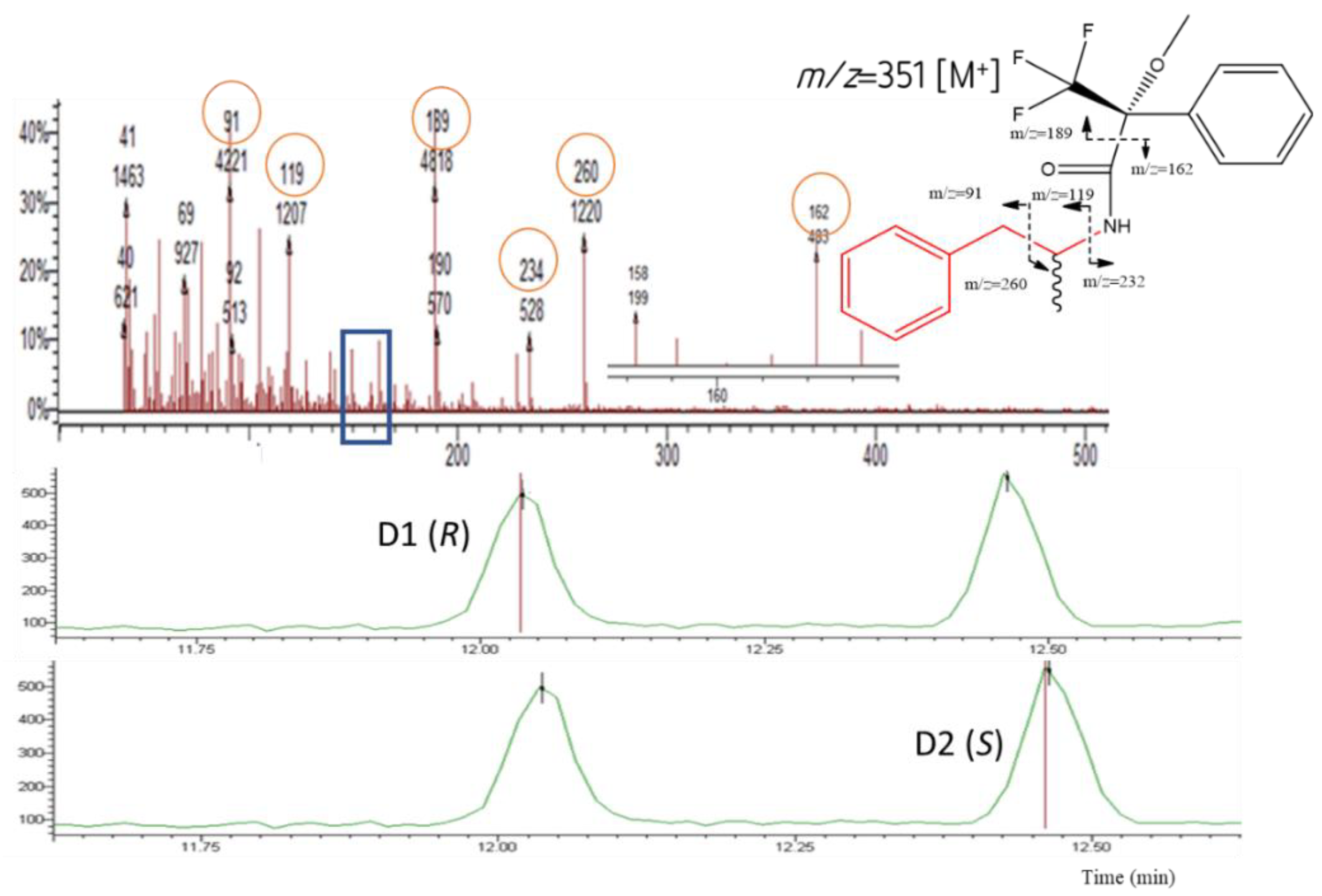
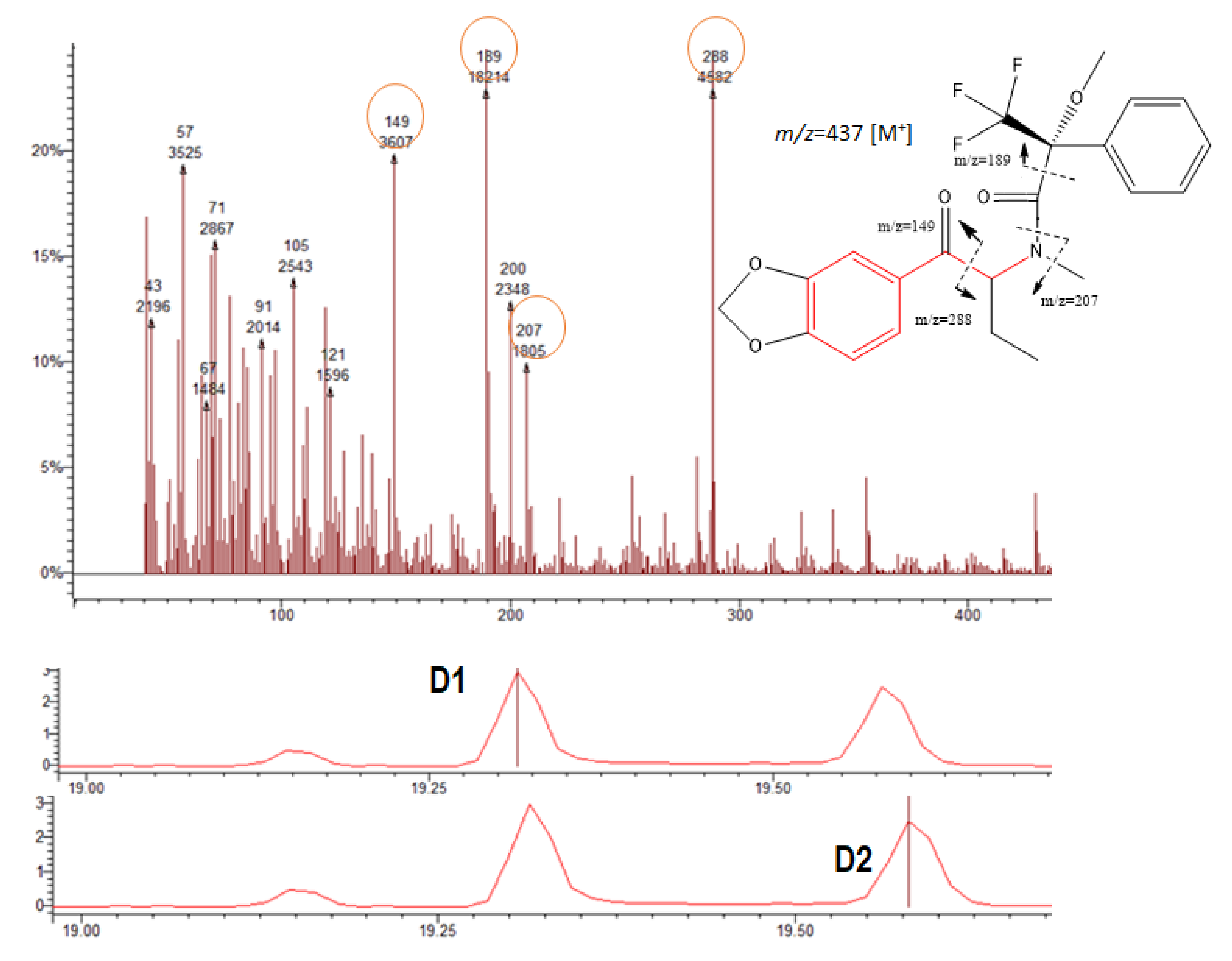
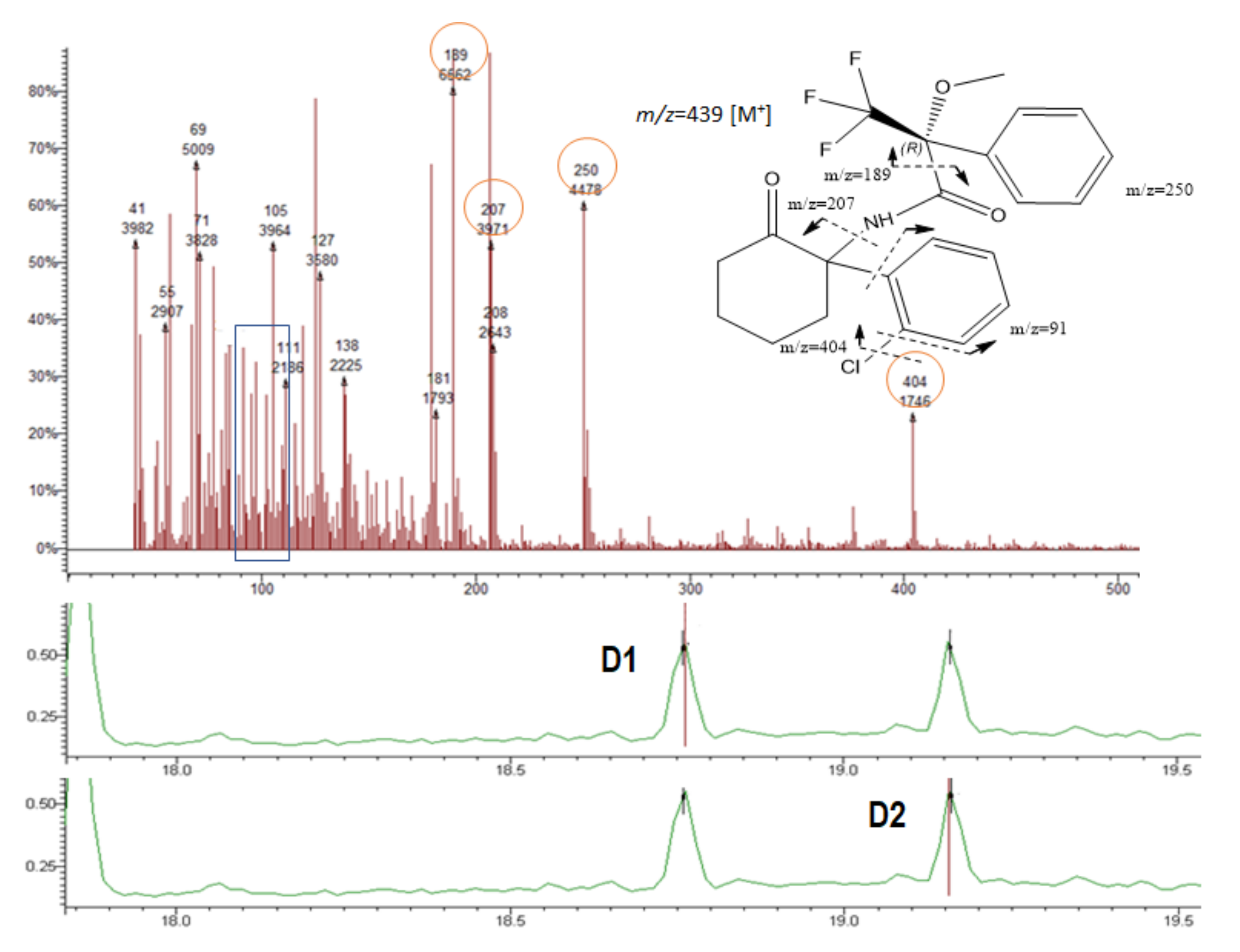
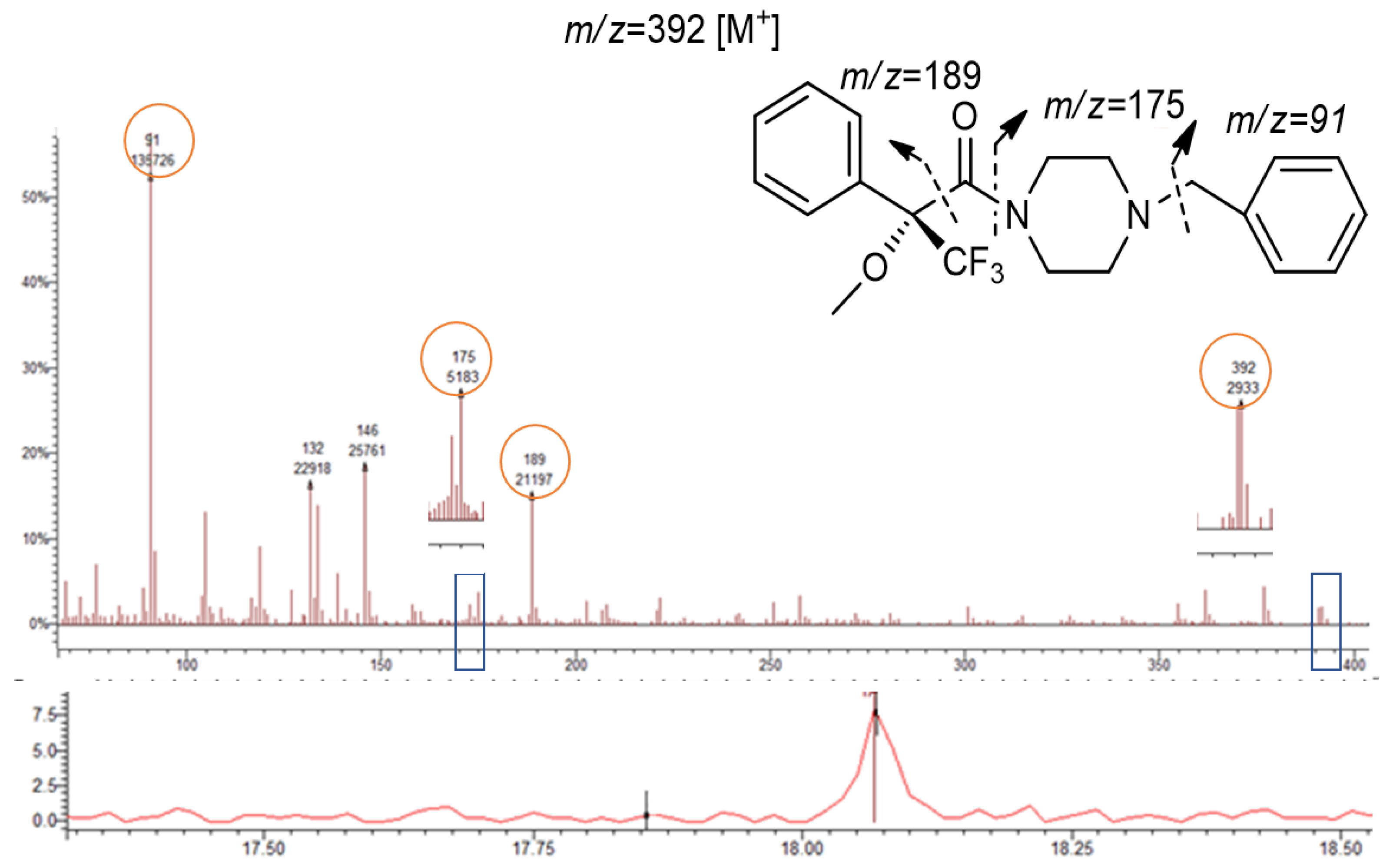
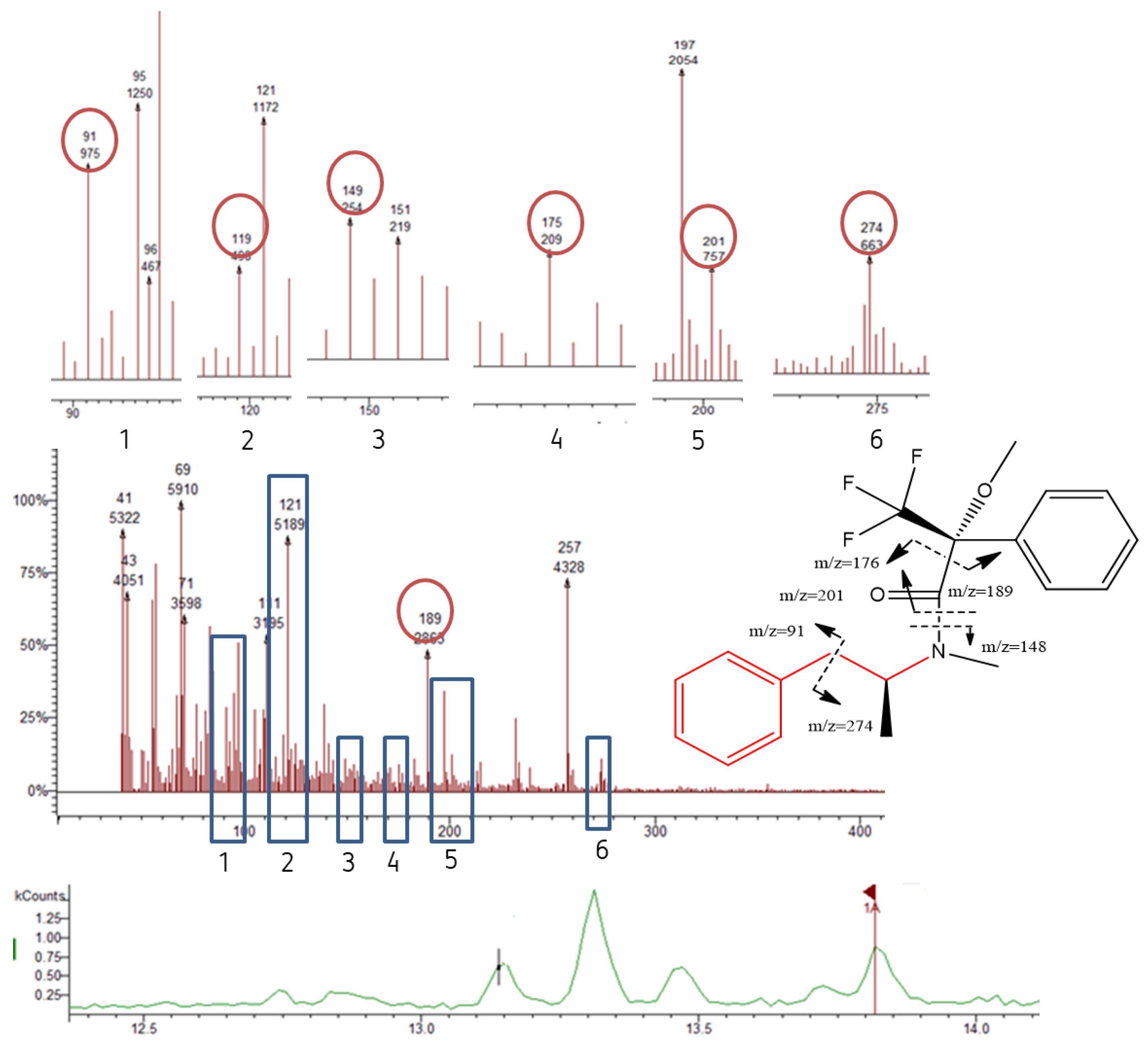
3.3. Mass Spectra of the Target Compound Diastereomers and Piperazine Derivatives
3.4. Method Validation
3.5. Application of the Method
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- EMCDDA. European Drug Report 2021: Trends and Developments; Publications Office of the European Union: Luxembourg, 2021. [Google Scholar]
- EMCDDA. European Drug Report 2020: Trends and Developments; European Monitoring Centre for Drugs and Drug Addiction: Lisbon, Portugal, 2020. [Google Scholar]
- EMCDDA. European Drug Report 2019: Trends and Developments; European Monitoring Centre for Drugs and Drug Addiction: Lisbon, Portugal, 2019. [Google Scholar]
- Government, P. Decreto-Lei n.o 54/2013. Diário da República 75. 2013. [Google Scholar]
- Sanganyado, E.; Lu, Z.; Liu, W. Application of enantiomeric fractions in environmental forensics: Uncertainties and inconsistencies. Environ. Res. 2020, 184, 109354. [Google Scholar] [CrossRef]
- Archer, E.; Castrignanò, E.; Kasprzyk-Hordern, B.; Wolfaardt, G.M. Wastewater-based epidemiology and enantiomeric profiling for drugs of abuse in South African wastewaters. Sci. Total Environ. 2018, 625, 792–800. [Google Scholar] [CrossRef]
- Gonçalves, R.; Ribeiro, C.; Cravo, S.; Cunha, S.C.; Pereira, J.A.; Fernandes, J.; Afonso, C.; Tiritan, M.E. Multi-residue method for enantioseparation of psychoactive substances and beta blockers by gas chromatography–mass spectrometry. J. Chromatogr. B 2019, 1125, 121731. [Google Scholar] [CrossRef] [PubMed]
- Rice, J.; Kannan, A.M.; Castrignanò, E.; Jagadeesan, K.; Kasprzyk-Hordern, B. Wastewater-based epidemiology combined with local prescription analysis as a tool for temporalmonitoring of drugs trends-A UK perspective. Sci. Total Environ. 2020, 735, 139433. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, C.; Santos, C.; Gonçalves, V.; Ramos, A.; Afonso, C.; Tiritan, M.E. Chiral Drug Analysis in Forensic Chemistry: An Overview. Molecules 2018, 23, 262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maia, A.S.; Castro, P.M.; Tiritan, M.E. Integrated liquid chromatography method in enantioselective studies: Biodegradation of ofloxacin by an activated sludge consortium. J. Chromatogr. B 2016, 1029, 174–183. [Google Scholar] [CrossRef]
- Maia, A.S.; Tiritan, M.E.; Castro, P.M. Enantioselective degradation of ofloxacin and levofloxacin by the bacterial strains Labrys portucalensis F11 and Rhodococcus sp. FP1. Ecotoxicol. Environ. Saf. 2018, 155, 144–151. [Google Scholar] [CrossRef]
- Ribeiro, A.R.; Maia, A.S.; Cass, Q.B.; Tiritan, M.E. Enantioseparation of chiral pharmaceuticals in biomedical and environmental analyses by liquid chromatography: An overview. J. Chromatogr. B 2014, 968, 8–21. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Pereira, A.; Ribeiro, C.; Teles, F.; Gonçalves, R.; MF Gonçalves, V.; Pereira, J.A.; Carrola, J.S.; Pires, C.; Tiritan, M.E. Ketamine and Norketamine: Enantioresolution and Enantioselective Aquatic Ecotoxicity Studies. Environ. Toxicol. Chem. 2021. [Google Scholar] [CrossRef]
- Ribeiro, C.; Gonçalves, R.; Tiritan, M. Separation of Enantiomers Using Gas Chromatography: Application in Forensic Toxicology, Food and Environmental Analysis. Crit. Rev. Anal. Chem. 2020, 61, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Coelho, M.M.; Ribeiro, A.R.L.; Sousa, J.C.; Ribeiro, C.; Fernandes, C.; Silva, A.M.; Tiritan, M.E. Dual enantioselective LC–MS/MS method to analyse chiral drugs in surface water: Monitoring in Douro River estuary. J. Pharm. Biom. Anal. 2019, 170, 89–101. [Google Scholar] [CrossRef]
- Ribeiro, A.R.L.; Maia, A.S.; Ribeiro, C.; Tiritan, M.E. Analysis of chiral drugs in environmental matrices: Current knowledge and trends in environmental, biodegradation and forensic fields. TrAC Trends Anal. Chem. 2020, 124, 115783. [Google Scholar] [CrossRef]
- Yu, L.; Wang, S.; Zeng, S. Chiral mobile phase additives in HPLC enantioseparations. Methods Mol. Biol. 2013, 970, 221–231. [Google Scholar] [CrossRef] [PubMed]
- Langa, I.; Gonçalves, R.; Tiritan, M.E.; Ribeiro, C. Wastewater analysis of psychoactive drugs: Non-enantioselective vs enantioselective methods for estimation of consumption. Forensic Sci. Int. 2021, 325, 110873. [Google Scholar] [CrossRef] [PubMed]
- ICH. ICH Harmonized Tripartite Guideline-Text on Validation of Analytical Procedures Methodology, Proceedings of the International Conference on Harmonization Committee; ICH Secretariat: Geneva, Switzerland, 1996; pp. 5–11. [Google Scholar]
- Shabir, G. Validation of HPLC methods for pharmaceutical analysis: Understanding the differences and similarities between validation requirements of the US Food and Drug Administration, the US Pharmacopoeia and the International Conference on Harmonization. J. Chromatogr. A 2003, 987, 57–66. [Google Scholar] [CrossRef]
- Kasprzyk-Hordern, B.; Baker, D.R. Estimation of community-wide drugs use via stereoselective profiling of sewage. Sci. Total Environ. 2012, 423, 142–150. [Google Scholar] [CrossRef] [Green Version]
- Paul, B.D.; Jemionek, J.; Lesser, D.; Jacobs, A.; Searles, D.A. Enantiomeric separation and quantitation of (+/−)-amphetamine, (+/−)-methamphetamine, (+/−)-MDA, (+/−)-MDMA, and (+/−)-MDEA in urine specimens by GC-EI-MS after derivatization with (R)-(−)- or (S)-(+)-alpha-methoxy-alpha-(trifluoromethy)phenylacetyl chloride (MTPA). J. Anal. Toxicol. 2004, 28, 449–455. [Google Scholar] [CrossRef] [Green Version]
- Jenske, R.; Vetter, W. Highly selective and sensitive gas chromatography-electron-capture negative-ion mass spectrometry method for the indirect enantioselective identification of 2- and 3-hydroxy fatty acids in food and biological samples. J. Chromatogr. A 2007, 1146, 225–231. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, A.R.; Maia, A.; Santos, M.; Tiritan, M.E.; Ribeiro, C.M.R. Occurrence of natural contaminants of emerging concern in the Douro River Estuary, Portugal. Arch. Environ. Contam. Toxicol. 2016, 70, 361–371. [Google Scholar] [CrossRef] [PubMed]
- Bordalo, A.A.; Teixeira, R.; Wiebe, W.J. A Water Quality Index applied to an international shared river basin: The case of the Douro River. Environ. Manag. 2006, 38, 910–920. [Google Scholar] [CrossRef]
- Castrignanò, E.; Yang, Z.; Bade, R.; Baz-Lomba, J.A.; Castiglioni, S.; Causanilles, A.; Covaci, A.; Gracia-Lor, E.; Hernandez, F.; Kinyua, J. Enantiomeric profiling of chiral illicit drugs in a pan-European study. Water Res. 2018, 130, 151–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subedi, B.; Kannan, K. Mass loading and removal of select illicit drugs in two wastewater treatment plants in New York State and estimation of illicit drug usage in communities through wastewater analysis. Environ. Sci Technol 2014, 48, 6661–6670. [Google Scholar] [CrossRef]
- Zheng, Q.-D.; Wang, Z.; Liu, C.-Y.; Yan, J.-H.; Pei, W.; Wang, Z.; Wang, D.-G. Applying a population model based on hydrochemical parameters in wastewater-based epidemiology. Sci. Total Environ. 2019, 657, 466–475. [Google Scholar] [CrossRef]
- Skees, A.J.; Foppe, K.S.; Loganathan, B.; Subedi, B. Contamination profiles, mass loadings, and sewage epidemiology of neuropsychiatric and illicit drugs in wastewater and river waters from a community in the Midwestern United States. Sci. Total Environ. 2018, 631, 1457–1464. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.T.; Thai, P.K.; Kaserzon, S.L.; O’Brien, J.W.; Eaglesham, G.; Mueller, J.F. Assessment of drugs and personal care products biomarkers in the influent and effluent of two wastewater treatment plants in Ho Chi Minh City, Vietnam. Sci. Total Environ. 2018, 631, 469–475. [Google Scholar] [CrossRef]
- Pacheco Ferreira, A. Illicit Drugs in Wastewater Treatment Plants A case study: Rio de Janeiro, Brazil. J. Chem. Health Risks 2019, 9, 191–202. [Google Scholar] [CrossRef]
- Evans, S.E.; Bagnall, J.; Kasprzyk-Hordern, B. Enantioselective degradation of amphetamine-like environmental micropollutants (amphetamine, methamphetamine, MDMA and MDA) in urban water. Environ. Pollut. 2016, 215, 154–163. [Google Scholar] [CrossRef]
- Xu, Z.; Du, P.; Li, K.; Gao, T.; Wang, Z.; Fu, X.; Li, X. Tracing methamphetamine and amphetamine sources in wastewater and receiving waters via concentration and enantiomeric profiling. Sci. Total Environ. 2017, 601, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Baker, D.R.; Kasprzyk-Hordern, B. Spatial and temporal occurrence of pharmaceuticals and illicit drugs in the aqueous environment and during wastewater treatment: New developments. Sci. Total Environ. 2013, 454, 442–456. [Google Scholar] [CrossRef]
- Gatidou, G.; Kinyua, J.; van Nuijs, A.L.; Gracia-Lor, E.; Castiglioni, S.; Covaci, A.; Stasinakis, A.S. Drugs of abuse and alcohol consumption among different groups of population on the Greek Island of Lesvos through sewage-based epidemiology. Sci. Total Environ. 2016, 563, 633–640. [Google Scholar] [CrossRef]
- Krizman, I.; Senta, I.; Ahel, M.; Terzic, S. Wastewater-based assessment of regional and temporal consumption patterns of illicit drugs and therapeutic opioids in Croatia. Sci. Total Environ. 2016, 566–567, 454–462. [Google Scholar] [CrossRef] [PubMed]
- Maia, A.S.; Ribeiro, A.R.; Castro, P.M.; Tiritan, M.E. Chiral analysis of pesticides and drugs of environmental concern: Biodegradation and enantiomeric fraction. Symmetry 2017, 9, 196. [Google Scholar] [CrossRef] [Green Version]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | m/z | QI | RT (Minutes) | |
|---|---|---|---|---|
| D1 | D2 | |||
| AMPd3 | 92; 119; 165; 189; 235; 263 | 235; 263 | 12.02 | 12.45 |
| AMP | 91; 119; 162; 189; 234; 260 | 162; 234; 260 | 12.04 * | 12.47 ** |
| MAMP | 91; 119; 148; 176; 189; 274 | 274 | 13.88 ** | 14.05 * |
| BPD | 105; 119; 189; 288 | 288 | 15.47 | 15.60 |
| 3-MMC | 119; 189; 274 | 274 | 15.60 | 15.89 |
| 3,4-DMMC | 105; 119; 133; 189; 200; 274 | 274 | 17.18 | 17.50 |
| 1-BP | 91; 175; 189; 392 | 392 | 18.06 | |
| MDMA | 119; 135; 162; 189; 274 | 162; 274 | 18.25 * | 18.39 ** |
| NK | 189; 207; 250; 404 | 206; 250 | 18.75 | 19.16 |
| BTL | 119; 149; 189; 207; 288 | 288 | 19.32 | 19.58 |
| 1,4-MPP | 91; 189; 207; 393; 408 | 408 | 20.61 | |
| PAS | Concentration Range (ng L−1) | Equation | r2 | LOD (ng L−1) | LOQ (ng L−1) | Recovery (%) | Accuracy (%) | Precision RSD (%) |
|---|---|---|---|---|---|---|---|---|
| AMP (R) | 50.0–300 | y = 0.0071 (±0.00037)x + 0.5269 (±0.068) | 0.9891 | 31.8 | 50.0 | 84.5 | 89.4–107.0 | 2.39–5.31 |
| AMP (S) | 50.0–300 | y = 0.0071(±0.00065)x + 0.563 (±0.12) | 0.9846 | 38.0 | 50.0 | 83.5 | 88.4–108.7 | 3.65–5.53 |
| MAMP (S) | 50.0– 300 | y = 0.0081(±0.00026)x − 0.0145 (±0.044) | 0.9968 | 18.0 | 50.0 | 83.7 | 107.7–109.9 | 0.49–3.54 |
| MAMP (R) | 50.0–300 | y = 0.0081(±0.00037) + 0.1966 (±0.063) | 0.9935 | 25.0 | 50.0 | 98.0 | 96.6–106.6 | 0.87–3.42 |
| BPD D1 | 125–425 | y = 0.0017 (±0.00088)x + 0.0773 (±0.027) | 0.9938 | 35.9 | 125 | 48.5 | 85.8–104.0 | 3.07–5.60 |
| BPD D2 | 125–425 | y = 0.0018 (±0.00087)x + 0.0609 (±0.019) | 0.992 | 40.8 | 125 | 43.5 | 90.1–103.2 | 0.40–3.73 |
| 3-MMC D1 | 250–575 | y = 0.001 (±0.0009)x − 0.1153 (±0.037) | 0.9887 | 89.5 | 250 | 19.7 | 82.9–94.9 | 2.27–4.86 |
| 3-MMC D2 | 250–575 | y = 0.001 (±0.0009)x − 0.0899 (±0.029) | 0.9928 | 71.1 | 250 | 18.6 | 82.4–94.0 | 1.00–6.94 |
| 3,4-DMMC D1 | 250–625 | y= 0.0018 (±0.0001)x − 0.2648 (±0.044) | 0.9928 | 70.3 | 250 | 55.7 | 89.5–90.9 | 1.02–4.41 |
| 3,4-DMMC D2 | 250–625 | y= 0.0018 (±0.0001)x − 0.2585 (±0.038) | 0.9919 | 74.3 | 250 | 50.2 | 83.2–91.6 | 3.88–7.83 |
| MDMA (R) | 75.0–375 | y = 0.0108 (±0.00051)x − 0.23 (±0.122) | 0.9909 | 52.0 | 75.0 | 86.4 | 111.0–115.2 | 1.97–4.92 |
| MDMA (S) | 75.0–375 | y = 0.0108 (±0.00048)x − 0.1765 (±0.089) | 0.9949 | 38.0 | 75.0 | 88.7 | 109.9–116.9 | 2.14–4.92 |
| NK D1 | 75.0–375 | y = 53.535 (± 1.98)x − 1227.7 (±256) | 0.9972 | 14.2 | 75.0 | 76.1 | 87.3–108.4 | 3.90–5.64 |
| NK D2 | 75.0–375 | y = 56.272 (±0.98)x − 1679.8 (±176) | 0.9903 | 26.4 | 75.0 | 74.2 | 87.7–107.9 | 4.04–7.37 |
| BTL D1 | 75.0–375 | y = 0.0037 (±0.00037)x + 0.325 (±0.036) | 0.9926 | 24.7 | 75.0 | 78.1 | 88.9–97.9 | 0.98–4.84 |
| BTL D2 | 75.0–375 | y = 0.0038 (±0.00049)x + 0.2954 (±0.023) | 0.9906 | 17.0 | 75.0 | 82.0 | 88.2–101.8 | 1.96–5.73 |
| 1-BP | 250–625 | y = 70.682 (±4.46)x − 7237.6 (±1892) | 0.9882 | 88.0 | 250 | 53.0 | 97.3–112.4 | 3.27–7.35 |
| 1,4-MPP | 75.0–250 | y = 44.495 (±2.18)x − 2729.9 (±353) | 0.9928 | 29.0 | 75.0 | - | 98.9–112.9 | 2.69–5.14 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Langa, I.; Tiritan, M.E.; Silva, D.; Ribeiro, C. Gas Chromatography Multiresidue Method for Enantiomeric Fraction Determination of Psychoactive Substances in Effluents and River Surface Waters. Chemosensors 2021, 9, 224. https://doi.org/10.3390/chemosensors9080224
Langa I, Tiritan ME, Silva D, Ribeiro C. Gas Chromatography Multiresidue Method for Enantiomeric Fraction Determination of Psychoactive Substances in Effluents and River Surface Waters. Chemosensors. 2021; 9(8):224. https://doi.org/10.3390/chemosensors9080224
Chicago/Turabian StyleLanga, Ivan, Maria Elizabeth Tiritan, Diana Silva, and Cláudia Ribeiro. 2021. "Gas Chromatography Multiresidue Method for Enantiomeric Fraction Determination of Psychoactive Substances in Effluents and River Surface Waters" Chemosensors 9, no. 8: 224. https://doi.org/10.3390/chemosensors9080224