In-Depth Analysis of the Plasma Proteome in ME/CFS Exposes Disrupted Ephrin-Eph and Immune System Signaling



Abstract
:
1. Introduction
2. Materials and Methods
2.1. Cohort and Blood Sampling
2.2. Proteomics Data Acquisition and Handling
2.3. Data Analysis
3. Results
3.1. Population Statistics
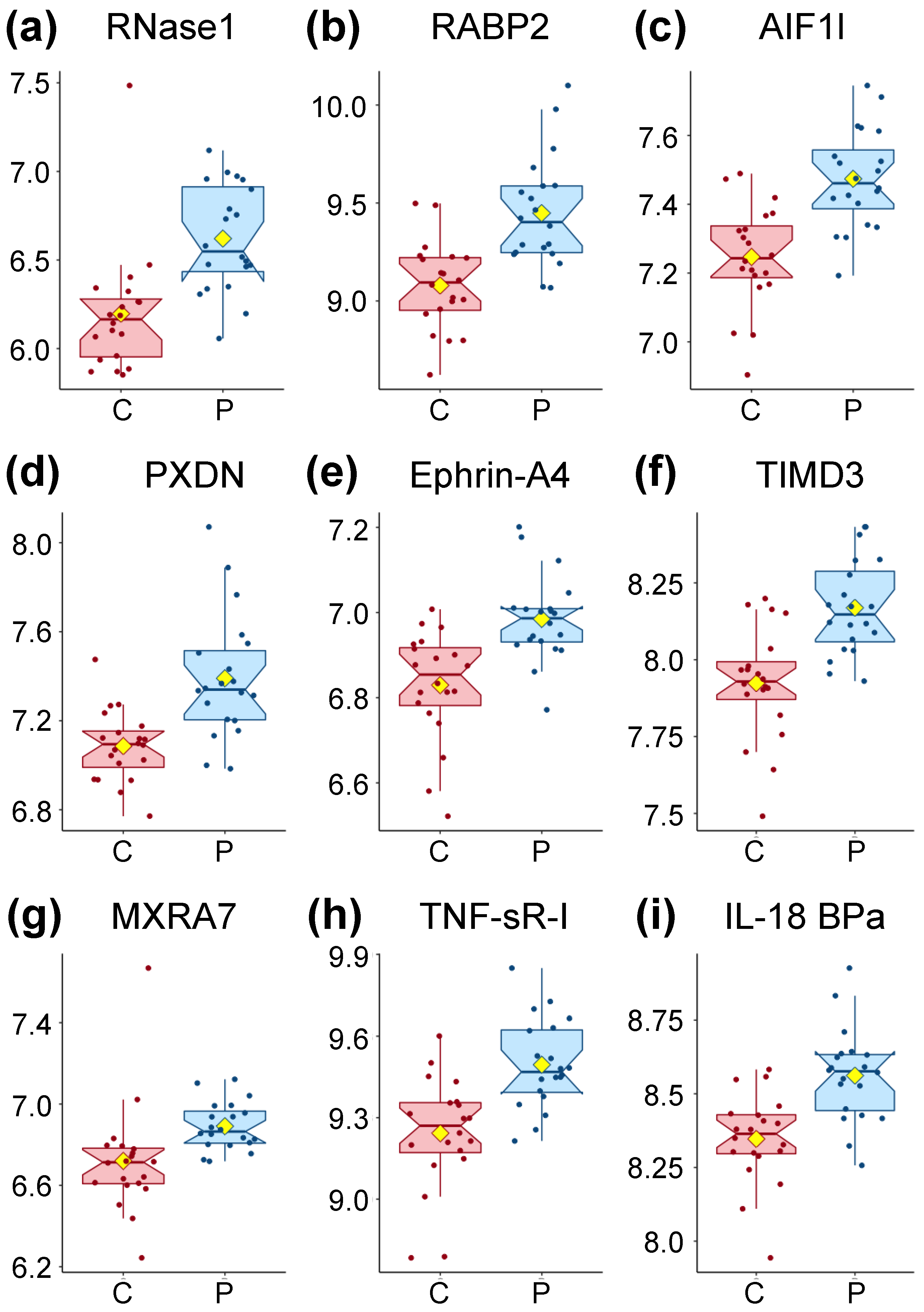
3.2. Nine Proteins Are Significantly Higher in ME/CFS Patients
3.3. Additional Proteins Emerging from a Relaxed q-Value Threshold
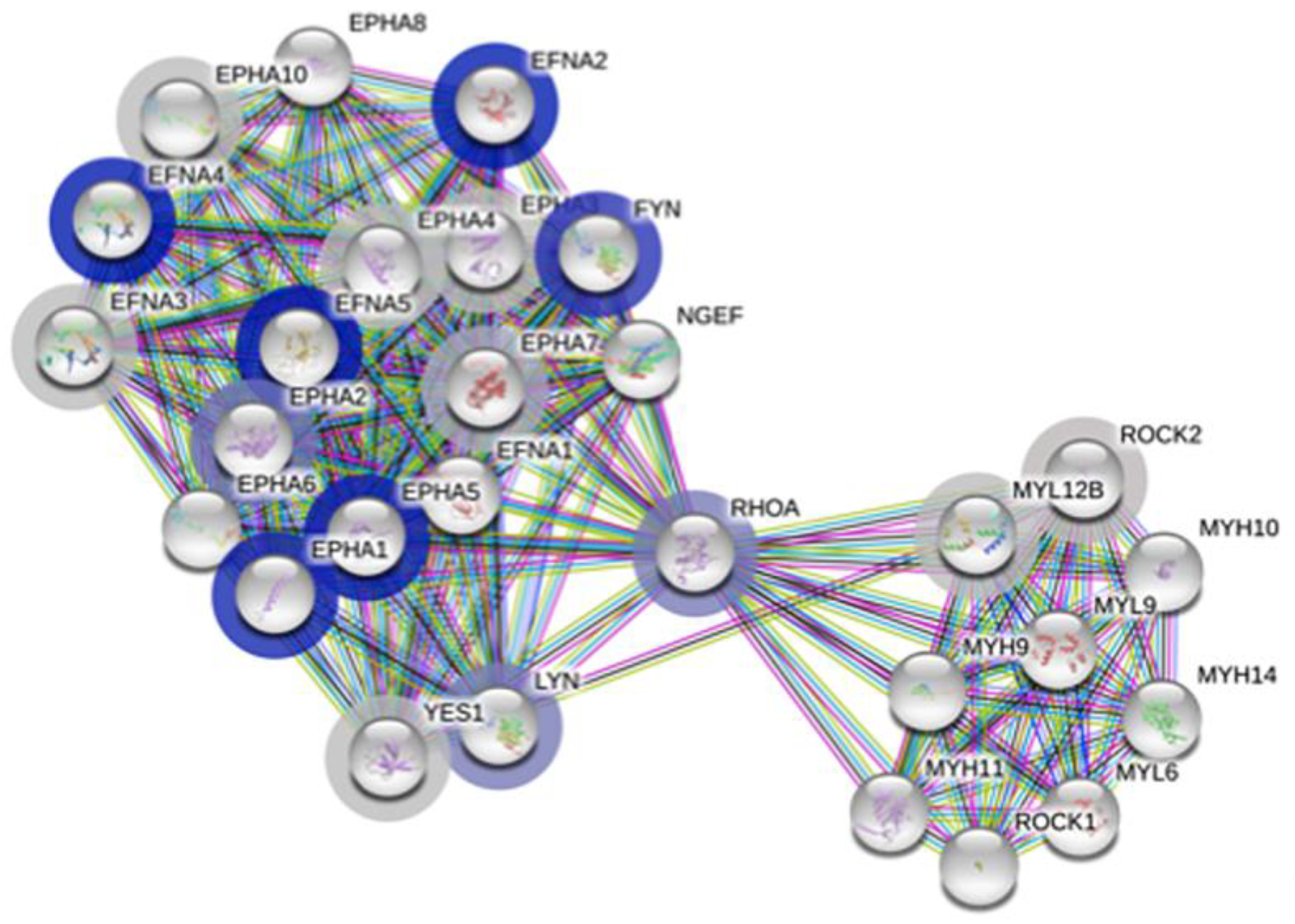
3.4. Protein–Protein Association Analysis Points Both to the Ephrin Family and Immune Metabolism
3.5. High Levels of Prediction Are Achieved Using Univariate ROC Curve Analysis
3.6. Highly Enriched Clusters Include Ephrin-Related Pathways and Glucose
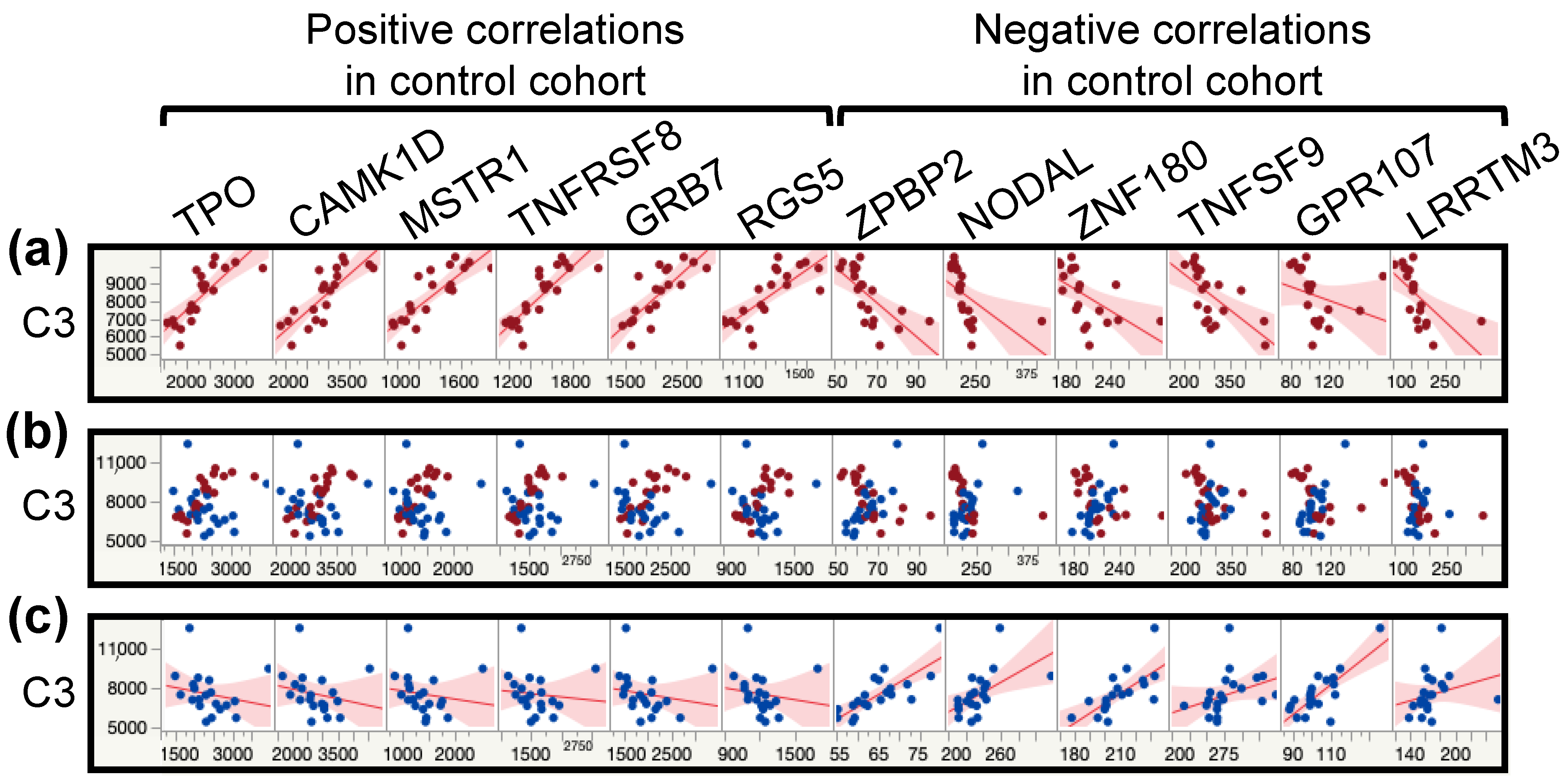
3.7. Protein–Protein Correlations Are Highly Disrupted in the Patient Cohort
4. Discussion
4.1. Most Significantly Different Proteins Are More Abundant in the Plasma of ME/CFS Patients
4.2. Much Evidence Implicates Disrupted Ephrin-Eph Signaling in Our Dataset
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bell, D.S.; Bell, D.S. The Doctor′s Guide to Chronic Fatigue Syndrome: Understanding, Treating, and Living with CFIDS; Addison-Wesley Pub. Co.: Boston, MA, USA, 1994; p. xxii. 275p. [Google Scholar]
- Institute of Medicine. Beyond Myalgic Encephalomyelitis/Chronic Fatigue Syndrome: Redefining An Illness; National Academies Press: Washington, DC, USA, 2015. [Google Scholar]
- Montoya, J.G.; Holmes, T.H.; Anderson, J.N.; Maecker, H.T.; Rosenberg-Hasson, Y.; Valencia, I.J.; Chu, L.; Younger, J.W.; Tato, C.M.; Davis, M.M. Cytokine signature associated with disease severity in chronic fatigue syndrome patients. Proc. Natl. Acad. Sci. USA 2017, 114, E7150–E7158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hornig, M.; Montoya, J.G.; Klimas, N.G.; Levine, S.; Felsenstein, D.; Bateman, L.; Peterson, D.L.; Gottschalk, C.G.; Schultz, A.F.; Che, X.; et al. Distinct plasma immune signatures in ME/CFS are present early in the course of illness. Sci. Adv. 2015, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naviaux, R.K.; Naviaux, J.C.; Li, K.; Bright, A.T.; Alaynick, W.A.; Wang, L.; Baxter, A.; Nathan, N.; Anderson, W.; Gordon, E. Metabolic features of chronic fatigue syndrome. Proc. Natl. Acad. Sci. USA 2016, 113, E5472–E5480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fluge, O.; Mella, O.; Bruland, O.; Risa, K.; Dyrstad, S.E.; Alme, K.; Rekeland, I.G.; Sapkota, D.; Rosland, G.V.; Fossa, A.; et al. Metabolic profiling indicates impaired pyruvate dehydrogenase function in myalgic encephalopathy/chronic fatigue syndrome. JCI Insight 2016, 1, e89376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Germain, A.; Ruppert, D.; Levine, S.M.; Hanson, M.R. Metabolic profiling of a myalgic encephalomyelitis/chronic fatigue syndrome discovery cohort reveals disturbances in fatty acid and lipid metabolism. Mol. Biosyst. 2017, 13, 371–379. [Google Scholar] [CrossRef] [Green Version]
- Armstrong, C.W.; McGregor, N.R.; Butt, H.L.; Gooley, P.R. Metabolism in chronic fatigue syndrome. Adv. Clin. Chem. 2014, 66, 121–172. [Google Scholar]
- Germain, A.; Ruppert, D.; Levine, S.M.; Hanson, M.R. Prospective biomarkers from plasma metabolomics of Myalgic Encephalomyelitis/Chronic Fatigue Syndrome implicate redox imbalance in disease symptomatology. Metabolites 2018, 8, 90. [Google Scholar] [CrossRef] [Green Version]
- Fletcher, M.A.; Zeng, X.R.; Barnes, Z.; Levis, S.; Klimas, N.G. Plasma cytokines in women with chronic fatigue syndrome. J. Transl. Med. 2009, 7, 96. [Google Scholar] [CrossRef] [Green Version]
- Milivojevic, M.; Che, X.; Bateman, L.; Cheng, A.; Garcia, B.A.; Hornig, M.; Huber, M.; Klimas, N.G.; Lee, B.; Lee, H.; et al. Plasma proteomic profiling suggests an association between antigen driven clonal B cell expansion and ME/CFS. PLoS ONE 2020, 15, e0236148. [Google Scholar] [CrossRef]
- Klimas, N.G.; Salvato, F.R.; Morgan, R.; Fletcher, M.A. Immunologic abnormalities in chronic fatigue syndrome. J. Clin. Microbiol. 1990, 28, 1403–1410. [Google Scholar] [CrossRef] [Green Version]
- Maher, K.J.; Klimas, N.G.; Fletcher, M.A. Chronic fatigue syndrome is associated with diminished intracellular perforin. Clin. Exp. Immunol. 2005, 142, 505–511. [Google Scholar] [CrossRef] [PubMed]
- Mandarano, A.H.; Maya, J.; Giloteaux, L.; Peterson, D.L.; Maynard, M.; Gottschalk, C.G.; Hanson, M.R. Myalgic encephalomyelitis/chronic fatigue syndrome patients exhibit altered T cell metabolism and cytokine associations. J. Clin. Investig. 2020, 130, 1491–1505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smits, B.; van den Heuvel, L.; Knoop, H.; Küsters, B.; Janssen, A.; Borm, G.; Bleijenberg, G.; Rodenburg, R.; van Engelen, B. Mitochondrial enzymes discriminate between mitochondrial disorders and chronic fatigue syndrome. Mitochondrion 2011, 11, 735–738. [Google Scholar] [CrossRef] [PubMed]
- Tomas, C.; Brown, A.; Strassheim, V.; Elson, J.L.; Newton, J.; Manning, P. Cellular bioenergetics is impaired in patients with chronic fatigue syndrome. PLoS ONE 2017, 12, e0186802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomas, C.; Brown, A.E.; Newton, J.L.; Elson, J.L. Mitochondrial complex activity in permeabilised cells of chronic fatigue syndrome patients using two cell types. PeerJ 2019, 7, e6500. [Google Scholar] [CrossRef] [Green Version]
- Fukuda, K.; Straus, S.E.; Hickie, I.; Sharpe, M.C.; Dobbins, J.G.; Komaroff, A. The chronic fatigue syndrome: A comprehensive approach to its definition and study. International Chronic Fatigue Syndrome Study Group. Ann. Intern. Med. 1994, 121, 953–959. [Google Scholar] [CrossRef] [PubMed]
- Germain, A.; Barupal, D.K.; Levine, S.M.; Hanson, M.R. Comprehensive circulatory metabolomics in ME/CFS reveals disrupted metabolism of acyl lipids and steroids. Metabolites 2020, 10, 34. [Google Scholar] [CrossRef] [Green Version]
- Brummelman, J.; Raeven, R.H.; Helm, K.; Pennings, J.L.; Metz, B.; van Eden, W.; van Els, C.A.; Han, W.G. Transcriptome signature for dampened Th2 dominance in acellular pertussis vaccine-induced CD4(+) T cell responses through TLR4 ligation. Sci. Rep. 2016, 6, 25064. [Google Scholar] [CrossRef] [Green Version]
- Chan, O.; Sherwin, R. Influence of VMH fuel sensing on hypoglycemic responses. Trends Endocrinol. Metab. 2013, 24, 616–624. [Google Scholar] [CrossRef] [Green Version]
- Chopra, N.; Choudhury, S.; Bhargava, S.; Wajid, S.; Ganguly, N.K. Potentials of "stem cell-therapy" in pancreatic cancer: An update. Pancreatology 2019, 19, 1034–1042. [Google Scholar] [CrossRef]
- Wong, A.P.; Bear, C.E.; Chin, S.; Pasceri, P.; Thompson, T.O.; Huan, L.J.; Ratjen, F.; Ellis, J.; Rossant, J. Directed differentiation of human pluripotent stem cells into mature airway epithelia expressing functional CFTR protein. Nat. Biotechnol. 2012, 30, 876–882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Y.; Ye, Y.; Bao, W.; Yang, Q.; Wang, J.; Liu, Z.; Shi, S. Genome-wide identification of genes essential for podocyte cytoskeletons based on single-cell RNA sequencing. Kidney Int. 2017, 92, 1119–1129. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Li, W.; Hu, Y.; Jiang, Y. Absence of AIF1L contributes to cell migration and a poor prognosis of breast cancer. Onco Targets Ther. 2018, 11, 5485–5498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yasuda-Yamahara, M.; Rogg, M.; Yamahara, K.; Maier, J.I.; Huber, T.B.; Schell, C. AIF1L regulates actomyosin contractility and filopodial extensions in human podocytes. PLoS ONE 2018, 13, e0200487. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.Z.; Liang, L. High expression of PXDN is associated with poor prognosis and promotes proliferation, invasion as well as migration in ovarian cancer. Ann. Diagn. Pathol. 2018, 34, 161–165. [Google Scholar] [CrossRef]
- Peterfi, Z.; Geiszt, M. Peroxidasins: Novel players in tissue genesis. Trends Biochem. Sci. 2014, 39, 305–307. [Google Scholar] [CrossRef]
- Liu, Z.; Xu, Q.; Yang, Q.; Cao, J.; Wu, C.; Peng, H.; Zhang, X.; Chen, J.; Cheng, G.; Wu, Y.; et al. Vascular peroxidase 1 is a novel regulator of cardiac fibrosis after myocardial infarction. Redox Biol. 2019, 22, 101151. [Google Scholar] [CrossRef]
- Jia, C.; Zhang, F.; Zhu, Y.; Qi, X.; Wang, Y. Public data mining plus domestic experimental study defined involvement of the old-yet-uncharacterized gene matrix-remodeling associated 7 (MXRA7) in physiopathology of the eye. Gene 2017, 632, 43–49. [Google Scholar] [CrossRef]
- Ning, J.; Shen, Y.; Wang, T.; Wang, M.; Liu, W.; Sun, Y.; Zhang, F.; Chen, L.; Wang, Y. Altered expression of matrix remodelling associated 7 (MXRA7) in psoriatic epidermis: Evidence for a protective role in the psoriasis imiquimod mouse model. Exp. Dermatol. 2018, 27, 1038–1042. [Google Scholar] [CrossRef]
- Jeon, C.; Sekhon, S.; Yan, D.; Afifi, L.; Nakamura, M.; Bhutani, T. Monoclonal antibodies inhibiting IL-12, -23, and -17 for the treatment of psoriasis. Hum. Vaccin. Immunother. 2017, 13, 2247–2259. [Google Scholar] [CrossRef] [Green Version]
- Leonardi, C.L. Antibodies in the Treatment of Psoriasis: IL-12/23 p40 and IL-17a. Semin. Cutan. Med. Surg. 2016, 35, S74–S77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, D.; Sun, Z.; Jin, Z.; Lei, L.; Liu, Y.; Hu, B.; Wang, B.; Shen, Y.; Wang, Y. Matrix remodeling associated 7 deficiency alleviates carbon tetrachloride-induced acute liver injury in mice. Front. Immunol. 2018, 9, 773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Z.; Shen, Y.; Yin, J.; Xi, F.; Xu, R.; Lin, D.; Saijilafu; Chen, J.; Wang, Y. Matrix remodeling associated 7 promotes differentiation of bone marrow mesenchymal stem cells toward osteoblasts. J. Cell. Physiol. 2019, 234, 18053–18064. [Google Scholar] [CrossRef] [PubMed]
- Dixon, K.O.; Das, M.; Kuchroo, V.K. Human disease mutations highlight the inhibitory function of TIM-3. Nat. Genet. 2018, 50, 1640–1641. [Google Scholar] [CrossRef]
- McDermott, M.F.; Aksentijevich, I.; Galon, J.; McDermott, E.M.; Ogunkolade, B.W.; Centola, M.; Mansfield, E.; Gadina, M.; Karenko, L.; Pettersson, T.; et al. Germline mutations in the extracellular domains of the 55 kDa TNF receptor, TNFR1, define a family of dominantly inherited autoinflammatory syndromes. Cell 1999, 97, 133–144. [Google Scholar] [CrossRef]
- Pucino, V.; Lucherini, O.M.; Perna, F.; Obici, L.; Merlini, G.; Cattalini, M.; La Torre, F.; Maggio, M.C.; Lepore, M.T.; Magnotti, F.; et al. Differential impact of high and low penetrance TNFRSF1A gene mutations on conventional and regulatory CD4+ T cell functions in TNFR1-associated periodic syndrome. J. Leukoc. Biol. 2016, 99, 761–769. [Google Scholar] [CrossRef]
- Yoshida, T.; Friehs, I.; Mummidi, S.; del Nido, P.J.; Addulnour-Nakhoul, S.; Delafontaine, P.; Valente, A.J.; Chandrasekar, B. Pressure overload induces IL-18 and IL-18R expression, but markedly suppresses IL-18BP expression in a rabbit model. IL-18 potentiates TNF-alpha-induced cardiomyocyte death. J. Mol. Cell. Cardiol. 2014, 75, 141–151. [Google Scholar] [CrossRef] [Green Version]
- Naftali, T.; Novick, D.; Gabay, G.; Rubinstein, M.; Novis, B. Interleukin-18 and its binding protein in patients with inflammatory bowel disease during remission and exacerbation. Isr. Med. Assoc. J. 2007, 9, 504–508. [Google Scholar]
- Muhl, H.; Bachmann, M. IL-18/IL-18BP and IL-22/IL-22BP: Two interrelated couples with therapeutic potential. Cell Signal. 2019, 63, 109388. [Google Scholar] [CrossRef]
- VanElzakker, M.B.; Brumfield, S.A.; Lara Mejia, P.S. Neuroinflammation and cytokines in Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS): A critical review of research methods. Front. Neurol. 2018, 9, 1033. [Google Scholar] [CrossRef]
- Chan, M.Y.; Efthymios, M.; Tan, S.H.; Pickering, J.W.; Troughton, R.; Pemberton, C.; Ho, H.H.; Prabath, J.F.; Drum, C.L.; Ling, L.H.; et al. Prioritizing candidates of post-myocardial infarction heart failure using plasma proteomics and single-cell transcriptomics. Circulation 2020, 142, 1408–1421. [Google Scholar] [CrossRef] [PubMed]
- Jeffrey, M.G.; Nathanson, L.; Aenlle, K.; Barnes, Z.M.; Baig, M.; Broderick, G.; Klimas, N.G.; Fletcher, M.A.; Craddock, T.J.A. Treatment avenues in Myalgic Encephalomyelitis/Chronic Fatigue Syndrome: A split-gender pharmacogenomic study of gene-expression modules. Clin. Ther. 2019, 41, 815–835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Condren, M.E.; Bradshaw, M.D. Ivacaftor: A novel gene-based therapeutic approach for cystic fibrosis. J. Pediatr. Pharm. Ther. 2013, 18, 8–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hisert, K.B.; Birkland, T.P.; Schoenfelt, K.Q.; Long, M.E.; Grogan, B.; Carter, S.; Liles, W.C.; McKone, E.F.; Becker, L.; Manicone, A.M.; et al. CFTR modulator therapy enhances peripheral blood monocyte contributions to immune responses in people with cystic fibrosis. Front. Pharm. 2020, 11, 1219. [Google Scholar] [CrossRef] [PubMed]
- Miller, A.C.; Comellas, A.P.; Hornick, D.B.; Stoltz, D.A.; Cavanaugh, J.E.; Gerke, A.K.; Welsh, M.J.; Zabner, J.; Polgreen, P.M. Cystic fibrosis carriers are at increased risk for a wide range of cystic fibrosis-related conditions. Proc. Natl. Acad. Sci. USA 2020, 117, 1621–1627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saint-Criq, V.; Gray, M.A. Role of CFTR in epithelial physiology. Cell. Mol. Life Sci. 2017, 74, 93–115. [Google Scholar] [CrossRef] [Green Version]
- Ji, H.; Zhang, X.; Oh, S.; Mayhew, C.N.; Ulm, A.; Somineni, H.K.; Ericksen, M.; Wells, J.M.; Khurana Hershey, G.K. Dynamic transcriptional and epigenomic reprogramming from pediatric nasal epithelial cells to induced pluripotent stem cells. J. Allergy Clin. Immunol. 2015, 135, 236–244. [Google Scholar] [CrossRef] [Green Version]
- Baraniuk, J.N.; Casado, B.; Maibach, H.; Clauw, D.J.; Pannell, L.K.; Hess, S.S. A Chronic Fatigue Syndrome-related proteome in human cerebrospinal fluid. BMC Neurol. 2005, 5, 22. [Google Scholar] [CrossRef] [Green Version]
- Kania, A.; Klein, R. Mechanisms of ephrin-Eph signalling in development, physiology and disease. Nat. Rev. Mol. Cell Biol. 2016, 17, 240–256. [Google Scholar] [CrossRef]
- Schutzer, S.E.; Angel, T.E.; Liu, T.; Schepmoes, A.A.; Clauss, T.R.; Adkins, J.N.; Camp, D.G.; Holland, B.K.; Bergquist, J.; Coyle, P.K.; et al. Distinct cerebrospinal fluid proteomes differentiate post-treatment lyme disease from chronic fatigue syndrome. PLoS ONE 2011, 6, e17287. [Google Scholar] [CrossRef]
- Darling, T.K.; Lamb, T.J. Emerging roles for Eph receptors and Ephrin ligands in immunity. Front. Immunol. 2019, 10, 1473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Boer, E.C.W.; van Gils, J.M.; van Gils, M.J. Ephrin-Eph signaling usage by a variety of viruses. Pharm. Res. 2020, 159, 105038. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, M.; Asprusten, T.T.; Godang, K.; Leegaard, T.M.; Osnes, L.T.; Skovlund, E.; Tjade, T.; Oie, M.G.; Wyller, V.B.B. Predictors of chronic fatigue in adolescents six months after acute Epstein-Barr virus infection: A prospective cohort study. Brain Behav. Immun. 2019, 75, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Wallace, H.L., 2nd; Natelson, B.; Gause, W.; Hay, J. Human herpesviruses in chronic fatigue syndrome. Clin. Diagn. Lab. Immunol. 1999, 6, 216–223. [Google Scholar]
- Eguchi, A.; Fukuda, S.; Kuratsune, H.; Nojima, J.; Nakatomi, Y.; Watanabe, Y.; Feldstein, A.E. Identification of actin network proteins, talin-1 and filamin-A, in circulating extracellular vesicles as blood biomarkers for human myalgic encephalomyelitis/chronic fatigue syndrome. Brain Behav. Immun. 2020, 84, 106–114. [Google Scholar] [CrossRef]
- Himanen, J.P.; Chumley, M.J.; Lackmann, M.; Li, C.; Barton, W.A.; Jeffrey, P.D.; Vearing, C.; Geleick, D.; Feldheim, D.A.; Boyd, A.W.; et al. Repelling class discrimination: Ephrin-A5 binds to and activates EphB2 receptor signaling. Nat. Neurosci. 2004, 7, 501–509. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gender (n) | Female | Controls | ME/CFS |
|---|---|---|---|
| 20 | 20 | ||
| Age | Mean +/− SD | 46 +/− 13.3 | 49.2 +/− 11.8 |
| Median (min-max) | 50.5 (27–66) | 52 (27–68) | |
| BMI | Mean +/− SD | 23.2 +/− 3.4 | 24 +/− 5.1 |
| Median (min-max) | 21.8 (18.1–29.3) | 22.6 (16.8–37.4) | |
| Type of onset | Gradual | NA | 35% |
| Sudden | NA | 65% | |
| Gut symptoms * | 5% | 55% | |
| Positive tilt table test ** (n = 12) | ND | 67% | |
| Bell’s disability scale *** | 10–20 | 0 | 7 |
| 30–40 | 0 | 7 | |
| 50–60 | 1 | 6 | |
| 90–100 | 19 | 0 | |
| SF-36 *** | Physical component summary (PCS) | 55.4 +/− 5.9 | 27 +/− 8.6 |
| Mental component summary (MCS) | 53.6 +/− 9.6 | 37.6 +/− 11.8 |
| Protein | Full Name | UniProt | EntrezGene | Fold Change | p-Value | q-Value |
|---|---|---|---|---|---|---|
| RNase1 | Ribonuclease pancreatic | P07998 | RNASE1 (6035) | 1.47 | 0.00002 | 0.035 |
| RABP2 | Cellular retinoic acid-binding protein 2 | P29373 | CRABP2 (1382) | 1.47 | 0.00002 | 0.035 |
| AIF1L | Allograft inflammatory factor 1-like | Q9BQI0 | AIFL1 (83543) | 1.25 | 0.00002 | 0.035 |
| PXDN | Peroxidasin homolog | Q92626 | PXDN (7837) | 1.39 | 0.00005 | 0.038 |
| Ephrin-A4 | Ephrin-A4 | P52798 | EFNA4 (1945) | 1.16 | 0.00005 | 0.038 |
| TIMD3 | Hepatitis A virus cellular receptor 2 | Q8TDQ0 | HAVCR2 (84868) | 1.1 | 0.00006 | 0.038 |
| MXRA7 | Matrix-remodeling-associated protein 7 | P84157 | MXRA7 (439921) | 1.14 | 0.00006 | 0.038 |
| TNF sR-I | Tumor necrosis factor receptor superfamily member 1A | P19438 | TNFRSF1A (7132) | 1.28 | 0.00007 | 0.038 |
| IL-18 BPa | Interleukin-18-binding protein | O95998 | IL18BP (10068) | 1.24 | 0.00007 | 0.038 |
| Term ID * | Term Description | Enrichment Score | Genes Mapped | Pathway Size | False Discovery (afc) |
|---|---|---|---|---|---|
| HSA-3928663 | EPHA-mediated growth cone collapse | 2.26367 | 17 | 28 | 0.00051 |
| CL:416 | Ephrin receptor activity, and Ephrin | 2.07704 | 20 | 23 | 0.00038 |
| CL:420 | Ephrin receptor activity, and Ephrin | 2.72701 | 13 | 16 | 0.00038 |
| IPR031328 | Ephrin | 3.36604 | 7 | 9 | 0.0049 |
| IPR019765 | Ephrin, conserved site | 3.36604 | 7 | 9 | 0.0049 |
| IPR001799 | Ephrin receptor-binding domain | 3.36604 | 7 | 9 | 0.0049 |
| IPR034252 | Ephrin-A ectodomain | 5.24701 | 4 | 6 | 0.0049 |
| Protein | AUC | Protein Ratio | EntrezGene | UniProt | AUC |
|---|---|---|---|---|---|
| RNase1 | 0.87 | FRDA/TIMD3 * | FXN/HAVCR2 * | Q16595/Q8TDQ0 | 0.95 |
| RABP2 | 0.87 | Layilin/CLC4D * | LAYN/CLEC4D * | Q6UX15/Q8WXI8 | 0.94 |
| AIF1L | 0.87 | RNase1/EKI1 | RNASE1/ETNK1 | P07998/Q9HBU6 | 0.94 |
| PXDN | 0.86 | BMPER/TNF sR-I | BMPER/TNFRSF1A | Q8N8U9/P19438 | 0.93 |
| Ephrin-A4 | 0.86 | EphA5/CFTR * | EPHA5/CFTR * | P54756/P13569 | 0.93 |
| TIMD3 | 0.85 | TIMD3/CFTR | HAVCR2/CFTR | Q8TDQ0/P13569 | 0.93 |
| MXRA7 | 0.85 | VIGLN/EphA5 | HDLBP/EPHA5 | Q00341/P54756 | 0.93 |
| TNF sR-I | 0.85 | Keratin-16/CFTR * | KRT16/CFTR * | P08779/P13569 | 0.93 |
| IL-18 BPa | 0.85 | P5CR2/Layilin | PYCR2/LAYN | Q96C36/Q6UX15 | 0.92 |
| Annotation Cluster | Enrichment Score | GO Term | Protein Count | p-Value | q-Value | |
|---|---|---|---|---|---|---|
| 1 | 14.3 | CC_GO:0005913 | Cell–cell adherens junction | 113 | 4.46 × 10−18 | 6.72 × 10−16 |
| MF_GO:0098641 | Cadherin binding involved in cell-cell adhesion | 98 | 1.59 × 10−14 | 3.49 × 10−12 | ||
| BP_GO:0098609 | Cell–cell adhesion | 90 | 2.46 × 10−12 | 9.64 × 10−10 | ||
| 2 | 6.5 | BP_GO:0038083 | Peptidyl-tyrosine autophosphorylation | 24 | 2.06 × 10−9 | 3.93 × 10−7 |
| MF_GO:0004715 | Nonmembrane spanning protein tyrosine kinase activity | 22 | 1.21 × 10−6 | 0.00009 | ||
| CC_GO:0031234 | Extrinsic component of cytoplasmic side of plasma membrane | 26 | 0.00001 | 0.0004 | ||
| 3 | 5.5 | BP_GO:0061621 | Canonical glycolysis | 17 | 1.65 × 10−7 | 0.00002 |
| BP_GO:0006096 | Glycolytic process | 18 | 3.70 × 10−6 | 0.0003 | ||
| BP_GO:0006094 | Gluconeogenesis | 19 | 0.00006 | 0.003 | ||
| 4 | 5.1 | BP_GO:0050919 | Negative chemotaxis | 21 | 1.37 × 10−8 | 2.17 × 10−6 |
| MF_GO:0045499 | Chemorepellent activity | 18 | 2.85 × 10−8 | 2.81 × 10−6 | ||
| BP_GO:0048843 | Negative regulation of axon extension involved in axon guidance | 15 | 8.89 × 10−6 | 0.0007 | ||
| MF_GO:0030215 | Semaphorin receptor binding | 13 | 0.00005 | 0.002 | ||
| BP_GO:0071526 | Semaphorin-plexin signaling pathway | 16 | 0.00006 | 0.003 | ||
| MF_GO:0038191 | Neuropilin binding | 10 | 0.0001 | 0.004 | ||
| BP_GO:0001755 | Neural crest cell migration | 16 | 0.003 | 0.07 | ||
| BP_GO:0008543 | Fibroblast growth factor receptor signaling pathway | 34 | 9.67 × 10−8 | 0.00001 | ||
| 5 | 5 | BP_GO:0048015 | Phosphatidylinositol-mediated signaling | 40 | 1.22 × 10−7 | 0.00002 |
| MF_GO:0046934 | Phosphatidylinositol-4,5-bisphosphate 3-kinase activity | 28 | 1.26 × 10−7 | 0.00001 | ||
| MF_GO:0005088 | Ras guanyl-nucleotide exchange factor activity | 41 | 2.52 × 10−7 | 0.00002 | ||
| BP_GO:0014066 | Regulation of phosphatidylinositol 3-kinase signaling | 32 | 3.27 × 10−7 | 0.00004 | ||
| BP_GO:0046854 | Phosphatidylinositol phosphorylation | 32 | 0.00003 | 0.002 | ||
| MF_GO:0005104 | Fibroblast growth factor receptor binding | 12 | 0.0002 | 0.006 | ||
| MF_GO:0016303 | 1-phosphatidylinositol-3-kinase activity | 13 | 0.03 | 0.35 | ||
| BP_GO:0036092 | Phosphatidylinositol-3-phosphate biosynthetic process | 14 | 0.04 | 0.46 | ||
| Protein | Negative Correlations in Controls Positive Correlations in ME/CFS | Protein | Positive Correlations in Controls Negative Correlations in ME/CFS |
|---|---|---|---|
| CGA FSHB | 347 (349) | CILP2 | 381 (386) |
| FIS1 | 281 (340) | CASC4 | 301 (308) |
| TNXB | 251 (331) | TMEM9 | 285 (287) |
| CGA LHB | 226 (230) | ENO3 | 280 (296) |
| RNF215 | 211 (242) | VWA2 | 267 (291) |
| RCN3 | 209 (306) | AMN | 222 (240) |
| RNF215.1 | 202 (242) | CLIC5 | 200 (205) |
| C3 | 195 (359) | CACNA2D3 | 193 (200) |
| GRB14 | 194 (218) | OBP2B | 183 (190) |
| FIGF | 144 (271) | THSD7A | 174 (231) |
| BRD2 | 129 (132) | C3 | 164 (359) |
| ANTXR1 | 124 (166) | TTC9B | 157 (257) |
| C1orf210 | 105 (182) | LAMC2 | 143 (151) |
| IGFBP1 | 101 (134) | PIANP | 129 (133) |
| TTC9B | 100 (257) | TRAPPC3 | 129 (181) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Germain, A.; Levine, S.M.; Hanson, M.R. In-Depth Analysis of the Plasma Proteome in ME/CFS Exposes Disrupted Ephrin-Eph and Immune System Signaling. Proteomes 2021, 9, 6. https://doi.org/10.3390/proteomes9010006
Germain A, Levine SM, Hanson MR. In-Depth Analysis of the Plasma Proteome in ME/CFS Exposes Disrupted Ephrin-Eph and Immune System Signaling. Proteomes. 2021; 9(1):6. https://doi.org/10.3390/proteomes9010006
Chicago/Turabian StyleGermain, Arnaud, Susan M. Levine, and Maureen R. Hanson. 2021. "In-Depth Analysis of the Plasma Proteome in ME/CFS Exposes Disrupted Ephrin-Eph and Immune System Signaling" Proteomes 9, no. 1: 6. https://doi.org/10.3390/proteomes9010006