
Multidisciplinary Approach to the Diagnosis and Therapy of Mycosis Fungoides
 ,
,  ,
,  , ,
, ,
Abstract
:1. Introduction
2. The Staging of Mycosis Fungoides

{kind=link}
{kind=link}
| TNMB Stages Definition | |
|---|---|
| Skin (T) | |
| T1 | Patches, papules, and or plaques covering <10% body surface area (BSA) T1a patch only T1b plaque ± patch |
| T2 | Patches, papules, and/or plaques covering ˃10% BSA T2a patch only T2b plaque ± patch |
| T3 | One or more tumors (at least one 1 cm diameter) solid or nodular lesion with evidence of depth and/or vertical growth |
| T4 | Confluence of erythema covering ˃80% BSA |
| NODE (N) | |
| N0 | No clinically abnormal peripheral lymph nodes, biopsy not required |
| N1 | Clinically abnormal peripheral lymph nodes, histopathology Dutch grade 1 or NCI LN0–2 (Table 2) N1a clone negative N1b clone positive |
| N2 | Clinically abnormal peripheral lymph nodes, histopathology Dutch grade 2 or NCI LN3 (Table 2) N2a clone negative N2b clone positive |
| N3 | Clinically abnormal peripheral lymph nodes, histopathology Dutch grade 3–4 (Table 2) or NCI LN4 (Table 3), clone positive or negative |
| Nx | Clinically abnormal peripheral lymph nodes, no histopathologic confirmation |
| VICERAL (M) | |
| M0 | No visceral organ involvement |
| M1 | Visceral involvement (must have pathology, and organ is to be specified) |
| BLOOD (B) | |
| B0 | Absence of significant blood involvement: <5% of blood lymphocytes are Sézary cells B0a clone negative B0b clone positive |
| B1 | Low blood tumor burden: >5% of peripheral blood lymphocytes are Sézary cells but does not meet criteria for B2 disease B1a clone negative B1b clone positive |
| B2 | High blood tumor burden: ˃1000/µL Sézary cells with positive clone in blood (matching clone in skin) or positive clone. |
| DUTCH GRADE SYSTEM | |
| Grade1 | Dermatopathic lymphadenopathy (DL) |
| Grade2 | Early involvement by mycosis fungoides (MF), presence of cerebriform nuclei larger than 7.5 μm |
| Grade3 | Partial effacement of lymph node architecture; many atypical cerebriform mononuclear cells (CMCs) |
| Grade4 | Complete effacement of lymph node architecture |
| NCI LN GRADE SYSTEM | |
| LN0 | No atypical lymphocytes |
| LN1 | Occasional and isolated atypical lymphocytes not arranged in clusters |
| LN2 | Many atypical lymphocytes or in 3–6 cell clusters |
| LN3 | Many atypical lymphocytes or in 3–6 cell clusters |
| LN4 | Partial/complete effacement of nodal architecture by atypical lymphocytes or frankly neoplastic cells |
| STAGE | T | N | M | B | 5-Year DSS (%) |
|---|---|---|---|---|---|
| IA | 1 | 0 | 0 | 0,1 | 98 |
| IB | 2 | 0 | 0 | 0,1 | 89 |
| IIA | 1,2 | 1,2 | 0 | 0,1 | 89 |
| IIB | 3 | 0–2 | 0 | 0,1 | 56 |
| IIIA | 4 | 0–2 | 0 | 0 | 54 |
| IIIB | 4 | 0–2 | 0 | 1 | 48 |
| IVA1 | 1–4 | 0–2 | 0 | 2 | 41 |
| IVA2 | 1–4 | 3 | 0 | 0–2 | 23 |
| IVB | 1–4 | 0–3 | 1 | 0–2 | 18 |
| Stage I Stage II |
|
| Stage III Stage IV |
|
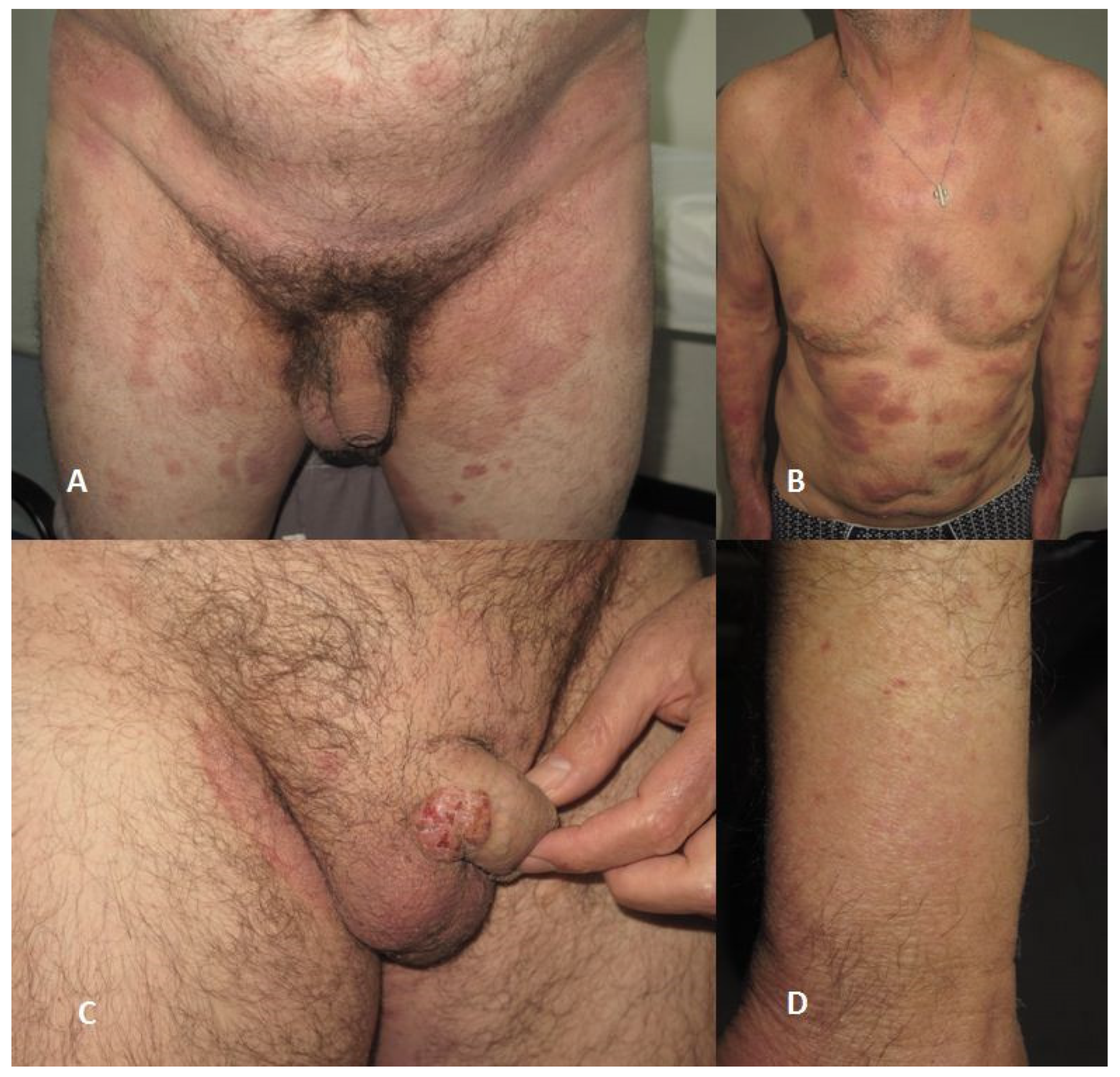
3. Clinical Features
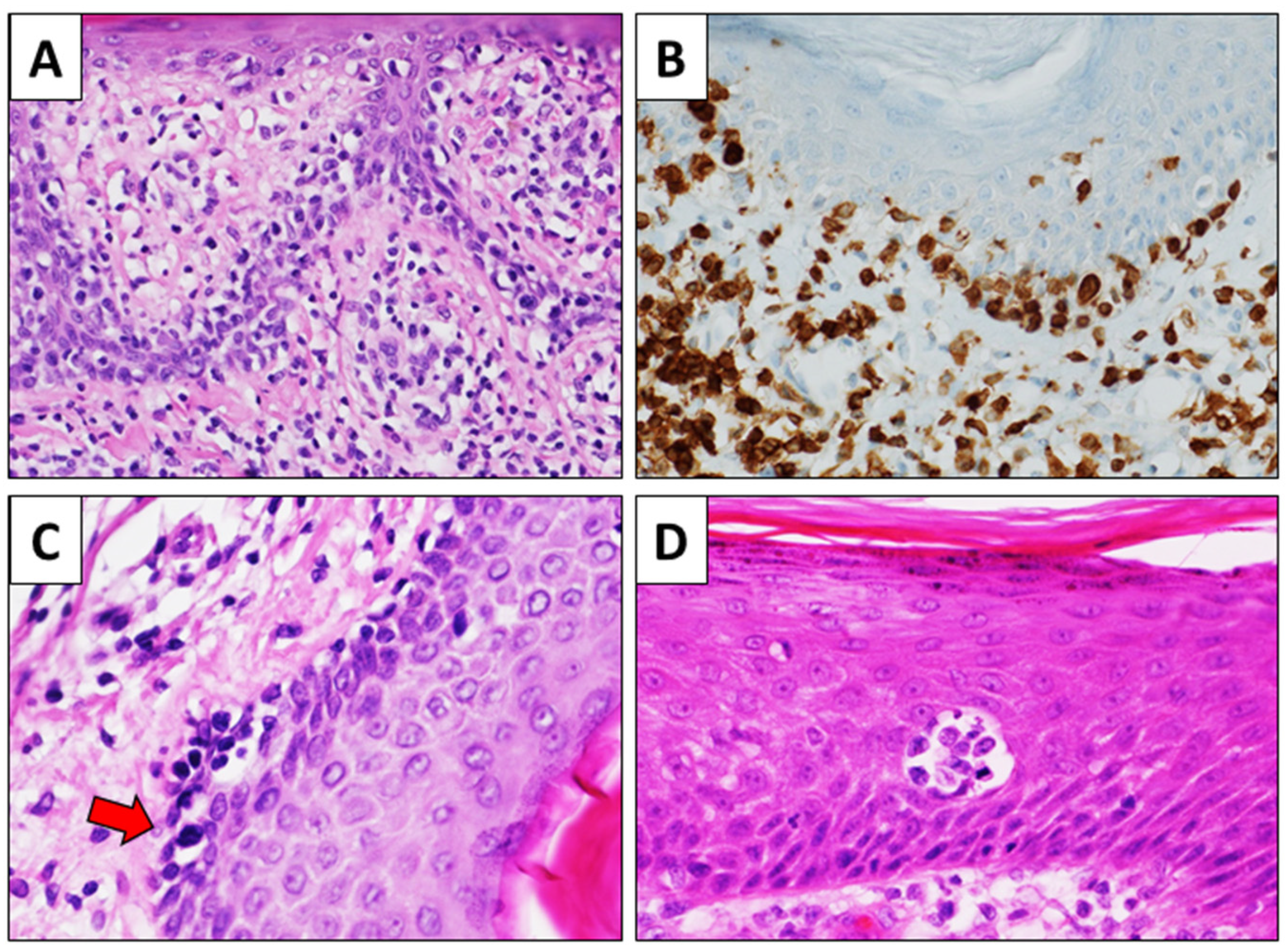
4. The Role of Histology in MF Diagnosis: Clues and Boundaries
4.1. Pathogenesis: New Insights and Molecular Markers
4.2. Staging
4.3. Treatment
- ECP is recommended as second line for the treatment of MF stage IA, refractory to skin-directed therapies, alone or in combination with skin-directed therapies;
- As second line for the treatment of MF stage IB, IIA, and III refractory to skin-directed therapies;
- As first-line for the treatment of MF stage IIB alone or in combination with skin-directed therapies;
- As first-line for the treatment of SS stage IV.
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mourad, A.; Gniadecki, R. Overall Survival in Mycosis Fungoides: A Systematic Review and Meta-Analysis. J. Investig. Dermatol. 2020, 140, 495–497.e5. [Google Scholar] [CrossRef]
- Sica, A.; Vitiello, P.; Sorriento, A.; Ronchi, A.; Calogero, A.; Sagnelli, C.; Troiani, T.; Fasano, M.; Dodaro, C.A.; Franco, R.; et al. Lymphomatoid papulosis. Minerva Medica 2020, 111, 166–172. [Google Scholar] [CrossRef]
- Sica, A.; Vitiello, P.; Ronchi, A.; Casale, B.; Calogero, A.; Sagnelli, E.; Costa Nachtigal, G.C.; Troiani, T.; Franco, R.; Argenziano, G.; et al. Primary Cutaneous Anaplastic Large Cell Lymphoma (pcALCL) in the Elderly and the Importance of Sport Activity Training. Int. J. Environ. Res. Public Health 2020, 17, 839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Talpur, R.; Singh, L.; Daulat, S.; Liu, P.; Seyfer, S.; Trynosky, T.; Wei, W.; Duvic, M. Long-term outcomes of 1263 patients with mycosis fungoides and Sezary Syndrome from 1982 to 2009. Clin. Cancer Res. 2012, 18, m5041-60. [Google Scholar]
- Bobrowicz, M.; Fassnacht, C.; Ignatova, D.; Chang, Y.-T.; Dimitriou, F.; Guenova, E. Pathogenesis and Therapy of Primary Cutaneous T-Cell Lymphoma: Collegium Internationale Allergologicum (CIA) Update 2020. Int. Arch. Allergy Immunol. 2020, 181, 733–745. [Google Scholar] [CrossRef] [PubMed]
- Pimpinelli, N.; Olsen, E.A.; Santucci, M.; Vonderheid, E.; Haeffner, A.C.; Stevens, S.; Burg, G.; Heald, P.W.; Cerroni, L. International Society for Cutaneous Lymphoma. Defining early mycosis fungoides. J. Am. Acad. Dermatol. 2005, 53, 1053–1063. [Google Scholar] [CrossRef] [PubMed]
- Puyol, R.M.; Gallardo, F. Cutaneous lymphomas. Part I: Mycosis Fungoides, Sezary Syndrome, and CD30+ Cutaneous lym-phoproliferative disorders. Actas Dermosifiliogr. 2021, 112, 14–23. [Google Scholar]
- Phyo, Z.H.; Shanbhag, S.; Rozati, S. Update on Biology of Cutaneous T-Cell Lymphoma. Front. Oncol. 2020, 10, 765. [Google Scholar] [CrossRef]
- Salhany, K.E.; Cousar, J.B.; Greer, J.P.; Casey, T.T.; Fields, J.P.; Collins, R.D. Transformation of cutaneous T cell lymphoma to large cell lymphoma. A clinicopathologic and immunologic study. Am. J. Pathol. 1988, 132, 265–277. [Google Scholar]
- Lansigan, F.; Horwitz, S.M.; Pinter-Brown, L.C.; Carson, K.R.; Shustov, A.R.; Rosen, S.T.; Pro, B.; Hsi, E.D.; Federico, M.; Gisselbrecht, C.; et al. Outcomes of Patients with Transformed Mycosis Fungoides: Analysis from a Prospective Multicenter US Cohort Study. Clin. Lymphoma Myeloma Leuk. 2020, 20, 744–748. [Google Scholar] [CrossRef]
- Nashan, D.; Faulhaber, D.; Ständer, S.; Luger, T.; Stadler, R. Mycosis fungoides: A dermatological masquerader. Br. J. Dermatol. 2007, 156, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Willemze, R.; Jaffe, E.S.; Burg, G.; Cerroni, L.; Berti, E.; Swerdlow, S.H.; Ralfkiaer, E.; Chimenti, S.; Diaz-Perez, J.L.; Duncan, L.M.; et al. WHO-EORTC Classification for Cutaneous Lymphomas. Blood 2005, 105, 3768–3785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willemze, R.; Cerroni, L.; Kempf, W.; Berti, E.; Facchetti, F.; Swerdlow, S.H.; Jaffe, E.S. The 2018 update of the WHO-EORTC classification for primary cutaneous lymphomas. Blood 2019, 133, 1703–1714. [Google Scholar] [CrossRef]
- Van Santen, S.; Roach, R.E.; van Doorn, R.; Horváth, B.; Bruijn, M.S.; Sanders, C.J.G.; de Pooter, J.C.; van Rossum, M.M.; de Haas, E.R.M.; Veraart, J.C.J.M.; et al. Clinical staging and prognostic factors in follicu-lotropic mycosis fungoides. JAMA Dermatol. 2016, 152, 992–1000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hodak, E.; Amitay-Laish, I.; Atzmony, L.; Prag-Naveh, H.; Yanichkin, N.; Barzilai, A.; Kershenovich, R.; Feinmesser, M. New insights into folliculotropic mycosis fungoides (FMF): A single-center experience. J. Am. Acad. Dermatol. 2016, 75, 347–355. [Google Scholar] [CrossRef]
- Feng, H.; Beasley, J.; Meehan, S.; Liebman, T.N. Folliculotropic Mycosis Fungoides. Dermatol. Online J. 2018, 15, 543–557. [Google Scholar] [CrossRef]
- Klemke, C.D.; Booken, N.; Weiss, C.; Nicolay, J.P.; Goerdt, S.; Felcht, M.; Géraud, C.; Kempf, W.; Assaf, C.; Ortonne, N.; et al. Histo-pathological and immunophenotypical criteria for the diagnosis of Sezary syndrome in differentiation from other erythrodermic skin diseases: A European Organisation for Research and Treatment of Cancer (EORTC) Cutaneous Lymphoma Task Force Study of 97 cases. Br. J. Dermatol. 2015, 173, 93–105. [Google Scholar]
- Boonk, S.E.; Zoutman, W.H.; Marie-Cardine, A.; van der Fits, L.; Out-Luiting, J.J.; Mitchell, T.J.; Tos, I.I.; Morris, S.L.; Moriarty, B.; Booken, N.; et al. Evaluation of Immuno-phenotypic and Molecular Biomarkers for Sezary Syndrome Using Standard Operating Procedures: A Multicenter Study of 59 Patients. J. Investig. Dermatol. 2016, 136, 1364–1372. [Google Scholar] [CrossRef] [Green Version]
- Carlesimo, M.; Tammaro, A.; Cox, C.; Mari, E.; Fidanza, L.; Narcisi, A.; Cacchi, C.; Camplone, G. A case of Ketron-Goodman Disease. Case Rep. Dermatol. 2009, 1, 39–43. [Google Scholar] [CrossRef]
- Shah, A.; Safaya, A. Granulomatous slack skin disease: A review, in comparison with mycosis fungoides. J. Eur. Acad. Dermatol. Venereol. 2012, 26, 1472–1478. [Google Scholar] [CrossRef]
- Ahn, C.S.; ALSayyah, A.; Sangüeza, O.P. Mycosis fungoides: An updated review of clinicopathologic variants. Am. J. Dermatopathol. 2014, 36, 933–948; quiz 949–951. [Google Scholar] [CrossRef] [PubMed]
- Caccavale, S.; Vitiello, P.; Franco, R.; Panarese, I.; Ronchi, A.; Sica, A.; Toncic, R.J.; Alfano, R.; Argenziano, G. Dermoscopic characterization of folliculotropic mycosis fungoides selectively localized on trunk and limbs. Int. J. Dermatol. 2019, 58, e187–e189. [Google Scholar] [CrossRef] [PubMed]
- Echols, K.F.; Bressler, L.; Armeson, K.; Maize, J.C., Sr. Syringotropic Mycosis Fungoides: A Variant of Folliculotropic Mycosis Fungoides or a Distinct Entity? Am. J. Dermatopathol. 2019, 41, 807–809. [Google Scholar] [CrossRef]
- Fatima, S.; Siddiqui, S.; Usman, M.; Ishtiaque, H.; Idrees, R.; Ahmed, Z.; Ahmed, A. Mycosis fungoides: A clinicopathological study of 60 cases from a tertiary care center. Indian J. Dermatol. 2020, 65, 123–129. [Google Scholar] [CrossRef] [PubMed]
- Cerroni, L. Mycosis fungoides-clinical and histopathologic features, differential diagnosis, and treatment. Semin. Cutan. Med. Surg. 2018, 37, 2–10. [Google Scholar] [CrossRef]
- Skov, A.G.; Gniadecki, R. Delay in the Histopathologic Diagnosis of Mycosis Fungoides. Acta Dermato-Venereol. 2015, 95, 472–475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Massone, C.; Kodama, K.; Kerl, H.; Cerroni, L. Histopathologic features of early (patch) lesions of mycosis fungoides: A mor-phologic study on 745 biopsy specimens from 427 patients. Am. J. Surg. Pathol. 2005, 29, 550–560. [Google Scholar] [CrossRef]
- Santucci, M.; Biggeri, A.; Feller, A.C.; Massi, D.; Burg, G. Efficacy of Histologic Criteria for Diagnosing Early Mycosis Fungoides. Am. J. Surg. Pathol. 2000, 24, 40–50. [Google Scholar] [CrossRef]
- Guitart, J.; Kennedy, J.; Ronan, S.; Chmiel, J.S.; Hsiegh, Y.-C.; Variakojis, D. Histologic criteria for the diagnosis of mycosis fungoides: Proposal for a grading system to standardize pathology reporting. J. Cutan. Pathol. 2001, 28, 174–183. [Google Scholar] [CrossRef]
- Wood, G.S.; Hong, S.R.; Sasaki, D.T.; Abel, E.A.; Hoppe, R.T.; Warnke, R.A. V B Morhenn Leu-8/CD7 antigen expression by CD31 T cells: Comparative analysis of skin and blood in mycosis fungoides/Sézary syndrome relative to normal blood values. J. Am. Acad. Dermatol. 1990, 22, 602–607. [Google Scholar] [CrossRef]
- Michie, S.A.; Abel, E.A.; Hoppe, R.T.; Warnke, R.A.; Wood, G.S. Discordant expression of antigens between intraepidermal and intradermal T cells in mycosis fungoides. Am. J. Pathol. 1990, 137, 1447–1451. [Google Scholar] [PubMed]
- Jankowska-Konsur, A.; Kobierzycki, C.; Grzegrzółka, J.; Piotrowska, A.; Gomulkiewicz, A.; Glatzel-Plucinska, N.; Reich, A.; Podhorska-Okołów, M.; Dzięgiel, P.; Szepietowski, J.C. Podoplanin Expression Correlates with Disease Progression in Mycosis Fungoides. Acta Derm. Venereol. 2017, 97, 235–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mascolo, M.; Romano, M.F.; Ilardi, G.; Romano, S.; Baldo, A.; Scalvenzi, M.; Argenziano, G.; Merolla, F.; Russo, D.; Varricchio, S.; et al. Expression of FK506-binding protein 51 (FKBP51) in Mycosis fungoides. J. Eur. Acad. Dermatol. Venereol. 2018, 32, 735–744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Comfere, N.; Sundram, U.; Hurley, M.Y.; Swick, B. Views of dermatopathologists about clonality assays in the diagnosis of cutaneous T-cell and B-cell lymphoproliferative disorders. J. Cutan. Pathol. 2018, 45, 39–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hodges, E.; Krishna, M.T.; Pickard, C.; Smith, J.L. Diagnostic role of tests for T cell receptor (TCR) genes. J. Clin. Pathol. 2003, 56, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Hsiao, P.F.; Hsiao, C.H.; Lin, Y.C.; Tseng, M.; Tsai, T.F.; Jee, S.H. Histopathologicmolecular correlation in early mycosis fun-goides using T-cell receptor gamma gene rearrangement by polymerase chain reaction with laser capture microdissection. J. FormosMedAssoc. 2007, 106, 265–272. [Google Scholar]
- Ponti, R.; Fierro, M.T.; Quaglino, P.; Bonello, L.; Francia di Celle, P.; Ortoncelli, M.; Fava, P.; Comessatti, A.; Novelli, M.; Bernengo, M.G. TCRγ-Chain Gene Rearrangement by PCR-Based GeneScan: Diagnostic Accuracy Improvement and Clonal Heterogeneity Analysis in Multiple Cutaneous T-Cell Lymphoma Samples. J. Investig. Dermatol. 2008, 128, 1030–1038. [Google Scholar] [CrossRef] [Green Version]
- Kirsch, I.R.; Watanabe, R.; O’Malley, J.T.; Williamson, D.W.; Scott, L.-L.; Elco, C.P.; Teague, J.E.; Gehad, A.; Lowry, E.L.; LeBoeuf, N.R.; et al. TCR sequencing facilitates diagnosis and identifies mature T cells as the cell of origin in CTCL. Sci. Transl. Med. 2015, 7, 308ra158. [Google Scholar] [CrossRef] [Green Version]
- Sufficool, K.E.; Lockwood, C.M.; Abel, H.J.; Hagemann, I.; Schumacher, J.A.; Kelley, T.W.; Duncavage, E.J. T-cell clonality assessment by next-generation sequencing improves detection sensitivity in mycosis fungoides. J. Am. Acad. Dermatol. 2015, 73, 228–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prochazkova, M.; Chevret, E.; Mainhaguiet, G.; Sobotka, J.; Vergier, B.; Belaud-Rotureau, M.-A.; Beylot-Barry, M.; Merlio, J.-P. Common chromosomal abnormalities in mycosis fungoides transformation. Genes, Chromosom. Cancer 2007, 46, 828–838. [Google Scholar] [CrossRef]
- Contassot, E.; Kerl, K.; Roques, S.; Shane, R.; Gaide, O.; Dupuis, M.; Rook, A.H.; French, L.E. Resistance to FasL and tumor necrosis factor-related apoptosis-inducing ligand-mediated apoptosis in Sezary syndrome T-cells associated with impaired death receptor and FLICE-inhibitory protein expression. Blood 2008, 111, 4780–4787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dereure, O.; Levi, E.; Kadin, M.E.; Vonderheid, E.C. Infrequent Fas Mutations but No Bax or p53 Mutations in Early Mycosis Fungoides: A Possible Mechanism for the Accumulation of Malignant T Lymphocytes in the Skin. J. Investig. Dermatol. 2002, 118, 949–956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dereure, O.; Portales, P.; Clot, J.; Guilhou, J.J. Decreased expression of Fas (APO-1/CD95) on peripheral blood CD41 T lym-phocytes in cutaneous T-cell lymphomas. Br. J. Dermatol. 2000, 143, 1205–1210. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Nihal, M.; Siddiqui, J.; Vonderheid, E.C.; Wood, G.S. Low FAS/CD95 Expression by CTCL Correlates With Reduced Sensitivity to Apoptosis That Can Be Restored by FAS Upregulation. J. Investig. Dermatol. 2009, 129, 1165–1173. [Google Scholar] [CrossRef] [Green Version]
- Sagnelli, C.; Sica, A.; Creta, M.; Borsetti, A.; Ciccozzi, M.; Sagnelli, E. Prevention of HBV Reactivation in Hemato-Oncologic Setting during COVID-19. Pathogens 2022, 11, 567. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.; Orchard, G.; Lillington, D.M.; Child, F.J.; Vonderheid, E.C.; Nowell, P.C.; Bagot, M.; Bensussan, A.; Russell-Jones, R.; Young, B.; et al. BCL2 and JUNB abnormalities in primary cutaneous lymphomas. Br. J. Dermatol. 2004, 151, 546–556. [Google Scholar] [CrossRef]
- Mao, X.; Orchard, G.; Lillington, D.M.; Russell-Jones, R.; Young, B.D.; Whittaker, S.J. Amplification and overexpression of JUNB is associated with primary cutaneous T-cell lymphomas. Blood 2003, 101, 1513–1519. [Google Scholar] [CrossRef] [Green Version]
- Mao, X.; Orchard, G.; Mitchell, T.J.; Oyama, N.; Russell-Jones, R.; Vermeer, M.H.; Willemze, R.; van Doorn, R.; Tensen, C.P.; Young, B.D.; et al. A genomic and expression study of AP-1 in primary cutaneous T-cell lymphoma: Evidence for dysregulated expression of JUNB and JUND in MF and SS. J. Cutan. Pathol. 2008, 35, 899–910. [Google Scholar] [CrossRef]
- Nielsen, M.; Kaestel, C.G.; Eriksen, K.W.; Woetmann, A.; Stokkedal, T.; Kaltoft, K.; Geiser, C.; Ropke, C.; Odum, N. Inhibition of constitutively activated Stat3 correlates with altered Bcl-2/Bax expression and induction of apoptosis in mycosis fungoides tumor cells. Leukemia 1999, 13, 735–738. [Google Scholar] [CrossRef] [Green Version]
- Sommer, V.H.; Clemmensen, O.J.; Nielsen, O.; Wasik, M.; Lovato, P.; Brender, C.; Eriksen, K.W.; Woetmann, A.; Kaestel, C.G.; Nissen, M.H.; et al. In vivo activation of STAT3 in cuta-neous T-cell lymphoma. Evidence for an antiapoptotic function of STAT3. Leukemia 2004, 18, 1288–1295. [Google Scholar] [CrossRef] [Green Version]
- Navas, I.C.; Ortiz-Romero, P.L.; Villuendas, R.; Martínez, P.; García, C.; Gómez, E.; Rodriguez, J.L.; García, D.; Vanaclocha, F.; Iglesias, L.; et al. p16I(NK4a) Gene Alterations Are Frequent in Lesions of Mycosis Fungoides. Am. J. Pathol. 2000, 156, 1565–1572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, X.; Orchard, G.; Vonderheid, E.C.; Nowell, P.C.; Bagot, M.; Bensussan, A.; Russell-Jones, R.; Young, B.D.; Whittaker, S.J. Heterogeneous Abnormalities of CCND1 and RB1 in Primary Cutaneous T-Cell Lymphomas Suggesting Impaired Cell Cycle Control in Disease Pathogenesis. J. Investig. Dermatol. 2006, 126, 1388–1395. [Google Scholar] [CrossRef] [PubMed]
- Rabenhorst, A.; Schlaak, M.; Heukamp, L.C.; Forster, A.; Theurich, S.; von Bergwelt-Baildon, M.; Buttner, R.; Kurschat, P.; Mauch, C.; Roers, A.; et al. Mast cells play a pro-tumorigenic role in primary cutaneous lymphoma. Blood 2012, 120, 2042–2054. [Google Scholar] [CrossRef] [Green Version]
- Vermee, R.M.H.; van Doorn, R.; Dukers, D.; Bekkenk, M.W.; Meijer, C.J.; Willemze, R. CD81 T cells in cutaneous T-cell lym-phoma. J. Am. Acad. Dermatol. 2014, 205, e14. [Google Scholar]
- Vermeer, M.H.; van Doorn, R.; Dukers, D.; Bekkenk, M.W.; Meijer, C.J.; Willemze, R. CD8+ T cells in cutaneous T-cell lymphoma: Expression of cytotoxic proteins, Fas Ligand, and killing inhibitory receptors and their relationship with clinical behavior. J. Clin. Oncol. 2001, 19, 4322–4329. [Google Scholar] [CrossRef]
- Goteri, G.; Filosa, A.; Mannello, B.; Stramazzotti, D.; Rupoli, S.; Leoni, P.; Fabris, G. Density of neoplastic lymphoid infiltrate, CD81 T cells, and CD1a1 dendritic cells in mycosis fungoides. J. Clin. Pathol. 2003, 56, 453–458. [Google Scholar] [CrossRef] [Green Version]
- Rea, B.; Haun, P.; Emerson, R.; Vignali, M.; Farooqi, M.; Samimi, S.; Elenitsas, R.; Kirsch, I.; Bagg, A. Role of high-throughput sequencing in the diagnosis of cutaneous T-cell lymphoma. J. Clin. Pathol. 2018, 71, 814–820. [Google Scholar] [CrossRef]
- Guastafierro, S.; Sica, A.; Parascandola, R.R.; Ferrara, M.G.; Di Martino, A.; Pezone, L.; Falcone, U. Clinical significance of serum triple monoclonal components: A report of 6 cases and a review of the literature. Leuk. Res. 2014, 38, 166–169. [Google Scholar] [CrossRef]
- Walia, R.; Yeung, C.C.S. An Update on Molecular Biology of Cutaneous T Cell Lymphoma. Front. Oncol. 2020, 9, 1558. [Google Scholar] [CrossRef]
- Dippel, E.; Assaf, C.; Hummel, M.; Schrag, H.J.; Stein, H.; Goerdt, S.; Orfanos, C.E. Clonal T-cell receptor gamma-chain gene rear-rangement by PCR based Gene Scan analysis in advanced cutaneous T-cell lymphoma: A critical evaluation. J. Pathol. 1999, 188, 146–154. [Google Scholar] [CrossRef]
- Schiller, P.I.; Flaig, M.J.; Puchta, U.; Kind, P.; Sander, C. Detection of clonal T cells in lichen planus. Arch. Dermatol. Res. 2000, 292, 568–569. [Google Scholar] [CrossRef]
- Thurber, S.E.; Zhang, B.; Kim, Y.H.; Schrijver, I.; Zehnder, J.; Kohler, S. T-cell clonality analysis in biopsy specimens from two different skin sites shows high specificity in the diagnosis of patients with suggested mycosis fungoides. J. Am. Acad. Dermatol. 2007, 57, 782–790. [Google Scholar] [CrossRef] [PubMed]
- Willemze, R.; Hodak, E.; Zinzani, P.L.; Specht, L.; Ladetto, M. Primary cutaneous lymphomas: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2018, 29, iv30–iv40. [Google Scholar] [CrossRef]
- Agar, N.S.; Wedgeworth, E.; Crichton, S.; Mitchell, T.J.; Cox, M.; Ferreira, S.; Robson, A.; Calonje, E.; Stefanato, C.H.; Wain, E.M.; et al. Survival outcomes and prognostic factors in MF/Sézary syndrome: Validation of the revised International Society for Cutaneous Lymphomas/European Organisation for Research and Treatment of Cancer staging proposal. J. Clin. Oncol. 2010, 28, 4730–4739. [Google Scholar] [CrossRef] [PubMed]
- Liner, K.; Brown, C.; McGirt, L.Y. Clinical potential of mechlorethamine gel for the topical treatment of mycosis fungoides-type cutaneous T-cell lymphoma: A review on current efficacy and safety data. Drug Des. Dev. Ther. 2018, 12, 241–254. [Google Scholar] [CrossRef] [Green Version]
- Lessin, S.R.; Duvic, M.; Guitart, J.; Pandya, A.G.; Strober, B.E.; Olsen, E.A. Topical chemotherapy in cutaneous T-cell lymphoma: Positive results of a randomized, controlled, multicenter trial testing the efficacy and safety of a novel mechlor-ethamine, 0.02%, gel in mycosis fungoides. JAMA Dermatol. 2013, 149, 25–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Quatrebarbes, J.; Estève, E.; Bagot, M.; Bernard, P.; Beylot-Barry, M.; Delaunay, M. Treatment of early-stage mycosis fungoides with twice-weekly applications of mechlorethamine and topical corticosteroids: A prospective study. Arch. Dermatol. 2005, 141, 1117–1120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scarisbrick, J.J.; Prince, H.M.; Vermeer, M.H.; Quaglino, P.; Horwitz, S.; Porcu, P.; Stadler, R.; Wood, G.S.; Beylot-Barry, M.; Pham-Ledard, A. Cutaneous Lymphoma International Consortium study of outcome in ad-vanced stages of MF and SS: Effect of specific prognostic markers on survival and development of a prognostic model. J. Clin. Oncol. 2015, 33, 3766–3773. [Google Scholar] [CrossRef]
- Lovgren, M.-L.; Scarisbrick, J.J. Update on skin directed therapies in mycosis fungoides. Chin. Clin. Oncol. 2019, 8, 7. [Google Scholar] [CrossRef]
- Creta, M.; Sica, A.; Napolitano, L.; Celentano, G.; La Rocca, R.; Capece, M.; Calogero, A.; Califano, G.; Vanni, L.; Mangiapia, F.; et al. Fournier’s Gangrene in Patients with Oncohematological Diseases: A Systematic Review of Published Cases. Healthcare 2021, 9, 1123. [Google Scholar] [CrossRef]
- Hughes, C.F.M.; Khot, A.; McCormack, C.; Lade, S.; Westerman, D.A.; Twigger, R.; Buelens, O.; Newland, K.; Tam, C.; Dickinson, M.; et al. Lack of durable disease control with chemotherapy for mycosis fungoides and Sézary syndrome: A comparative study of systemic therapy. Blood 2015, 125, 71–81. [Google Scholar] [CrossRef]
- Sica, A.; Vitiello, P.; Caccavale, S.; Sagnelli, C.; Calogero, A.; Dodaro, C.A.; Pastore, F.; Ciardiello, F.; Argenziano, G.; Reginelli, A.; et al. Primary cutaneous DLBCL non-GCB type: Challenges of a rare case. Open Med. 2020, 15, 119–125. [Google Scholar] [CrossRef] [Green Version]
- Ronchi, A.; Marino, F.Z.; Vitiello, P.; Caccavale, S.; Argenziano, G.; Crisci, S.; Franco, R.; Sica, A. A case of primary cutaneous B-cell lymphoma with immature features in an old man. Diffuse large B-cell lymphoma with immature features or B-cell lymphoblastic lymphoma? J. Cutan. Pathol. 2021, 48, 535–540. [Google Scholar] [CrossRef] [PubMed]
- Hanel, W.; Briski, R.; Ross, C.W.; Anderson, T.F.; Kaminski, M.S.; Hristov, A.C.; Wilcox, R.A. A retrospective comparative outcome analysis following systemic therapy in Mycosis fungoides and Sezary syndrome. Am. J. Hematol. 2016, 91, E491–E495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alberti-Violetti, S.; Talpur, R.; Schlichte, M.; Sui, D.; Duvic, M. Advanced-Stage Mycosis Fungoides and Sézary Syndrome: Survival and Response to Treatment. Clin. Lymphoma Myeloma Leuk. 2015, 15, e105–e112. [Google Scholar] [CrossRef]
- Shipman, A.R.; Scarisbrick, J. New Treatment Options for Mycosis Fungoides. Indian J. Dermatol. 2016, 61, 119. [Google Scholar] [CrossRef]
- Sica, A.; Sagnelli, C.; Casale, B.; Svanera, G.; Creta, M.; Calogero, A.; Franco, R.; Sagnelli, E.; Ronchi, A. How Fear of COVID-19 Can Affect Treatment Choices for Anaplastic Large Cell Lymphomas ALK+ Therapy: A Case Report. Healthcare 2021, 9, 135. [Google Scholar] [CrossRef] [PubMed]
- Trautinger, F.; Eder, J.; Assaf, C.; Bagot, M.; Cozzio, A.; Dummer, R.; Gniadecki, R.; Klemke, C.-D.; Ortiz-Romero, P.L.; Papadavid, E.; et al. European Organisation for Research and Treatment of Cancer consensus recommendations for the treatment of mycosis fungoides/Sézary syndrome—Update 2017. Eur. J. Cancer 2017, 77, 57. [Google Scholar] [CrossRef] [Green Version]
- Whittaker, S.; Hoppe, R.; Prince, H.M. How I treat mycosis fungoides and Sézary syndrome. Blood 2016, 127, 3142–3153. [Google Scholar] [CrossRef] [Green Version]
- Vitiello, P.; Sica, A.; Ronchi, A.; Caccavale, S.; Franco, R.; Argenziano, G. Primary Cutaneous B-Cell Lymphomas: An Update. Front. Oncol. 2020, 10, 651. [Google Scholar] [CrossRef]
- Dummer, R.; Duvic, M.; Scarisbrick, J.; Olsen, E.A.; Rozati, S.M.; Eggmann, N.; Goldinger, S.; Hutchinson, K.; Geskin, L.; Illidge, T.; et al. Final results of a multicenter phase II study of the purine nucleoside phosphorylase (PNP) inhibitor forodesine in patients with advanced cutaneous t-cell lymphomas (CTCL) (Mycosis fungoides and Sézary syndrome). Ann. Oncol. 2014, 25, 1807–1812. [Google Scholar] [CrossRef] [PubMed]
- Rothlin, C.V.; Ghosh, S.; Zuniga, E.I.; Oldstone, M.B.A.; Lemke, G. TAM receptors are pleiotropic inhibitors of the innate im-mune response. Cell 2007, 131, 1124–1136. [Google Scholar] [CrossRef] [Green Version]
- Lemke, G.; Rothlin, C.V. Immunobiology of the TAM receptors. Nat. Rev. Immunol. 2008, 8, 327–336. [Google Scholar] [CrossRef] [Green Version]
- Maeda, A.; Schwarz, A.; Kernebeck, K.; Gross, N.; Aragane, Y.; Peritt, D.; Schwarz, T.A. Intravenous Infusion of Syngeneic Apoptotic Cells by Photopheresis Induces Antigen-Specific Regulatory T Cells. J. Immunol. 2005, 174, 5968–5976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gatza, E.; Rogers, C.E.; Clouthier, S.G.; Lowler, K.P.; Tawara, I.; Liu, C.; Reddy, P.; Ferrara, J.L.M. Extracorporeal photopheresis reverses experimental graft-versus-host disease through regulatory T cells. Blood 2008, 112, 1515–1521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halperin, E.C.; Perez, C.A.; Brady, L.W. Perez and Brady’s Principles and Practice of Radiation Oncology; Lippincott Williams & Wilkins: Philadelphia, PN, USA, 2008. [Google Scholar]
- NCCN Clinical Practice Guidelines in Oncology. Available online: https://www.nccn.org/professionals/physician_gls/default.aspx (accessed on 14 February 2023).
- Huen, A.O.; Rook, A.H. Toll receptor agonist therapy of skin cancer and cutaneous T-cell lymphoma. Curr. Opin. Oncol. 2014, 26, 237–244. [Google Scholar] [CrossRef]
- Rook, A.H.; Gelfand, J.M.; Wysocka, M.; Troxel, A.B.; Benoit, B.; Surber, C.; Elenitsas, R.; Buchanan, M.A.; Leahy, D.S.; Watanabe, R. Topical resiquimod can induce disease re-gression and enhance T-cell effector functions in cutaneous T-cell lymphoma. Blood 2015, 126, 1452–1461. [Google Scholar] [CrossRef] [Green Version]
- Sica, A.; Sagnelli, C.; Papa, A.; Ciccozzi, M.; Sagnelli, E.; Calogero, A.; Martinelli, E.; Casale, B. An Anecdotal Case Report of Chronic Lymphatic Leukemia with del(11q) Treated with Ibrutinib: Artificial Nourishment and Physical Activity Program. Int. J. Environ. Res. Public Health 2020, 17, 1929. [Google Scholar] [CrossRef] [Green Version]
- Scholtz, W. Ueber den Einfluss der Röntgenstrahlen auf die Haut in gesundem und krankem Zustande. Arch. Dermatol. Res. 1902, 59, 421–446. [Google Scholar] [CrossRef]
- Jamieson, W.A. Mycosis Fungoides, and Its Treatment by the X-Rays. Trans. Med. Chir. Soc. Edinb. 1903, 22, 15–24. [Google Scholar]
- Marsh, J.P. A case of mycosis fungoides symptomatically cured by means of X-rays. Am. J. Med. Sci. 1903, 126, 314–318. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.H.; Nisce, L.Z.; D’Angio, G.J. Dose-Time Fractionation Study in Patients with Mycosis Fungoides and Lymphoma Cutis. Radiology 1976, 119, 439–442. [Google Scholar] [CrossRef]
- Hoppe, R.T. Mycosis fungoides: Radiation therapy. Dermatol. Ther. 2003, 16, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Sica, A.; Casale, B.; Spada, A.; Di Dato, M.; Sagnelli, C.; Calogero, A.; Buonavolontà, P.; Salzano, A.; Martinelli, E.; Saracco, E.; et al. Differential diagnosis: Retroperitoneal fibrosis and oncological diseases. Open Med. 2019, 15, 22–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yahalom, J.; Illidge, T.; Specht, L.; Hoppe, R.T.; Li, Y.-X.; Tsang, R.; Wirth, A.; International Lymphoma Radiation Oncology Group. Modern Radiation Therapy for Extranodal Lymphomas: Field and Dose Guidelines From the International Lymphoma Radiation Oncology Group. Int. J. Radiat. Oncol. Biol. Phys. 2015, 92, 11–31. [Google Scholar] [CrossRef]
- Rook, A.H.; Suchin, K.R.; Kao, D.M.; Yoo, E.K.; Maceyet, W.H.; DeNardo, B.J.; Bromely, P.G.; Geng, Y.; Junkins-Hopkins, J.M.; Lessin, S.R. Photopheresis: Clinical Applications and Mechanism of Action. J. Investig. Dermatol. Symp. Proc. 1999, 4, 85–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zic, J.A.; Miller, J.L.; Stricklin, G.P.; King, L.E., Jr. The north american experience with photopheresis. Ther. Apher. 1999, 3, 50–62. [Google Scholar] [CrossRef]
- Yoo, E.K.; Rook, A.H.; Elenitsas, R.; Gasparro, F.P.; Vowels, B.R. Apoptosis induction of ultraviolet light A and photochemo-therapy in cutaneous T-cell lymphoma: Relevance to mechanism of therapeutic action. J. Investig. Dermatol. 1996, 107, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Heshmati, F. Mechanisms of action of extracorporeal photochemotherapy. Transfus. Apher. Sci. 2003, 29, 61–70. [Google Scholar] [CrossRef]
- Sica, A.; Casale, D.; Rossi, G.; Casale, B.; Ciccozzi, M.; Fasano, M.; Ciotti, M.; Sagnelli, E.; Papa, A.; Sagnelli, C. The impact of the SARS-CoV-2 infection, with special reference to the haematological setting. J. Med. Virol. 2020, 93, 223–233. [Google Scholar] [CrossRef]
- Szodoraya, P.; Pappb, G.; Nakkenc, B.; Harangid, M.; Zeher, M. The molecular and clinical rationale of extracorporeal pho-tochemotherapy in autoimmune diseases, malignancies and transplantation. Autoimmun. Rev. 2010, 9, 459–464. [Google Scholar] [CrossRef] [PubMed]
- Capece, M.; Creta, M.; Calogero, A.; La Rocca, R.; Napolitano, L.; Barone, B.; Sica, A.; Fusco, F.; Santangelo, M.; Dodaro, C.; et al. Does Physical Activity Regulate Prostate Carcinogenesis and Prostate Cancer Outcomes? A Narrative Review. Int. J. Environ. Res. Public Health 2020, 17, 1441. [Google Scholar] [CrossRef] [Green Version]
- Kleinclauss, F.; Perruche, S.; Masson, E.; Bittencourt, M.; Biichle, S.; Remy-Martin, J.-P.; Bittard, H.; Chalopin, J.-M.; Seilles, E.; Tiberghien, P.; et al. Intravenous apoptotic spleen cell infusion induces a TGF-b-dependent regulatory T-cell expansion. Cell Death Diff. 2006, 13, 41–52. [Google Scholar] [CrossRef] [Green Version]
- Sica, A.; Sagnelli, C.; Vitiello, P.; Franco, R.; Argenziano, G.; Ciccozzi, M.; Sagnelli, E.; Ronchi, A. Rescue Therapy of Refractory Diffuse Large B-Cell Lymphomas BCL2 with Venetoclax: Case Report. Chemotherapy 2020, 65, 161–165. [Google Scholar] [CrossRef]
- Albert, M.L. Death-defying immunity: Do apoptotic cells influence antigen processing and presentation? Nat. Rev. Immunol. 2004, 4, 223–231. [Google Scholar] [CrossRef] [PubMed]
- Spisek, R.; Gasova, Z.; Bartunkova, J. Maturation state of dendritic cells during the extracorporeal photopheresis and its rel-evance for the treatment of chronic graft-versus-host disease. Transfusion 2006, 46, 55–65. [Google Scholar] [CrossRef] [PubMed]
- Berger, C.; Xu, A.; Hanlon, D.; Lee, C.; Schechner, J.; Glusac, E.; Christensen, I.; Snyder, E.; Holloway, V.; Tigelaar, R.; et al. Induction of human tumor-loaded dendritic cells. Int. J. Cancer 2001, 4, 438–447. [Google Scholar] [CrossRef]
- Hanlon, D.J.; Berger, C.L.; Edelson, R.L. Photoactivated 8-methoxypsoralen treatment causes a peptide-dependent increase in antigen display by transformed lymphocytes. Int. J. Cancer 1998, 78, 70–75. [Google Scholar] [CrossRef]
- Lutz, M.B.; Schuler, G. Immature, semi-mature and fully mature dendritic cells: Which signals induce tolerance and immunity? Trends Immunol. 2002, 23, 445–449. [Google Scholar] [CrossRef]
- Reginelli, A.; Urraro, F.; Sangiovanni, A.; Russo, G.M.; Russo, C.; Grassi, R.; Agostini, A.; Belfiore, M.P.; Cellina, M.; Floridi, C.; et al. Extranodal Lymphomas: A pictorial review for CT and MRI classification. Acta Biomed. 2020, 91, 34–42. [Google Scholar] [CrossRef]
- Zackheim, H.S.; McCalmont, T.H. Mycosis fungoides: The great imitator. J. Am. Acad. Dermatol. 2002, 47, 914–918. [Google Scholar] [CrossRef] [PubMed] [Green Version]


| Agent [Ref.] | Trial | CTCL Indication | Approval Year (FDA) | ORR%/CR%/PR% |
|---|---|---|---|---|
| Romidepsin [80,81] | Pivotal/supportive | Relapsed/refractory CTCL | 2009 | 33.8/5.6/28.2 |
| Denileukin diftitox [73,74,75,76] | Pivotal | Persistent/recurrent CTCL | 1999 | 44/10/34 |
| Bexarotene [82,83,84,85] | Pivotal | Refractory CTCL | 1999 | 45 |
| Vorinostat [80,81] | Pivotal/Supportive | Progressive/persistent/recurrent CTCL | 2007 | 29.5 |
| Brentuximab Vedotin [73,74,75,76] | Randomized trial vs methotrexate or bexarotene(ALCANZA) | pcALCL or CD30-expressing MF refractory | 2017 | 56.3 |
| Mogamolizumab [77,78,79] | Randomized trial vs vorinostat(MAVORIC) | Relapsed or refractory MF or SS | 2018 | 28 |
| Pembrolizumab [73,74,75,76] | Multicenter Phase II study | Advanced relapsed or refractory MF/SS | - | 37.5/8.3/29.2 |
| Cobomarsen(MRG-106) [77,78,79] | Phase II trialRandomized Trial(SOLAR) vs vorinostat | Progressive/recurrent/persistent MF | 2020 orfan drug | |
| Alemtuzumab [73,74,75,76] | Phase II Study | Advanced/relapsed MF or SS | - | 51.1/17.9/33.3 |
| IPH4102 (anti-KIR3DL2 antibody) [73,74,75,76] | Phase I clinical trial | Relapsed/refractory MF/SS | - | |
| Pralatrexate [63,86,87] | Phase I clinical trial | Advanced/relapsed PTCL | 2009 | 44.8/3.4/41.4 |
| Forodesine [81] | Multicenter phase I–II study iv/os | Advanced/relapsed CTCL | - | ORR 31% iv 27% os |
| Duvelisib [73,74,75,76] | Open label phase I study | Relapsed or refractory CTCL | - | 31.6/0/31.6 |
| Lenalidomide [73,74,75,76] | Open label multicenter phase II study | Advanced/refractory MF/SS | - | PR 28% |
| Gemcitabine [63,86,87] | Multicenter phase II study | Advanced/refractory CTCL | - | 75/21.8/53.1 |
| Pegylated liposomal doxorubicin [63,86,87] | Multicenter study | Advanced relapsed/recurrent mf/ss | - | 56/20/36 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vitiello, P.; Sagnelli, C.; Ronchi, A.; Franco, R.; Caccavale, S.; Mottola, M.; Pastore, F.; Argenziano, G.; Creta, M.; Calogero, A.; et al. Multidisciplinary Approach to the Diagnosis and Therapy of Mycosis Fungoides. Healthcare 2023, 11, 614. https://doi.org/10.3390/healthcare11040614
Vitiello P, Sagnelli C, Ronchi A, Franco R, Caccavale S, Mottola M, Pastore F, Argenziano G, Creta M, Calogero A, et al. Multidisciplinary Approach to the Diagnosis and Therapy of Mycosis Fungoides. Healthcare. 2023; 11(4):614. https://doi.org/10.3390/healthcare11040614
Chicago/Turabian StyleVitiello, Paola, Caterina Sagnelli, Andrea Ronchi, Renato Franco, Stefano Caccavale, Maria Mottola, Francesco Pastore, Giuseppe Argenziano, Massimiliano Creta, Armando Calogero, and et al. 2023. "Multidisciplinary Approach to the Diagnosis and Therapy of Mycosis Fungoides" Healthcare 11, no. 4: 614. https://doi.org/10.3390/healthcare11040614