
Enhanced Electrophoretic Depletion of Sodium Dodecyl Sulfate with Methanol for Membrane Proteome Analysis by Mass Spectrometry
Abstract
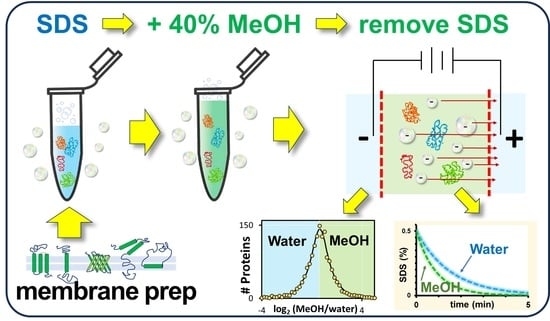
:
1. Introduction
2. Materials and Methods
2.1. Membrane Protein Isolation
2.2. Membrane Protein Resolubilization with Methanol
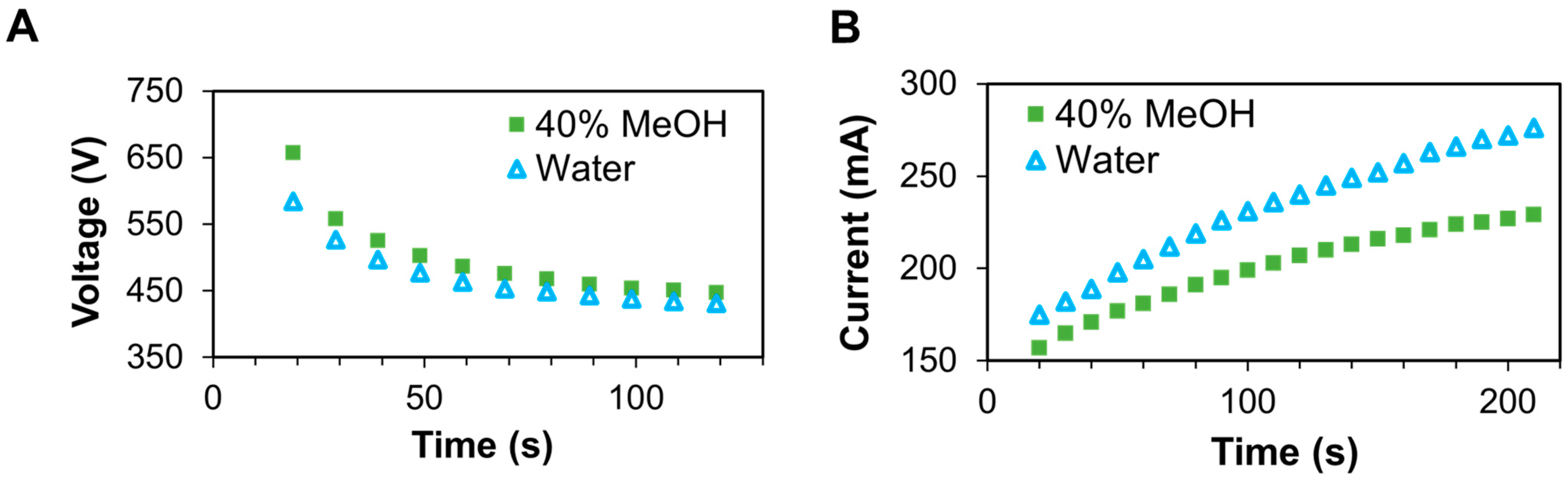
2.3. Transmembrane Electrophoresis
2.4. SDS Colorimetric Assay by MBAS
2.5. Protein Digestion
2.6. CMC Determination
2.7. SDS—Protein Binding Ratio in Methanol
2.8. LC-MS/MS
2.9. Data Analysis
3. Results
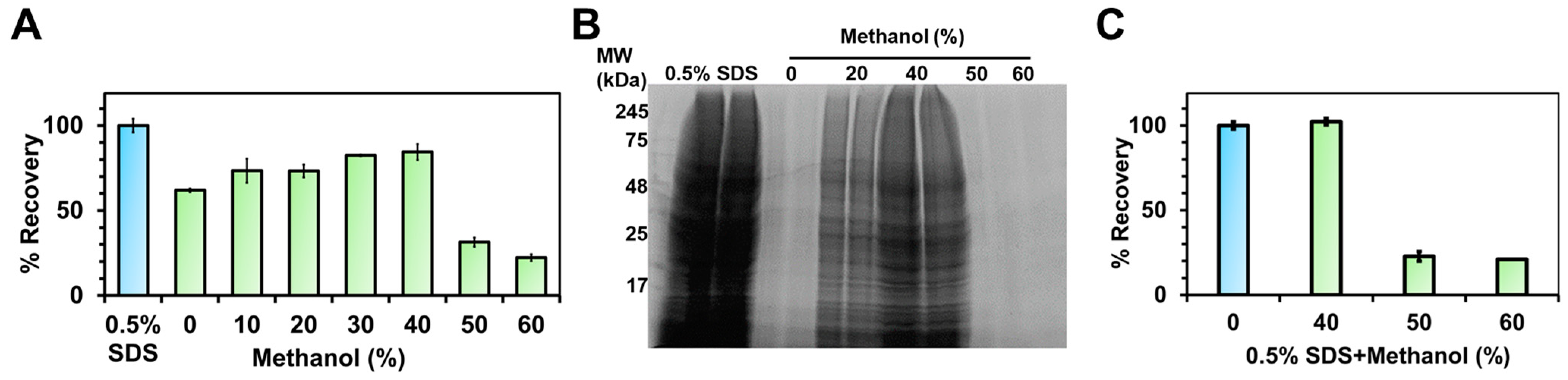
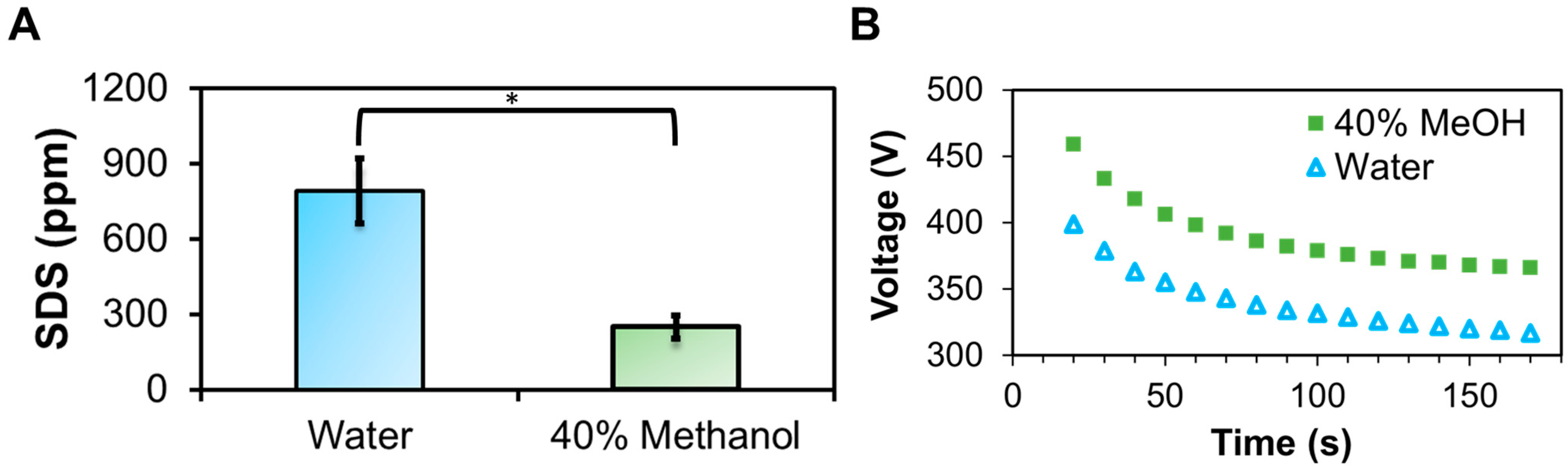
3.1. Enhanced Membrane Protein Recovery with 40% Methanol
3.2. Methanol Increases the CMC and Lowers the SDS to Protein Binding Energy
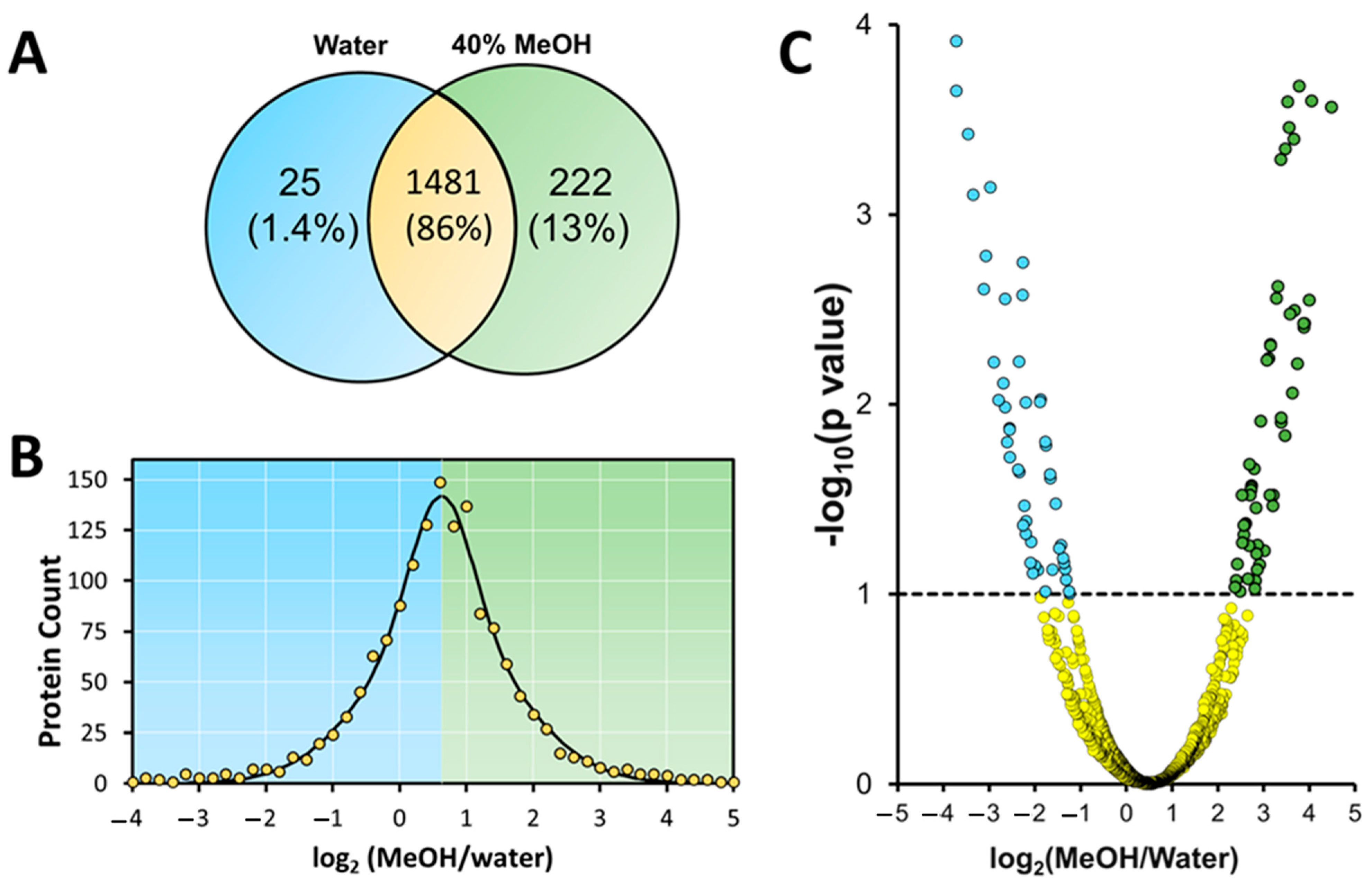
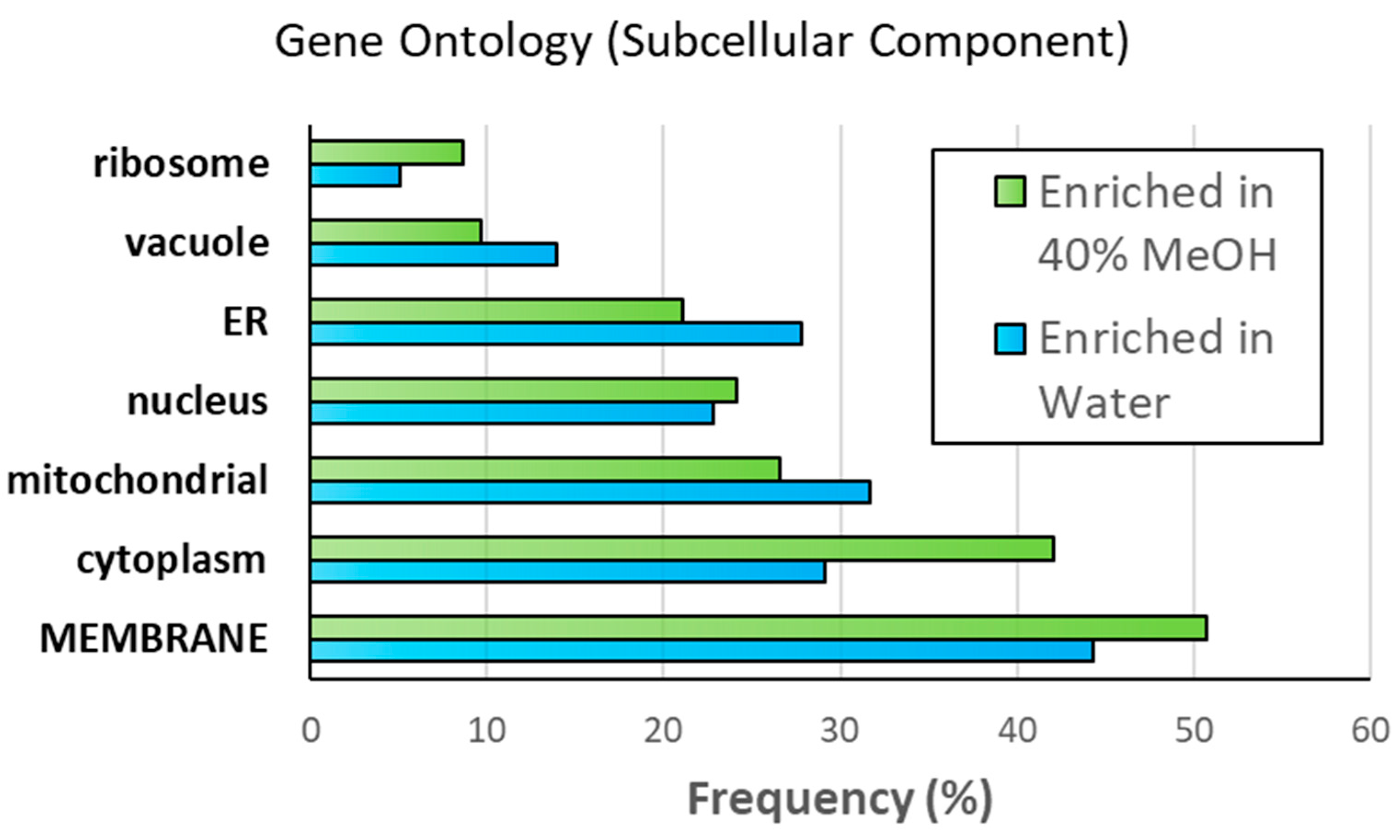
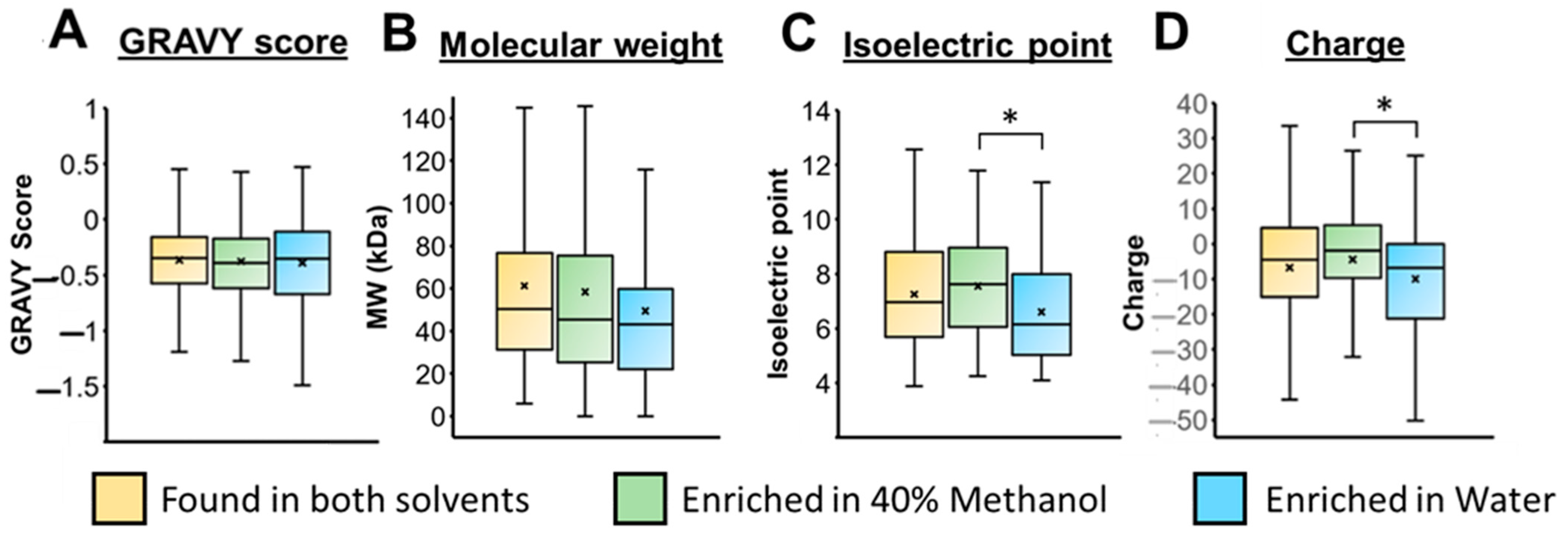
3.3. Mass Spectrometry Analysis of Samples Containing 40% Methanol vs. No Methanol
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bakheet, T.; Doig, A. Properties and identification of human protein drug targets. Bioinformatics 2009, 25, 451–457. [Google Scholar] [CrossRef]
- John, D. G-protein-coupled receptors: New approaches to maximise the impact of GPCRS in drug discovery. Expert Opin. Ther. Targets 2004, 8, 165–170. [Google Scholar]
- Chen, X.; Cui, D.; Liu, C.C.; Li, H. Microfluidic chip for blood cell separation and collection based on crossflow filtration. Sens. Actuators B Chem. 2008, 130, 216–221. [Google Scholar] [CrossRef]
- Modise, C.; Shan, H.; Neufeld, R.; Vidic, R. Evaluation of permeate flux rate and membrane fouling in dead-end microfiltration of primary sewage effluent. Environ. Eng. Sci. 2005, 22, 427–439. [Google Scholar] [CrossRef]
- Lin, Y.; Huo, L.; Liu, Z.; Li, J.; Liu, Y.; He, Q.; Liang, S. Sodium laurate, a novel protease- and mass spectrometry-compatible detergent for mass spectrometry-based membrane proteomics. PLoS ONE 2013, 8, e59779. [Google Scholar] [CrossRef] [PubMed]
- Kadiyala, C.S.; Tomechko, R.; Miyagi, M. Perfluorooctanoic acid for shotgun proteomics. PLoS ONE 2010, 5, e15332. [Google Scholar] [CrossRef] [PubMed]
- Masuda, T.; Tomita, M.; Ishihama, Y. Phase transfer surfactant-aided trypsin digestion for membrane proteome analysis. J. Proteome Res. 2008, 7, 731–740. [Google Scholar] [CrossRef]
- Garavito, R.M.; Ferguson-Miller, S. Detergents as tools in membrane biochemistry. J. Biol. Chem. 2001, 276, 32403–32406. [Google Scholar] [CrossRef] [PubMed]
- Le Maire, M.; Champeil, P.; Møller, J.V. Interaction of membrane proteins and lipids with solubilizing detergents. Biochim. Biophys. Acta 2000, 1508, 86–111. [Google Scholar] [CrossRef]
- Arribas, J.; Castano, J. Kinetic studies of the differential effect of detergents on the peptidase activities of the multicatalytic proteinase from rat liver. Biol. Chem. 1990, 265, 13969–13973. [Google Scholar] [CrossRef]
- Kawasaki, H.; Suzuki, K. Separation of peptides dissolved in a sodium dodecyl sulfate solution by reversed-phase liquid chromatography: Removal of sodium dodecyl sulfate from peptides using an ion-exchange precolumn. Anal. Biochem. 1990, 186, 264–268. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.; Gregorich, R.; Chen, J.; Hwang, L.; Guner, H.; Yu, D.; Zhang, J.; Ge, Y. New Mass-Spectrometry-Compatible degradable surfactant for tissue proteomics. J. Proteome Res. 2015, 14, 1587–1599. [Google Scholar] [CrossRef]
- Brown, R.B.; Audet, J. Current techniques for single-cell lysis. J. R. Soc. Interface 2008, 5, S131–S138. [Google Scholar] [CrossRef] [PubMed]
- Bereman, M.S.; Egertson, J.; MacCoss, M. Comparison between procedures using SDS for shotgun proteomic analyses of complex samples. Proteomics 2011, 11, 2931–2935. [Google Scholar] [CrossRef] [PubMed]
- Blonder, J.; Goshe, M.B.; Moore, R.J.; Pasa-Tolic, L.; Masselon, C.D.; Lipton, M.S.; Smith, R.D. Enrichment of integral membrane proteins for proteomic analysis using liquid chromatography-tandem mass spectrometry. J. Proteome Res. 2002, 1, 351–360. [Google Scholar] [CrossRef]
- Blonder, J.; Chan, K.; Issaq, H.; Veenstra, T. Identification of membrane proteins from mammalian cell/tissue using methanol-facilitated solubilization and tryptic digestion coupled with 2D-LC-MS/MS. Nat. Protoc. 2007, 1, 2784–2790. [Google Scholar] [CrossRef]
- Park, H.; Min, Y. Distinction between the Influence of Dielectric Constant and of Methanol Concentration on Trypsin-Catalyzed Hydrolysis and Methanolysis. J. Microbiol. Biotechnol. 1998, 8, 656–662. [Google Scholar]
- Wall, J.; Crowell, J.; Simms, A.; Liu, F.; Doucette, A.A. Implications of partial tryptic digestion in organic–aqueous solvent systems for bottom-up Proteome Analysis. Anal. Chim. Acta 2011, 703, 194–203. [Google Scholar] [CrossRef]
- Bansal, V.; Delgado, Y.; Fasoli, E.; Ferrer, A.; Griebenow, K.; Secundo, F.; Barletta, G. Effect of prolonged exposure to organic solvents on the active site environment of subtilisin Carlsberg. J. Mol. Catal. B Enzym. 2010, 64, 38–44. [Google Scholar] [CrossRef]
- Nickerson, J.L.; Doucette, A.A. Rapid and quantitative protein precipitation for proteome analysis by mass spectrometry. J. Proteome Res. 2020, 19, 2035–2042. [Google Scholar] [CrossRef]
- Mellanby, J. The precipitation of the proteins of horse serum. J. Physiol. 1907, 36, 288–333. [Google Scholar] [CrossRef]
- Polson, C.; Sarkar, P.; Incledon, B.; Raguvaran, V.; Grant, R. Optimization of protein precipitation based upon effectiveness of protein removal and ionization effect in liquid chromatography–tandem mass spectrometry. J. Chromatogr. B 2003, 785, 263–275. [Google Scholar] [CrossRef]
- Bruce, S.J.; Tavazzi, I.; Parisod, V.; Rezzi, S.; Kochhar, S.; Guy, P.A. Investigation of human blood plasma sample preparation for performing metabolomics using ultrahigh performance liquid chromatography/mass spectrometry. Anal. Chem. 2009, 81, 3285–3296. [Google Scholar] [CrossRef] [PubMed]
- Varnavides, G.; Madern, M.; Anrather, D.; Hartl, N.; Reiter, W.; Hartl, M. In search of a universal method: A comparative survey of bottom-up proteomics sample preparation methods. J. Proteome Res. 2022, 21, 2397–2411. [Google Scholar] [CrossRef]
- Wisniewski, J.R.; Zougman, A.; Nagaraj, N.; Mann, M. Universal sample preparation method for proteome analysis. Nat. Methods 2009, 6, 359–362. [Google Scholar] [CrossRef]
- Lin, Y.-H.; Eguez, R.V.; Torralba, M.G.; Singh, H.; Golusinski, P.; Golusinski, W.; Masternak, M.; Nelson, K.E.; Freire, M.; Yu, Y. Self-assembled STrap for global proteomics and salivary biomarker discovery. J. Proteome Res. 2019, 18, 1907–1915. [Google Scholar] [CrossRef]
- Hughes, C.S.; Moggridge, S.; Müller, T.; Sorensen, P.H.; Morin, G.B.; Krijgsveld, J. Single Pot, Solid-phase-enhanced sample preparation for proteomics experiments. Nat. Protoc. 2019, 14, 68–85. [Google Scholar] [CrossRef]
- Crowell, A.M.J.; MacLellan, D.L.; Doucette, A.A. A Two-stage spin cartridge for integrated protein precipitation, digestion and SDS removal in a comparative bottom-up proteomics workflow. J. Proteomics 2015, 118, 140–150. [Google Scholar] [CrossRef] [PubMed]
- Doellinger, J.; Schneider, A.; Hoeller, M.; Lasch, P. Sample Preparation by Easy Extraction and Digestion (SPEED)—A Universal, Rapid, and Detergent-free Protocol for Proteomics based on Acid Extraction. Mol. Cell Proteom. 2020, 19, 209–222. [Google Scholar] [CrossRef] [PubMed]
- Kachuk, C.; Stephen, K.; Doucette, A. Comparison of sodium dodecyl sulfate depletion techniques for proteome analysis by mass spectrometry. J. Chrom A. 2015, 1418, 158–166. [Google Scholar] [CrossRef]
- Unterlander, N.; Doucette, A. Membrane-based SDS depletion ahead of peptide and protein analysis by mass spectrometry. Proteomics 2018, 18, e1700025. [Google Scholar] [CrossRef]
- Kachuk, C.; Faulkner, M.; Liu, F.; Doucette, A. Automated SDS depletion for mass spectrometry of intact membrane proteins though transmembrane electrophoresis. J. Proteome Res. 2016, 15, 2634–2642. [Google Scholar] [CrossRef]
- Jakubec, P.; Doucette, A. Automated electrokinetic platform for high-throughput sodium dodecyl sulfate depletion ahead of proteome analysis by mass spectrometry. Anal. Chem. 2020, 93, 14042–14047. [Google Scholar] [CrossRef]
- Towbin, H.; Staehelin, T.; Gordon, J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets. Proc. Natl. Acad. Sci. USA 1979, 6, 4350–4354. [Google Scholar] [CrossRef]
- Mozdzanowski, J.; Speicher, D.W. Microsequence analysis of electroblotted proteins: I. Comparison of electroblotting recoveries using different types of PVDF membranes. Anal. Biochem. 1992, 207, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Bergman, L.W. Growth and maintenance of yeast. Methods Mol. Biol. 2001, 177, 9–14. [Google Scholar]
- Wu, F.; Sun, D.; Wang, N.; Gong, Y.; Li, L. Comparison of surfactant-assisted shotgun methods using acid labile surfactants and sodium dodecyl sulfate for membrane proteome analysis. Anal. Chim. Acta 2001, 698, 36–43. [Google Scholar] [CrossRef]
- George, A.; Graham, W. Optimization of the methylene blue assay for anionic surfactants added to estuarine and marine water. Environ. Toxicol. Chem. 1999, 18, 2232–2236. [Google Scholar] [CrossRef] [PubMed]
- Shevchenko, A.; Tomas, H.; Havli, J.; Olsen, J.V.; Mann, M. In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat. Protoc. 2006, 1, 2856–2860. [Google Scholar] [CrossRef] [PubMed]
- Perinelli, D.R.; Cespi, M.; Lorusso, N.; Palmieri, G.; Blasi, P. Surfactant self-assembling and critical micelle concentration: One approach fits all? Langmuir 2020, 36, 5745–5753. [Google Scholar] [CrossRef]
- Rharbi, Y.; Winnik, M.A. Salt effects on solute exchange in sodium dodecyl sulfate micelles. J. Am. Chem. Soc. 2002, 124, 2082–2083. [Google Scholar] [CrossRef] [PubMed]
- Sherman, B.T.; Qiu, H.; Jiao, X.; Lane, B.; Imamichi, L.; Chang, W. DAVID: A web server for functional enrichment analysis and functional annotation of gene lists (2021 update). Nucleic Acids Res. 2022, 50, W216–W221. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID Bioinformatics Resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, J.A.; Tanford, C. Binding of dodecyl sulfate to proteins at high binding ratios. Possible implications for the state of proteins in biological membranes. Proc. Natl. Acad. Sci. USA 1970, 66, 1002–1007. [Google Scholar] [CrossRef] [PubMed]
- Moore, S.; Hess, S.; Jorgenson, J.W. Extraction, enrichment, solubilization, and digestion techniques for membrane proteomics. J. Proteome Res. 2016, 15, 1243–1252. [Google Scholar] [CrossRef]
- Niraula, T.P.; Bhattarai, A.; Chatterjee, S.K. Critical micelle concentration of sodium dodecyl sulphate in pure water and in methanol-water mixed solvent media in presence and absence of KCl by surface tension and viscosity methods. Bibechana 2014, 11, 103–112. [Google Scholar] [CrossRef]
- Niraula, T.P.; Chatterjee, S.K.; Bhattarai, A. Micellization of sodium dodecyl sulphate in presence and absence of alkali metal halides at different temperatures in water and methanol-water mixtures. J. Mol. Liq. 2018, 250, 287–294. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Water | 40% MeOH | Ratio (MeOH/H2O) | p-Value |
|---|---|---|---|---|
| Protein recovery (%) | 57.1 ± 4 A | 76.2 ± 7 | 1.3 ± 0.2 | 0.02 |
| Time to 100 ppm (min) | 8.8 ± 0.9 | 2.8 ± 0.5 | 0.31 ± 0.1 | 5 × 10−4 |
| Temperature (°C) | 41 ± 4 | 35 ± 3 | 0.85 ± 0.1 | 0.11 |
| Decay constant (min−1) at 250 mA | 0.65 ± 0.001 | 1.13 ± 0.001 | 1.73 ± 0.2 | 6 × 10−4 |
| Decay constant (min−1) at 350 V | 0.99 ± 0.02 | 1.31 ± 0.1 | 1.32 ± 0.08 | 0.002 |
| Resistivity (µS) | 0.0015 ± 0.0002 | 0.0025 ± 0.001 | 1.63 ± 0.01 | 1 × 10−5 |
| Solvent | Water | 40% Methanol | Ratio (MeOH/H2O) |
|---|---|---|---|
| CMC (mM) | 8.4 ± 0.1 A | 25.7 ± 1.3 | 3.1 ± 0.2 |
| SDS-protein binding (g/g) | 1.4 ± 0.1 | 0.30 ± 0.05 | 0.2 ± 0.04 |
| Decay constant with no proteins (min−1) | 0.91 ± 0.02 | 1.41 ± 0.04 | 1.55 ± 0.06 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Said, H.H.; Doucette, A.A. Enhanced Electrophoretic Depletion of Sodium Dodecyl Sulfate with Methanol for Membrane Proteome Analysis by Mass Spectrometry. Proteomes 2024, 12, 5. https://doi.org/10.3390/proteomes12010005
Said HH, Doucette AA. Enhanced Electrophoretic Depletion of Sodium Dodecyl Sulfate with Methanol for Membrane Proteome Analysis by Mass Spectrometry. Proteomes. 2024; 12(1):5. https://doi.org/10.3390/proteomes12010005
Chicago/Turabian StyleSaid, Hammam H., and Alan A. Doucette. 2024. "Enhanced Electrophoretic Depletion of Sodium Dodecyl Sulfate with Methanol for Membrane Proteome Analysis by Mass Spectrometry" Proteomes 12, no. 1: 5. https://doi.org/10.3390/proteomes12010005