
The Relative Distance Prediction of Transmembrane Protein Surface Residue Based on Improved Residual Networks

Abstract
:1. Introduction
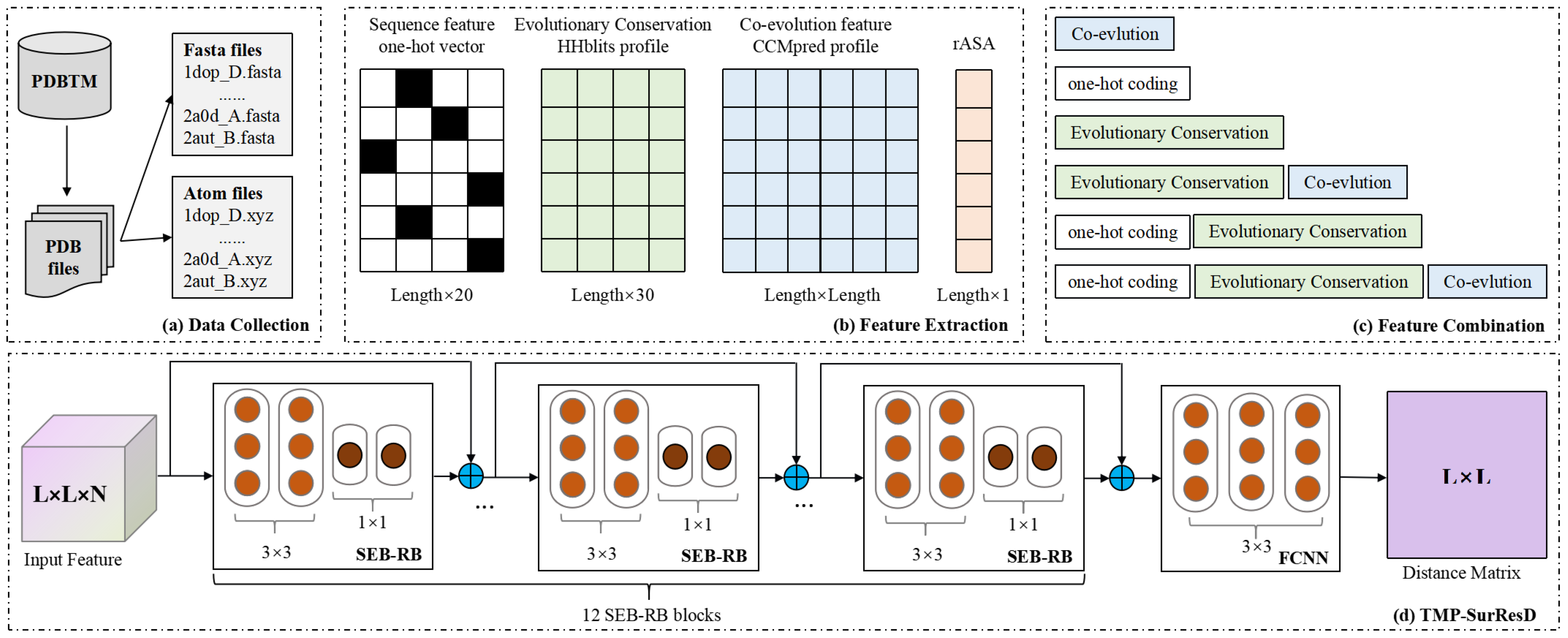
2. Materials and Methods
2.1. Benchmark Dataset
2.2. Protein Sequence Descriptors
2.2.1. Sequence Encoding
2.2.2. Evolutionary Conservation
2.2.3. Co-Evolutionary Information
2.2.4. Relative Solution Accessibility
2.3. The Representation of Residue Distance
2.3.1. Feature Combination
2.3.2. Label Matrix Generation
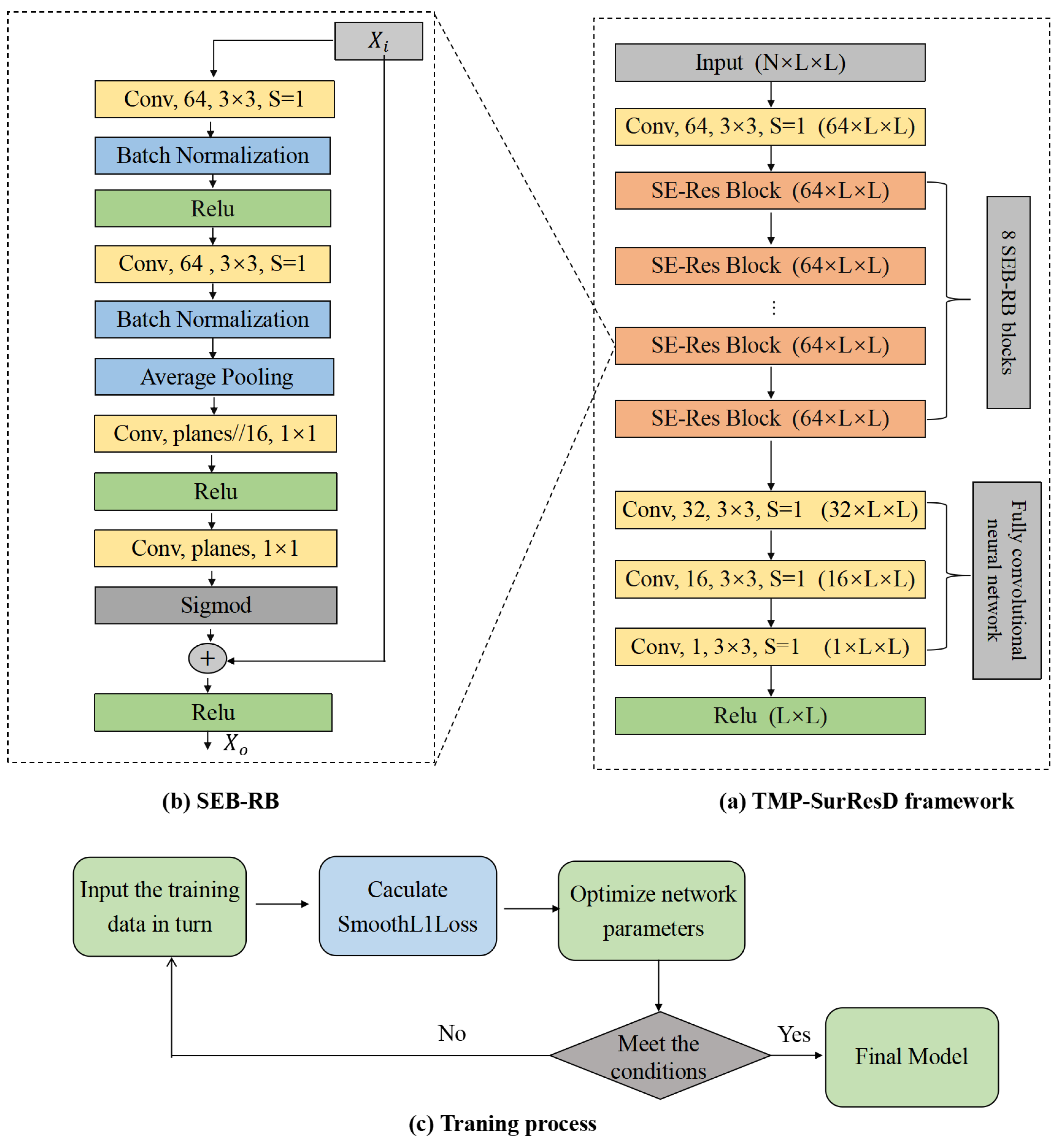
2.4. Deep Learning Model Details
2.4.1. Model Design
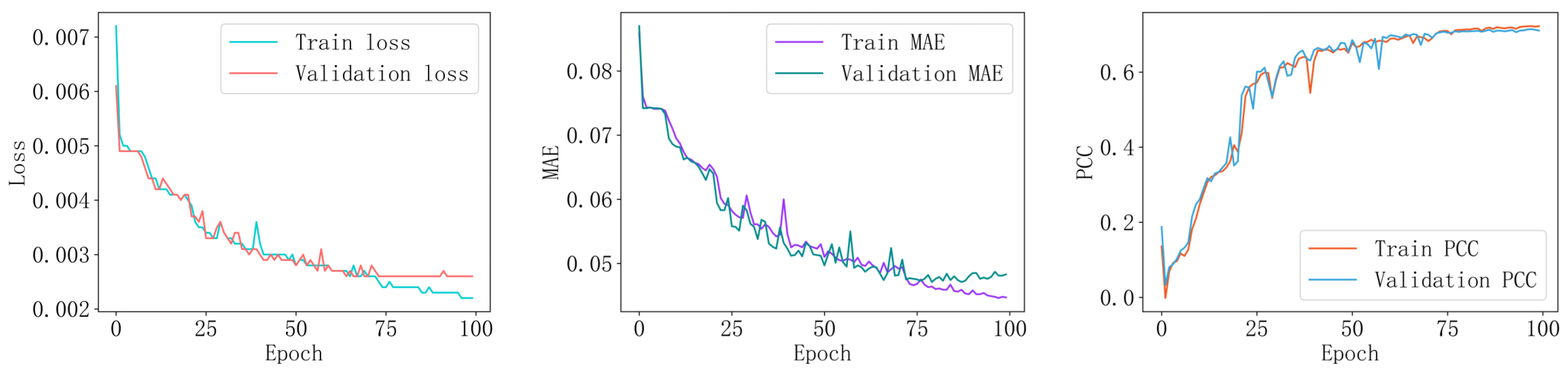
2.4.2. Model Training
2.5. Performance Metrics
3. Results
3.1. Characteristic Validity Analysis
3.2. Network Structure Analysis
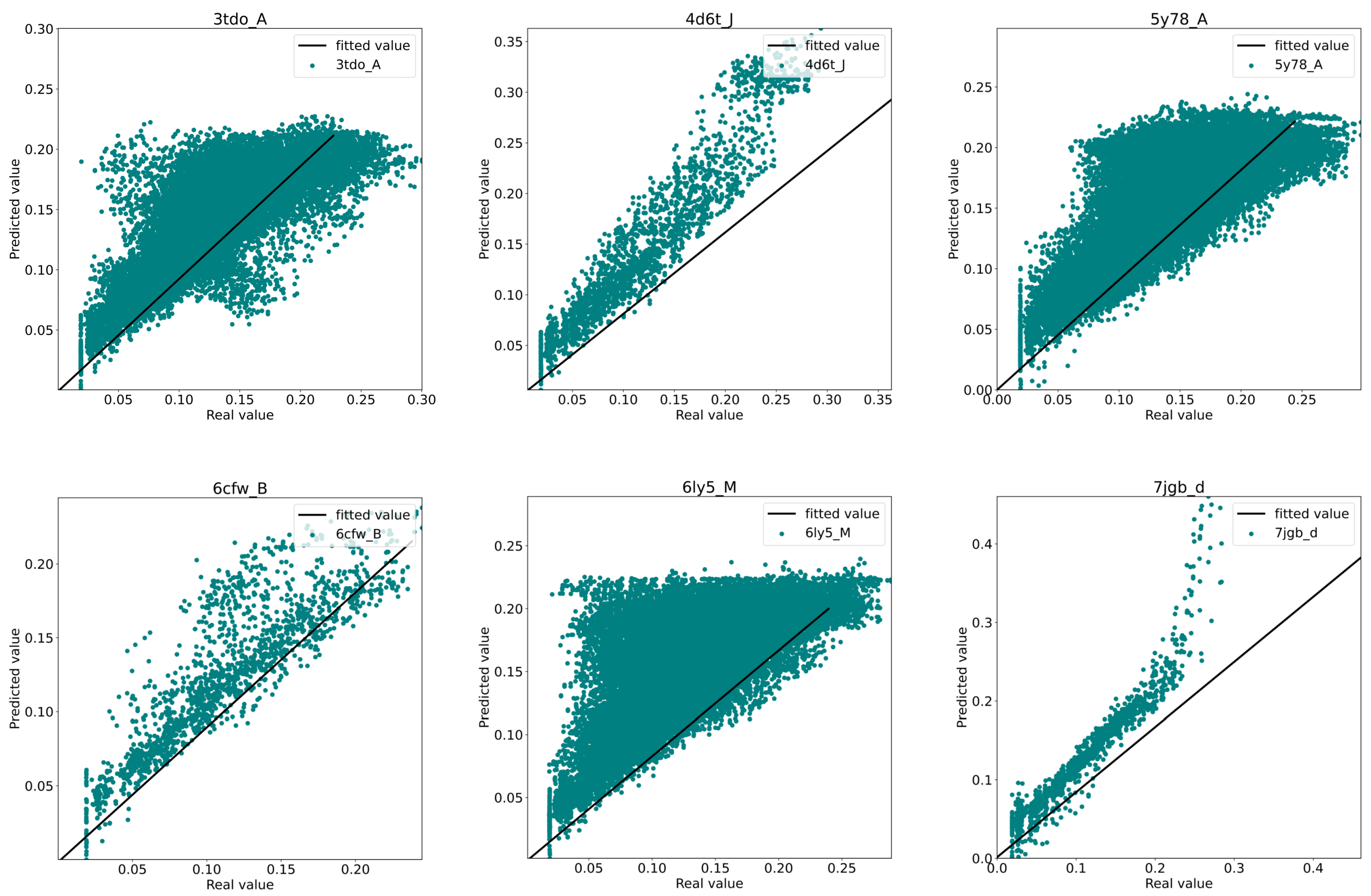
3.3. Model Performance Analysis
3.4. The Setting of Residue-Residue Distances Threshold
3.5. Comparison with Residue Contact Prediction Models
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
Abbreviations
| TMP | transmembrane protein |
| RB | the residual block |
| SEB | the Squeeze-and-Excitation block |
| PCC | Pearson correlation coefficient |
| MCC | Matthews correlation coefficient |
| SurTrain | the training set |
| SurValid | the validation set |
| SurTest | the test set |
| OH | the one-hot encoding |
| EC | evolutionary conservation |
| MSA | multiple sequence alignment |
| rASA | the relative solvent accessible surface area |
| PSSM | position-specific scoring matrix |
| CCM | CCMpred |
| FCNN | full convolutional neural network |
| MAE | mean absolute error |
| MAE | mean square error |
| ReLU | rectified linear activation function |
| ELU | exponential linear unit |
| CNN | convolutional neural network |
References
- Qu, J.; Yin, S.S.; Wang, H. Prediction of Metal Ion Binding Sites of Transmembrane Proteins. Comput. Math. Methods Med. 2021, 2021, 2327832. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Flynn, A.D. Drugging Membrane Protein Interactions. Annu. Rev. Biomed. Eng. 2016, 18, 51–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaucha, J.; Heinzinger, M.; Kulandaisamy, A.; Kataka, E.; Salvádor, L.; Popov, P.; Rost, B.; Gromiha, M.M.; Zhorov, B.S.; Frishman, D. Mutations in transmembrane proteins: Diseases, evolutionary insights, prediction and comparison with globular proteins. Briefings Bioinform. 2021, 22, bbaa132. [Google Scholar] [CrossRef] [PubMed]
- Mashayekhi, V.; Mocellin, O.; Fens, M.H.A.M.; Krijger, G.C.; Brosens, L.A.A.; Oliveira, S. Targeting of promising transmembrane proteins for diagnosis and treatment of pancreatic ductal adenocarcinoma. Theranostics 2021, 11, 9022–9037. [Google Scholar] [CrossRef]
- Liu, Z.; Gong, Y.; Guo, Y.; Zhang, X.; Lu, C.; Zhang, L.; Wang, H. TMP- SSurface2: A Novel Deep Learning-Based Surface Accessibility Predictor for Transmembrane Protein Sequence. Front. Genet. 2021, 12, 656140. [Google Scholar] [CrossRef]
- Lu, C.; Liu, Z.; Zhang, E.; He, F.; Ma, Z.; Wang, H. MPLs-Pred: Predicting Membrane Protein-Ligand Binding Sites Using Hybrid Sequence-Based Features and Ligand-Specific Models. Int. J. Mol. Sci. 2019, 20, 3120. [Google Scholar] [CrossRef] [Green Version]
- Kovalenko, O.; Metcalf, D.; DeGrado, W.; Hemler, M. Structural organization and interactions of transmembrane domains in tetraspanin proteins. BMC Struct. Biol. 2005, 5, 11. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Sun, S.; Li, Z.; Zhang, R.; Xu, J. Accurate De Novo Prediction of Protein Contact Map by Ultra-Deep Learning Model. PLoS Comput. Biol. 2016, 13, e1005324. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Huang, Y.; Bei, Z.; Ju, Z.; Meng, J.; Hao, M.; Zhang, J.; Zhang, H.; Xi, W. Inter-Residue Distance Prediction from Duet Deep Learning Models. Front. Genet. 2022, 13, 887491. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, Y.; Ma, Z. In silico Prediction of Human Secretory Proteins in Plasma Based on Discrete Firefly Optimization and Application to Cancer Biomarkers Identification. Front. Genet. 2019, 10, 542. [Google Scholar] [CrossRef] [Green Version]
- Rose, P.W.; Beran, B.; Bi, C.; Bluhm, W.; Dimitropoulos, D.; Goodsell, D.S.; Prlić, A.; Quesada, M.; Quinn, G.B.; Westbrook, J.D.; et al. The RCSB Protein Data Bank: Redesigned web site and web services. Nucleic Acids Res. 2011, 39, D392–D401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Yang, Y.; Yu, J.; Wang, X.; Zhao, D.; Xu, D.; Sun, P. DMCTOP: Topology Prediction of Alpha-Helical Transmembrane Protein Based on Deep Multi-Scale Convolutional Neural Network. In Proceedings of the 2019 IEEE International Conference on Bioinformatics and Biomedicine (BIBM), San Diego, CA, USA, 18–21 November 2019; pp. 36–43. [Google Scholar]
- Tunyasuvunakool, K.; Adler, J.; Wu, Z.; Green, T.; Zielinski, M.; Zídek, A.; Bridgland, A.; Cowie, A.; Meyer, C.; Laydon, A.; et al. Highly accurate protein structure prediction for the human proteome. Nature 2021, 596, 590–596. [Google Scholar] [CrossRef] [PubMed]
- Hönigschmid, P.; Frishman, D. Accurate prediction of helix interactions and residue contacts in membrane proteins. J. Struct. Biol. 2016, 194, 112–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Shen, H.B. MemBrain-contact 2.0: A new two-stage machine learning model for the prediction enhancement of transmembrane protein residue contacts in the full chain. Bioinformatics 2018, 34, 230–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, S.; Oruç, T.; Mead, L.; Rehman, M.F.; Thomas, C.M.; Butterworth, S.; Winn, P.J. DeepCDpred: Inter-residue distance and contact prediction for improved prediction of protein structure. PLoS ONE 2019, 14, e0205214. [Google Scholar] [CrossRef] [Green Version]
- Ding, W.; Gong, H. Predicting the Real-Valued Inter-Residue Distances for Proteins. Adv. Sci. 2020, 7, 2001314. [Google Scholar] [CrossRef]
- Du, Z.; Peng, Z.; Yang, J. Toward the assessment of predicted inter-residue distance. Bioinformatics 2022, 38, 962–969. [Google Scholar] [CrossRef]
- Wu, T.; Guo, Z.; Hou, J.; Cheng, J. DeepDist: Real-value inter-residue distance prediction with deep residual convolutional network. BMC Bioinform. 2020, 22, 30. [Google Scholar]
- Kuhlman, B.; Bradley, P. Advances in protein structure prediction and design. Nat. Rev. Mol. Cell Biol. 2019, 20, 681–697. [Google Scholar] [CrossRef] [PubMed]
- Senior, A.W.; Evans, R.; Jumper, J.M.; Kirkpatrick, J.; Sifre, L.; Green, T.; Qin, C.; Zídek, A.; Nelson, A.W.R.; Bridgland, A.; et al. Improved protein structure prediction using potentials from deep learning. Nature 2020, 577, 706–710. [Google Scholar] [CrossRef]
- Ju, F.; Zhu, J.; Shao, B.; Kong, L.; Liu, T.Y.; Zheng, W.; Bu, D. CopulaNet: Learning residue co-evolution directly from multiple sequence alignment for protein structure prediction. Nat. Commun. 2020, 12, 2535. [Google Scholar] [CrossRef]
- Russakovsky, O.; Deng, J.; Su, H.; Krause, J.; Satheesh, S.; Ma, S.; Huang, Z.; Karpathy, A.; Khosla, A.; Bernstein, M.S.; et al. ImageNet Large Scale Visual Recognition Challenge. Int. J. Comput. Vis. 2015, 115, 211–252. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.; Shen, L.; Sun, G. Squeeze-and-Excitation Networks. In Proceedings of the 2018 IEEE/CVF Conference on Computer Vision and Pattern Recognition, Salt Lake City, UT, USA, 18–22 June 2018; pp. 7132–7141. [Google Scholar]
- Kozma, D.; Simon, I.; Tusnády, G.E. PDBTM: Protein Data Bank of transmembrane proteins after 8 years. Nucleic Acids Res. 2013, 41, D524–D529. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Niu, B.; Gao, Y.; Fu, L.; Li, W. CD-HIT Suite: A web server for clustering and comparing biological sequences. Bioinformatics 2010, 26, 680–682. [Google Scholar] [CrossRef] [PubMed]
- Luo, F.; Wang, M.; Liu, Y.; Zhao, X.; Li, A. DeepPhos: Prediction of protein phosphorylation sites with deep learning. Bioinformatics 2019, 35, 2766–2773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawashima, S.; Ogata, H.; Kanehisa, M. AAindex: Amino Acid Index Database. Nucleic Acids Res. 1999, 27, 368–369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, H.; Chou, K.C. PseAAC: A flexible web server for generating various kinds of protein pseudo amino acid composition. Anal. Biochem. 2008, 373, 386–388. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Gong, Y.; Bao, Y.; Guo, Y.; Wang, H.; Lin, G.N. TMPSS: A Deep Learning-Based Predictor for Secondary Structure and Topology Structure Prediction of Alpha-Helical Transmembrane Proteins. Front. Bioeng. Biotechnol. 2020, 8, 629937. [Google Scholar] [CrossRef]
- Lim, S.; Lu, Y.; Cho, C.Y.; Sung, I.; Kim, J.; Kim, Y.; Park, S.; Kim, S. A review on compound-protein interaction prediction methods: Data, format, representation and model. Comput. Struct. Biotechnol. J. 2021, 19, 1541–1556. [Google Scholar] [CrossRef]
- Ding, H.; Li, D. Identification of mitochondrial proteins of malaria parasite using analysis of variance. Amino Acids 2015, 47, 329–333. [Google Scholar] [CrossRef]
- ElAbd, H.; Bromberg, Y.; Hoarfrost, A.; Lenz, T.L.; Franke, A.; Wendorff, M. Amino acid encoding for deep learning applications. BMC Bioinform. 2020, 21, 235. [Google Scholar] [CrossRef]
- Zeng, B.; Hönigschmid, P.; Frishman, D. Residue co-evolution helps predict interaction sites in α-helical membrane proteins. J. Struct. Biol. 2019, 206 2, 156–169. [Google Scholar] [CrossRef]
- Remmert, M.; Biegert, A.; Hauser, A.; Söding, J. HHblits: Lightning-fast iterative protein sequence searching by HMM-HMM alignment. Nat. Methods 2012, 9, 173–175. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Zheng, W.; Mortuza, S.M.; Li, Y.; Zhang, Y. DeepMSA: Constructing deep multiple sequence alignment to improve contact prediction and fold-recognition for distant-homology proteins. Bioinformatics 2020, 36, 2105–2112. [Google Scholar] [CrossRef] [PubMed]
- de Juan, D.; Pazos, F.; Valencia, A. Emerging methods in protein co-evolution. Nat. Reviews. Genet. 2013, 14, 249–261. [Google Scholar] [CrossRef]
- Seemayer, S.; Gruber, M.; Söding, J. CCMpred—Fast and precise prediction of protein residue–residue contacts from correlated mutations. Bioinformatics 2014, 30, 3128–3130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haldane, A.; Levy, R.M. Influence of multiple-sequence-alignment depth on Potts statistical models of protein covariation. Phys. Rev. E 2019, 99, 032405. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Wang, S. AcconPred: Predicting Solvent Accessibility and Contact Number Simultaneously by a Multitask Learning Framework under the Conditional Neural Fields Model. BioMed Res. Int. 2015, 2015, 678764. [Google Scholar] [CrossRef] [Green Version]
- Jeong, J.C.; Lin, X.; Chen, X.W. On Position-Specific Scoring Matrix for Protein Function Prediction. IEEE/ACM Trans. Comput. Biol. Bioinform. 2011, 8, 308–315. [Google Scholar] [CrossRef]
- Eigen, D.; Puhrsch, C.; Fergus, R. Depth Map Prediction from a Single Image using a Multi-Scale Deep Network. In Advances in Neural Information Processing Systems 27 (NIPS 2014); MIT Press: Cambridge, MA, USA, 2014; pp. 2366–2374. [Google Scholar]
- Laina, I.; Rupprecht, C.; Belagiannis, V.; Tombari, F.; Navab, N. Deeper Depth Prediction with Fully Convolutional Residual Networks. In Proceedings of the 2016 Fourth International Conference on 3D Vision (3DV), Stanford, CA, USA, 25–28 October 2016; pp. 239–248. [Google Scholar]
- Chen, S.; Tang, M.; Kan, J. Monocular image depth prediction without depth sensors: An unsupervised learning method. Appl. Soft Comput. 2020, 97, 106804. [Google Scholar] [CrossRef]
- Adhikari, B. DEEPCON: Protein Contact Prediction using Dilated Convolutional Neural Networks with Dropout. bioRxiv 2019. [Google Scholar] [CrossRef]
- Jones, D.T.; Kandathil, S.M. High precision in protein contact prediction using fully convolutional neural networks and minimal sequence features. Bioinformatics 2018, 34, 3308–3315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nair, V.; Hinton, G.E. Rectified Linear Units Improve Restricted Boltzmann Machines. In Proceedings of the International Conference on Machine Learning, Haifa, Israel, 21–24 June 2010. [Google Scholar]
- He, K.; Zhang, X.; Ren, S.; Sun, J. Deep Residual Learning for Image Recognition. In Proceedings of the 2016 IEEE Conference on Computer Vision and Pattern Recognition (CVPR), Santiago, Chile, 26 June–1 July 2016; pp. 770–778. [Google Scholar]
- Paszke, A.; Gross, S.; Massa, F.; Lerer, A.; Bradbury, J.; Chanan, G.; Killeen, T.; Lin, Z.; Gimelshein, N.; Antiga, L.; et al. PyTorch: An Imperative Style, High-Performance Deep Learning Library. arXiv 2019, arXiv:1912.01703. [Google Scholar]
- Kingma, D.P.; Ba, J. Adam: A Method for Stochastic Optimization. arXiv 2015, arXiv:1412.6980. [Google Scholar]
- DeGhett, V.J. Effective use of Pearson’s product-moment correlation coefficient: An additional point. Anim. Behav. 2014, 98, e1–e2. [Google Scholar] [CrossRef]
- Gromiha, M.M.; Selvaraj, S. Inter-residue interactions in protein folding and stability. Prog. Biophys. Mol. Biol. 2004, 86, 235–277. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Kim, S. Environment-dependent residue contact energies for proteins. Proc. Natl. Acad. Sci. USA 2000, 97, 2550–2555. [Google Scholar] [CrossRef] [Green Version]
- Latek, D.; Kolinski, A. Contact prediction in protein modeling: Scoring, folding and refinement of coarse-grained models. BMC Struct. Biol. 2008, 8, 36. [Google Scholar] [CrossRef] [Green Version]
- Lo, A.; Chiu, Y.Y.; Rødland, E.A.; Lyu, P.C.; Sung, T.Y.; Hsu, W.L. Predicting helix–helix interactions from residue contacts in membrane proteins. Bioinformatics 2009, 25, 996–1003. [Google Scholar] [CrossRef] [Green Version]
- Morcos, F.; Pagnani, A.; Lunt, B.; Bertolino, A.; Marks, D.S.; Sander, C.; Zecchina, R.; Onuchic, J.N.; Hwa, T.; Weigt, M. Direct-coupling analysis of residue coevolution captures native contacts across many protein families. Proc. Natl. Acad. Sci. USA 2011, 108, E1293–E1301. [Google Scholar] [CrossRef] [Green Version]
- Kamisetty, H.; Ovchinnikov, S.; Baker, D. Assessing the utility of coevolution-based residue–residue contact predictions in a sequence- and structure-rich era. Proc. Natl. Acad. Sci. USA 2013, 110, 15674–15679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Jang, R.; Zhang, Y.; Shen, H. High-accuracy prediction of transmembrane inter-helix contacts and application to GPCR 3D structure modeling. Bioinformatics 2013, 29 20, 2579–2587. [Google Scholar] [CrossRef] [Green Version]
- Jones, D.T.; Singh, T.; Kosciólek, T.; Tetchner, S.J. MetaPSICOV: Combining coevolution methods for accurate prediction of contacts and long range hydrogen bonding in proteins. Bioinformatics 2014, 31, 999–1006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaján, L.; Hopf, T.A.; Kalaš, M.; Marks, D.S.; Rost, B. FreeContact: Fast and free software for protein contact prediction from residue co-evolution. BMC Bioinform. 2014, 15, 85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Huang, Q.; Bei, Z.; Wei, Y.; Floudas, C.A. COMSAT: Residue contact prediction of transmembrane proteins based on support vector machines and mixed integer linear programming. Proteins Struct. Funct. Bioinform. 2016, 84, 332–348. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Jin, Q.Y.; Zhang, B.; Shen, H. R2C: Improving ab initio residue contact map prediction using dynamic fusion strategy and Gaussian noise filter. Bioinformatics 2016, 32 16, 2435–2443. [Google Scholar] [CrossRef]
- Hanson, J.; Paliwal, K.K.; Litfin, T.; Yang, Y.; Zhou, Y. Accurate prediction of protein contact maps by coupling residual two-dimensional bidirectional long short-term memory with convolutional neural networks. Bioinformatics 2018, 34, 4039–4045. [Google Scholar] [CrossRef]
- Fang, C.; Jia, Y.; Hu, L.; Lu, Y.; Wang, H. IMPContact: An Interhelical Residue Contact Prediction Method. BioMed Res. Int. 2020, 2020, 4569037. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.; Frishman, D. DeepHelicon: Accurate prediction of inter-helical residue contacts in transmembrane proteins by residual neural networks. J. Struct. Biol. 2020, 212, 107574. [Google Scholar] [CrossRef]
- Li, Y.; Hu, J.; Zhang, C.; Yu, D.J.; Zhang, Y. ResPRE: High-accuracy protein contact prediction by coupling precision matrix with deep residual neural networks. Bioinformatics 2019, 35, 4647–4655. [Google Scholar] [CrossRef]
- Zhang, H.; Bei, Z.; Xi, W.; Hao, M.; Ju, Z.; Saravanan, K.M.; Zhang, H.; Guo, N.; Wei, Y. Evaluation of residue-residue contact prediction methods: From retrospective to prospective. PLoS Comput. Biol. 2021, 17, e1009027. [Google Scholar] [CrossRef] [PubMed]
- Zimmer, J.; Nam, Y.; Rapoport, T.A. Structure of a complex of the ATPase SecA and the protein-translocation channel. Nature 2008, 455, 936–943. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.T.; Buchan, D.W.A.; Cozzetto, D.; Pontil, M. PSICOV: Precise structural contact prediction using sparse inverse covariance estimation on large multiple sequence alignments. Bioinformatics 2012, 28, 184–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
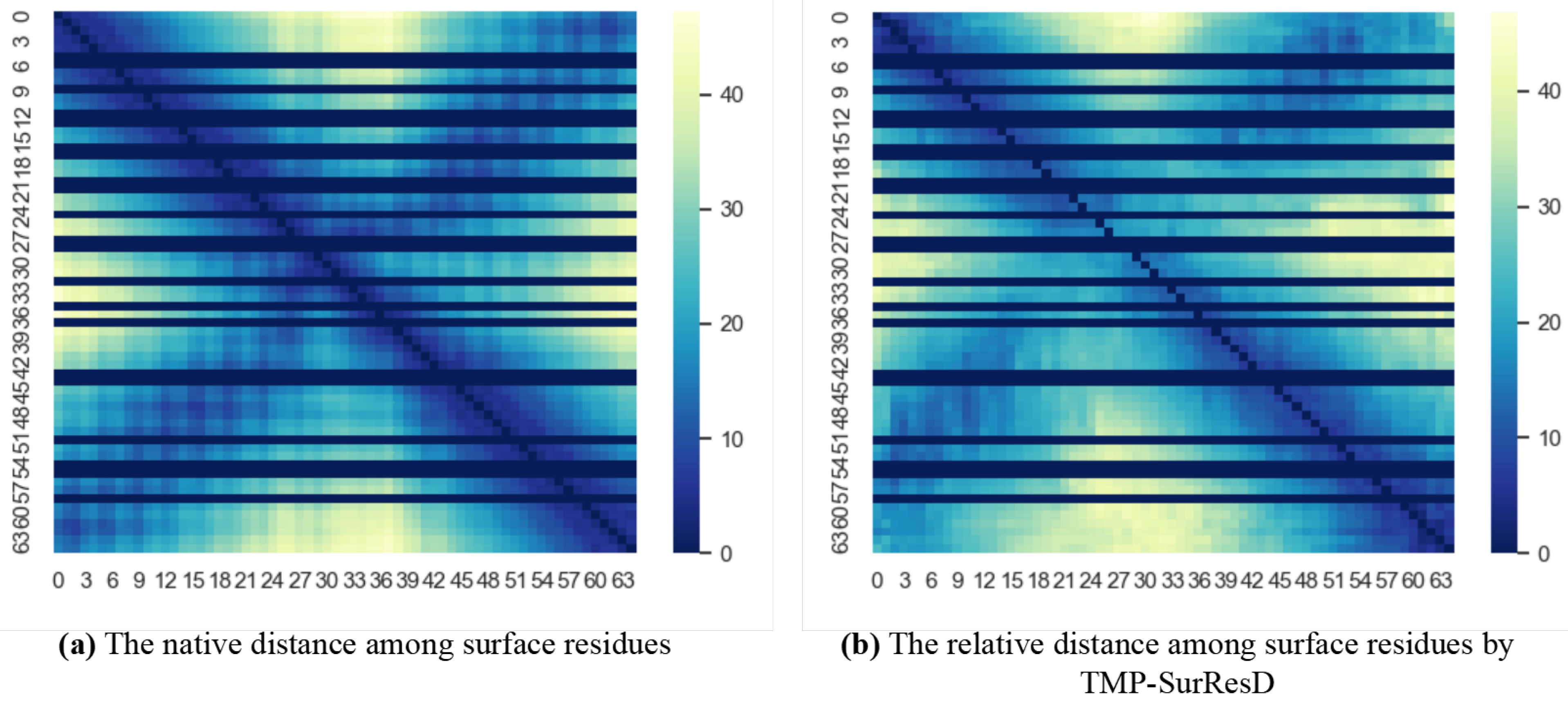{kind=link}
| Features | TraMAE | TraMSE | TraPCC | ValMAE | ValMSE | ValPCC |
|---|---|---|---|---|---|---|
| OH | 0.0358 | 0.1254 | 0.7920 | 0.0759 | 0.0102 | 0.2016 |
| HHM | 0.0637 | 0.0079 | 0.3378 | 0.0645 | 0.0081 | 0.3321 |
| CCM | 0.0387 | 0.0036 | 0.7706 | 0.0447 | 0.0049 | 0.7315 |
| OH+HHM | 0.0602 | 0.0067 | 0.3483 | 0.0698 | 0.0089 | 0.3256 |
| OH+CCM | 0.0306 | 0.0018 | 0.8586 | 0.0566 | 0.0067 | 0.5888 |
| HHM+CCM | 0.0472 | 0.0048 | 0.6983 | 0.0506 | 0.0062 | 0.6825 |
| OH+HHM+CCM | 0.0422 | 0.0038 | 0.7253 | 0.0504 | 0.0059 | 0.6864 |
| SEB-RB Blocks | TraMAE | TraMSE | TraPCC | ValMAE | ValMSE | ValPCC |
|---|---|---|---|---|---|---|
| 5 | 0.0925 | 0.0163 | 0.1757 | 0.0921 | 0.0137 | 0.2166 |
| 6 | 0.0470 | 0.0048 | 0.6965 | 0.0492 | 0.0055 | 0.6877 |
| 7 | 0.0482 | 0.0051 | 0.6932 | 0.0490 | 0.0054 | 0.6837 |
| 8 | 0.0472 | 0.0048 | 0.6983 | 0.0506 | 0.0062 | 0.6825 |
| 9 | 0.0471 | 0.0047 | 0.6972 | 0.0493 | 0.0058 | 0.6980 |
| 10 | 0.0483 | 0.0051 | 0.6932 | 0.0484 | 0.0054 | 0.6884 |
| 11 | 0.0463 | 0.0047 | 0.7091 | 0.0474 | 0.0052 | 0.7013 |
| 12 | 0.0447 | 0.0045 | 0.7222 | 0.0483 | 0.0052 | 0.7105 |
| 13 | 0.0458 | 0.0046 | 0.7173 | 0.0480 | 0.0055 | 0.6973 |
| 14 | 0.0433 | 0.0041 | 0.7243 | 0.0481 | 0.0055 | 0.6991 |
| 15 | 0.0470 | 0.0048 | 0.7030 | 0.0481 | 0.0054 | 0.6942 |
| 16 | 0.0449 | 0.0044 | 0.7225 | 0.0489 | 0.0056 | 0.6997 |
| 17 | 0.0425 | 0.0040 | 0.7335 | 0.0464 | 0.0052 | 0.7104 |
| Layers | TraMAE | TraMSE | TraPCC | ValMAE | ValMSE | ValPCC |
|---|---|---|---|---|---|---|
| 1 | 0.0438 | 0.0042 | 0.7170 | 0.0478 | 0.0054 | 0.6911 |
| 2 | 0.0474 | 0.0048 | 0.6952 | 0.0509 | 0.0062 | 0.6726 |
| 3 | 0.0447 | 0.0045 | 0.7222 | 0.0483 | 0.0052 | 0.7105 |
| 4 | 0.0456 | 0.0046 | 0.7161 | 0.0470 | 0.0054 | 0.7061 |
| Function | TraMAE | TraMSE | TraPCC | ValMAE | ValMSE | ValPCC |
|---|---|---|---|---|---|---|
| ELU | 0.0502 | 0.0054 | 0.6628 | 0.0504 | 0.0056 | 0.6602 |
| ReLU | 0.0447 | 0.0045 | 0.7222 | 0.0483 | 0.0052 | 0.7105 |
| Features | MAE | MSE | PCC |
|---|---|---|---|
| CCM | 0.0473 | 0.0054 | 0.7238 |
| HHM+CCM | 0.0504 | 0.0055 | 0.6999 |
| OH+HHM+CCM | 0.0522 | 0.0063 | 0.6878 |
| Threshold | ACC | Precision | Recall | F1 | MCC |
|---|---|---|---|---|---|
| 5.5 | 0.9825 | 0.9055 | 0.1725 | 0.2755 | 0.3667 |
| 6 | 0.9777 | 0.9434 | 0.1837 | 0.2950 | 0.3887 |
| 6.5 | 0.9737 | 0.9607 | 0.1951 | 0.3128 | 0.4066 |
| 7 | 0.9725 | 0.9666 | 0.2219 | 0.3483 | 0.4372 |
| 7.5 | 0.9728 | 0.9682 | 0.2658 | 0.4021 | 0.4814 |
| 8 | 0.9736 | 0.9656 | 0.3221 | 0.4654 | 0.5313 |
| 8.5 | 0.9728 | 0.9619 | 0.3631 | 0.5089 | 0.5651 |
| 9 | 0.9697 | 0.9620 | 0.3794 | 0.5274 | 0.5788 |
| 9.5 | 0.9684 | 0.9655 | 0.4039 | 0.5534 | 0.5997 |
| 10 | 0.9645 | 0.9676 | 0.4065 | 0.5576 | 0.6020 |
| 10.5 | 0.9594 | 0.9674 | 0.3973 | 0.5498 | 0.5940 |
| 11 | 0.9546 | 0.9647 | 0.3895 | 0.5422 | 0.5859 |
| Model | ProNum | ACC | Precision | Recall | F1 | MCC |
|---|---|---|---|---|---|---|
| PSICOV | 128 | 0.0011 | 0.9062 | 0.0011 | 0.0022 | 0.0000 |
| Freecontact | 178 | 0.0612 | 0.9831 | 0.0612 | 0.0959 | 0.0000 |
| DEEPCON | 178 | 0.0024 | 0.9944 | 0.0024 | 0.0047 | 0.0000 |
| TMP-SurResD | 178 | 0.9736 | 0.9656 | 0.3221 | 0.4654 | 0.5313 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, Q.; Guo, Y.; Jiang, J.; Qu, J.; Zhang, L.; Wang, H. The Relative Distance Prediction of Transmembrane Protein Surface Residue Based on Improved Residual Networks. Mathematics 2023, 11, 642. https://doi.org/10.3390/math11030642
Chen Q, Guo Y, Jiang J, Qu J, Zhang L, Wang H. The Relative Distance Prediction of Transmembrane Protein Surface Residue Based on Improved Residual Networks. Mathematics. 2023; 11(3):642. https://doi.org/10.3390/math11030642
Chicago/Turabian StyleChen, Qiufen, Yuanzhao Guo, Jiuhong Jiang, Jing Qu, Li Zhang, and Han Wang. 2023. "The Relative Distance Prediction of Transmembrane Protein Surface Residue Based on Improved Residual Networks" Mathematics 11, no. 3: 642. https://doi.org/10.3390/math11030642