Recent Advances in Computational Protocols Addressing Intrinsically Disordered Proteins



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
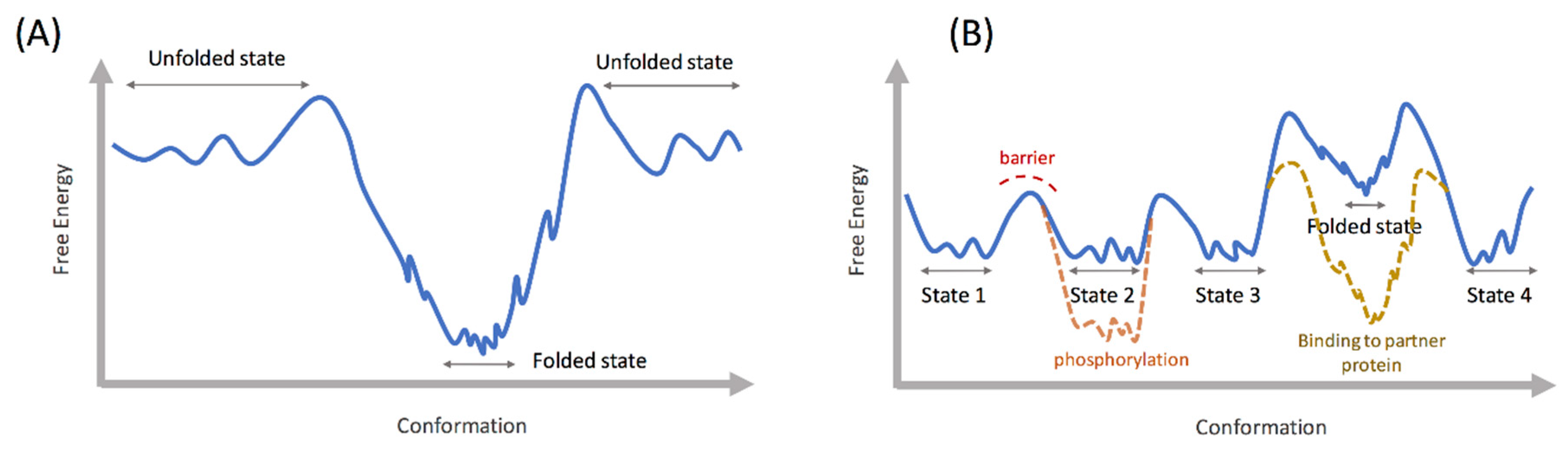
2. Energy Landscape of Intrinsically Disordered Proteins
3. Structure Prediction and Conformational Dynamics
3.1. Conformational Selection Based Methods
3.2. Molecular Dynamics Based Methods
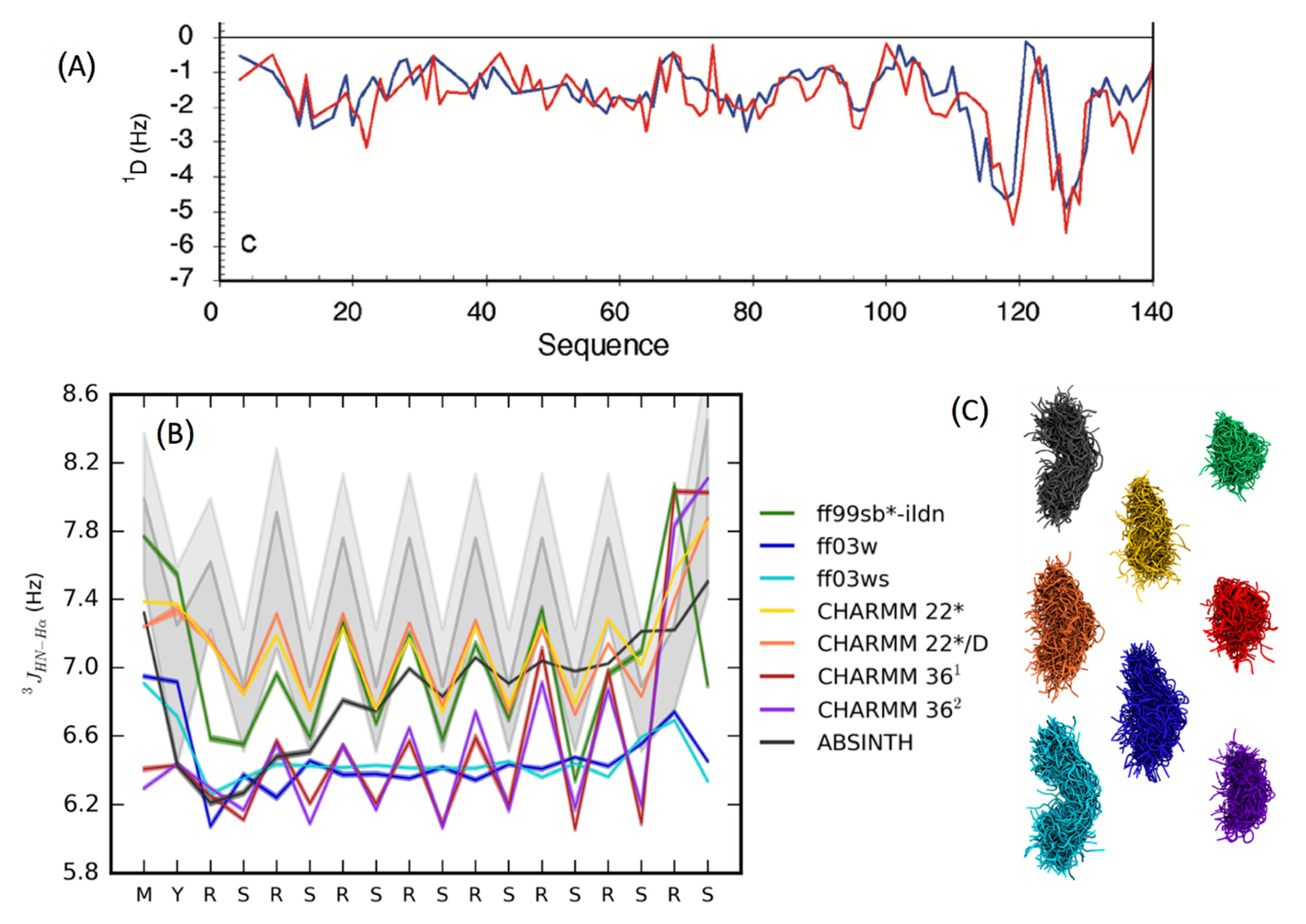
3.3. Force-Field Development for Intrisically Disordered Proteins
3.4. Coarse Grain and Multiscale Methods
4. Protein–Protein Interaction Involving Intrinsically Disordered Proteins
4.1. Disorder Retained upon Binding: Fuzzy Complexes
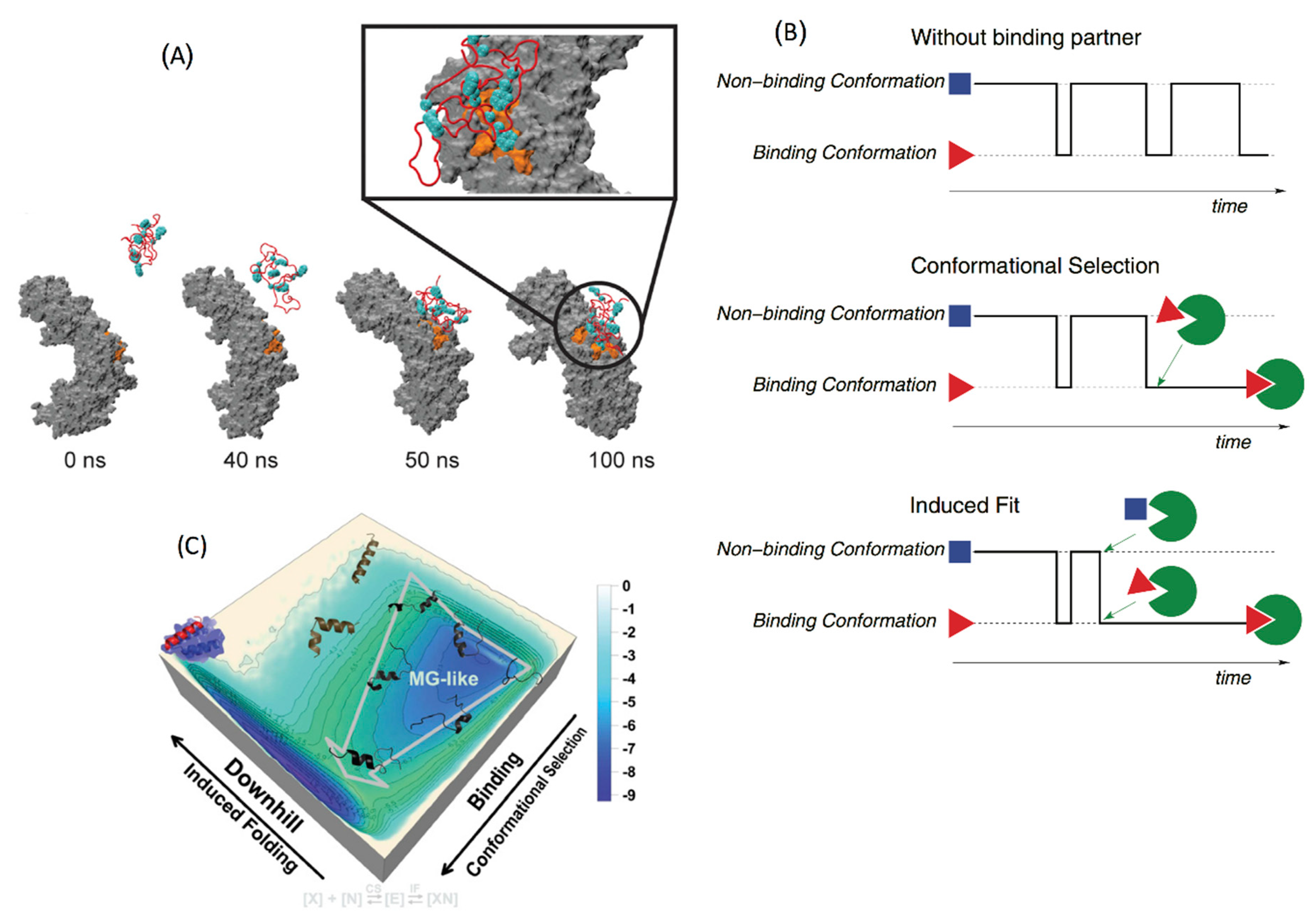
4.2. Conformational Selection versus Induced Fit
4.3. Liquid–Liquid Phase Separation and Aggregation
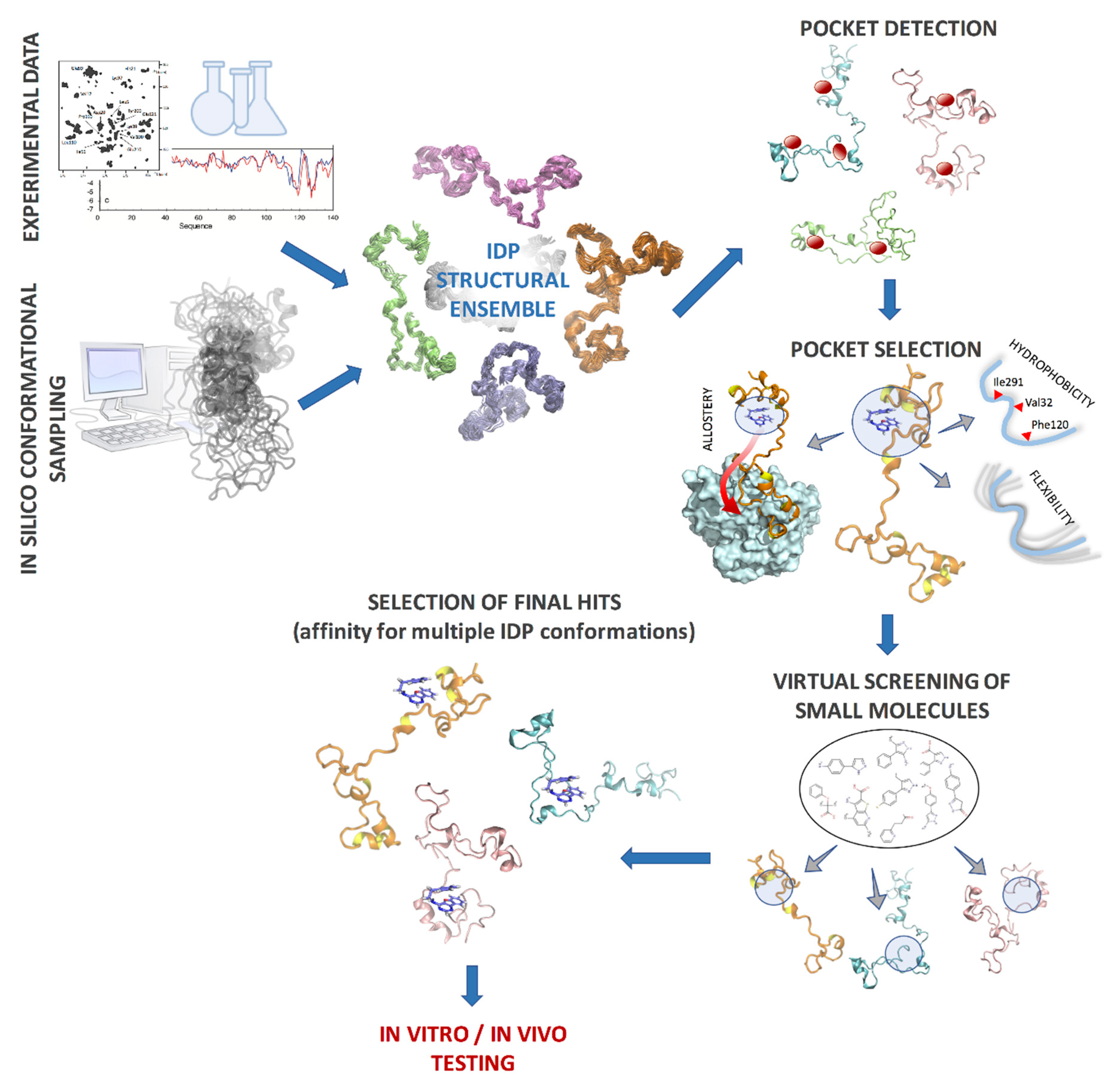
5. Intrinsically Disordered Proteins as Therapeutic Targets
6. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Wright, P.E.; Dyson, H.J. Intrinsically disordered proteins in cellular signalling and regulation. Nat. Rev. Mol. Cell. Biol. 2015, 16, 18–29. [Google Scholar] [CrossRef]
- Babu, M.M. The contribution of intrinsically disordered regions to protein function, cellular complexity, and human disease. Biochem. Soc. Trans. 2016, 44, 1185–1200. [Google Scholar] [CrossRef] [PubMed]
- Oldfield, C.J.; Dunker, A.K. Intrinsically disordered proteins and intrinsically disordered protein regions. Annu. Rev. Biochem. 2014, 83, 553–584. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N.; Oldfield, C.J.; Dunker, A.K. Intrinsically disordered proteins in human diseases: introducing the D2 concept. Annu. Rev. Biophys. 2008, 37, 215–246. [Google Scholar] [CrossRef]
- Mollica, L.; Bessa, L.M.; Hanoulle, X.; Jensen, M.R.; Blackledge, M.; Schneider, R. Binding Mechanisms of Intrinsically Disordered Proteins: Theory, Simulation, and Experiment. Front. Mol. Biosci. 2016, 3, 52. [Google Scholar] [CrossRef] [PubMed]
- Borgia, A.; Borgia, M.B.; Bugge, K.; Kissling, V.M.; Heidarsson, P.O.; Fernandes, C.B.; Sottini, A.; Soranno, A.; Buholzer, K.J.; Nettels, D.; et al. Extreme disorder in an ultrahigh-affinity protein complex. Nature 2018, 555, 61–66. [Google Scholar] [CrossRef]
- Jia, D.; Jolly, M.K.; Kulkarni, P.; Levine, H. Phenotypic Plasticity and Cell Fate Decisions in Cancer: Insights from Dynamical Systems Theory. Cancers (Basel) 2017, 9, 70. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. Intrinsically Disordered Proteins and Their “Mysterious” (Meta)Physics. Front. Phys-Lausanne 2019, 7, 10. [Google Scholar] [CrossRef]
- Dunker, A.K.; Babu, M.M.; Barbar, E.; Blackledge, M.; Bondos, S.E.; Dosztanyi, Z.; Dyson, H.J.; Forman-Kay, J.; Fuxreiter, M.; Gsponer, J.; et al. What’s in a name? Why these proteins are intrinsically disordered: Why these proteins are intrinsically disordered. Intrinsically Disord. Proteins 2013, 1, e24157. [Google Scholar] [CrossRef]
- O’Brien, E.P.; Morrison, G.; Brooks, B.R.; Thirumalai, D. How accurate are polymer models in the analysis of Forster resonance energy transfer experiments on proteins? J. Chem. Phys. 2009, 130, 124903. [Google Scholar] [CrossRef]
- Zheng, W.W.; Zerze, G.H.; Borgia, A.; Mittal, J.; Schuler, B.; Best, R.B. Inferring properties of disordered chains from FRET transfer efficiencies. J. Chem. Phys. 2018, 148, 123329. [Google Scholar] [CrossRef]
- Zheng, W.W.; Best, R.B. An Extended Guinier Analysis for Intrinsically Disordered Proteins. J. Mol. Biol. 2018, 430, 2540–2553. [Google Scholar] [CrossRef]
- Vitalis, A.; Wang, X.L.; Pappu, R.V. Quantitative characterization of intrinsic disorder in polyglutamine: Insights from analysis based on polymer theories. Biophys. J. 2007, 93, 1923–1937. [Google Scholar] [CrossRef]
- Piana, S.; Donchev, A.G.; Robustelli, P.; Shaw, D.E. Water Dispersion Interactions Strongly Influence Simulated Structural Properties of Disordered Protein States. J. Phys. Chem. B. 2015, 119, 5113–5123. [Google Scholar] [CrossRef]
- Song, J.H.; Gomes, G.N.; Shi, T.F.; Gradinaru, C.C.; Chan, H.S. Conformational Heterogeneity and FRET Data Interpretation for Dimensions of Unfolded Proteins. Biophys. J. 2017, 113, 1012–1024. [Google Scholar] [CrossRef] [PubMed]
- Riback, J.A.; Bowman, M.A.; Zmyslowski, A.M.; Knoverek, C.R.; Jumper, J.M.; Hinshaw, J.R.; Kaye, E.B.; Freed, K.F.; Clark, P.L.; Sosnick, T.R. Innovative scattering analysis shows that hydrophobic disordered proteins are expanded in water. Science 2017, 358, 238–241. [Google Scholar] [CrossRef] [PubMed]
- Schuler, B.; Soranno, A.; Hofmann, H.; Nettels, D. Single-Molecule FRET Spectroscopy and the Polymer Physics of Unfolded and Intrinsically Disordered Proteins. Annu. Rev. Biophys. 2016, 45, 207–231. [Google Scholar] [CrossRef] [PubMed]
- Best, R.B. Computational and theoretical advances in studies of intrinsically disordered proteins. Curr. Opin. Struct. Biol. 2017, 42, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Varadi, M.; Vranken, W.; Guharoy, M.; Tompa, P. Computational approaches for inferring the functions of intrinsically disordered proteins. Front Mol. Biosci. 2015, 2, 45. [Google Scholar] [CrossRef]
- Frauenfelder, H.; Sligar, S.G.; Wolynes, P.G. The energy landscapes and motions of proteins. Science 1991, 254, 1598–1603. [Google Scholar] [CrossRef] [PubMed]
- Wei, G.; Xi, W.; Nussinov, R.; Ma, B. Protein Ensembles: How Does Nature Harness Thermodynamic Fluctuations for Life? The Diverse Functional Roles of Conformational Ensembles in the Cell. Chem. Rev. 2016, 116, 6516–6551. [Google Scholar] [CrossRef] [PubMed]
- Fisher, C.K.; Stultz, C.M. Constructing ensembles for intrinsically disordered proteins. Curr. Opin. Struct. Biol. 2011, 21, 426–431. [Google Scholar] [CrossRef] [PubMed]
- Chebaro, Y.; Ballard, A.J.; Chakraborty, D.; Wales, D.J. Intrinsically disordered energy landscapes. Sci. Rep. 2015, 5, 10386. [Google Scholar] [CrossRef]
- He, Y.; Chen, Y.; Mooney, S.M.; Rajagopalan, K.; Bhargava, A.; Sacho, E.; Weninger, K.; Bryan, P.N.; Kulkarni, P.; Orban, J. Phosphorylation-induced Conformational Ensemble Switching in an Intrinsically Disordered Cancer/Testis Antigen. J. Biol. Chem. 2015, 290, 25090–25102. [Google Scholar] [CrossRef] [PubMed]
- Bah, A.; Vernon, R.M.; Siddiqui, Z.; Krzeminski, M.; Muhandiram, R.; Zhao, C.; Sonenberg, N.; Kay, L.E.; Forman-Kay, J.D. Folding of an intrinsically disordered protein by phosphorylation as a regulatory switch. Nature 2015, 519, 106–109. [Google Scholar] [CrossRef]
- Ma, B.Y.; Nussinov, R. Simulations as analytical tools to understand protein aggregation and predict amyloid conformation. Curr. Opin. Chem. Biol. 2006, 10, 445–452. [Google Scholar] [CrossRef]
- Tycko, R. Solid-State NMR Studies of Amyloid Fibril Structure. Annu. Rev. Phys. Chem. 2011, 62, 279–299. [Google Scholar] [CrossRef] [PubMed]
- Granata, D.; Baftizadeh, F.; Habchi, J.; Galvagnion, C.; De Simone, A.; Camilloni, C.; Laio, A.; Vendruscolo, M. The inverted free energy landscape of an intrinsically disordered peptide by simulations and experiments. Sci. Rep. 2015, 5, 15449. [Google Scholar] [CrossRef]
- Lincoff, J.; Sasmal, S.; Head-Gordon, T. Comparing generalized ensemble methods for sampling of systems with many degrees of freedom. J. Chem. Phys. 2016, 145, 174107. [Google Scholar] [CrossRef]
- Qiao, Q.; Bowman, G.R.; Huang, X.H. Dynamics of an Intrinsically Disordered Protein Reveal Metastable Conformations That Potentially Seed Aggregation. J. Am. Chem. Soc. 2013, 135, 16092–16101. [Google Scholar] [CrossRef] [PubMed]
- Das, P.; Matysiak, S.; Mittal, J. Looking at the Disordered Proteins through the Computational Microscope. ACS Central Sci. 2018, 4, 534–542. [Google Scholar] [CrossRef]
- Robustelli, P.; Piana, S.; Shaw, D.E. Developing a molecular dynamics force field for both folded and disordered protein states. Proc. Natl. Acad. Sci. USA 2018, 115, E4758–E4766. [Google Scholar] [CrossRef] [PubMed]
- Feldman, H.J.; Hogue, C.W. A fast method to sample real protein conformational space. Proteins 2000, 39, 112–131. [Google Scholar] [CrossRef]
- Feldman, H.J.; Hogue, C.W. Probabilistic sampling of protein conformations: new hope for brute force? Proteins 2002, 46, 8–23. [Google Scholar] [CrossRef] [PubMed]
- Ozenne, V.; Bauer, F.; Salmon, L.; Huang, J.R.; Jensen, M.R.; Segard, S.; Bernado, P.; Charavay, C.; Blackledge, M. Flexible-meccano: a tool for the generation of explicit ensemble descriptions of intrinsically disordered proteins and their associated experimental observables. Bioinformatics 2012, 28, 1463–1470. [Google Scholar] [CrossRef]
- Nodet, G.; Salmon, L.; Ozenne, V.; Meier, S.; Jensen, M.R.; Blackledge, M. Quantitative description of backbone conformational sampling of unfolded proteins at amino acid resolution from NMR residual dipolar couplings. J. Am. Chem. Soc. 2009, 131, 17908–17918. [Google Scholar] [CrossRef] [PubMed]
- Krzeminski, M.; Marsh, J.A.; Neale, C.; Choy, W.Y.; Forman-Kay, J.D. Characterization of disordered proteins with ENSEMBLE. Bioinformatics 2013, 29, 398–399. [Google Scholar] [CrossRef] [PubMed]
- Theobald, D.L.; Wuttke, D.S. Accurate structural correlations from maximum likelihood superpositions. PLoS Comput. Biol. 2008, 4, e4310. [Google Scholar] [CrossRef]
- Marchetti, J.; Monzon, A.M.; Tosatto, S.C.E.; Parisi, G.; Fornasari, M.S. Ensembles from Ordered and Disordered Proteins Reveal Similar Structural Constraints during Evolution. J. Mol. Biol. 2019, 431, 1298–1307. [Google Scholar] [CrossRef]
- Mitsutake, A.; Sugita, Y.; Okamoto, Y. Generalized-ensemble algorithms for molecular simulations of biopolymers. Biopolymers 2001, 60, 96–123. [Google Scholar] [CrossRef]
- Sugita, Y.; Okamoto, Y. Replica-exchange molecular dynamics method for protein folding. Chem. Phys. Lett. 1999, 314, 141–151. [Google Scholar] [CrossRef]
- Piana, S.; Laio, A. A bias-exchange approach to protein folding. J. Phys. Chem. B. 2007, 111, 4553–4559. [Google Scholar] [CrossRef]
- Brown, S.; Head-Gordon, T. Cool walking: a new Markov chain Monte Carlo sampling method. J. Comput. Chem. 2003, 24, 68–76. [Google Scholar] [CrossRef]
- Patel, S.; Vierling, E.; Tama, F. Replica exchange molecular dynamics simulations provide insight into substrate recognition by small heat shock proteins. Biophys. J. 2014, 106, 2644–2655. [Google Scholar] [CrossRef]
- Zerze, G.H.; Miller, C.M.; Granata, D.; Mittal, J. Free energy surface of an intrinsically disordered protein: comparison between temperature replica exchange molecular dynamics and bias-exchange metadynamics. J. Chem. Theory. Comput. 2015, 11, 2776–2782. [Google Scholar] [CrossRef]
- Sasmal, S.; Lincoff, J.; Head-Gordon, T. Effect of a Paramagnetic Spin Label on the Intrinsically Disordered Peptide Ensemble of Amyloid-β. Biophys. J. 2017, 113, 1002–1011. [Google Scholar] [CrossRef]
- Pande, V.S.; Beauchamp, K.; Bowman, G.R. Everything you wanted to know about Markov State Models but were afraid to ask. Methods 2010, 52, 99–105. [Google Scholar] [CrossRef]
- Buch, I.; Harvey, M.J.; Giorgino, T.; Anderson, D.P.; De Fabritiis, G. High-throughput all-atom molecular dynamics simulations using distributed computing. J. Chem. Inf. Model 2010, 50, 397–403. [Google Scholar] [CrossRef] [PubMed]
- Stanley, N.; Esteban-Martin, S.; De Fabritiis, G. Kinetic modulation of a disordered protein domain by phosphorylation. Nat. Commun. 2014, 5, 5272. [Google Scholar] [CrossRef] [PubMed]
- Deneroff, M.M.; Shaw, D.E.; Dror, R.O.; Kuskin, J.S.; Larson, R.H.; Salmon, J.K.; Young, C. Anton: A specialized ASIC for molecular dynamics. In Proceedings of the 2008 IEEE Hot Chips 20 Symposium (HCS), Stanford, CA, USA, 24–26 August 2008; pp. 1–34. [Google Scholar]
- Shaw, D.E.; Grossman, J.P.; Bank, J.A.; Batson, B.; Butts, J.A.; Chao, J.C.; Deneroff, M.M.; Dror, R.O.; Even, A.; Fenton, C.H.; et al. Anton 2: Raising the Bar for Performance and Programmability in a Special-Purpose Molecular Dynamics Supercomputer. In Proceedings of the SC 14: Proceedings of the International Conference for High Performance Computing, Networking, Storage and Analysis, New Orleans, LA, USA, 16–21 November 2014; pp. 41–53. [Google Scholar]
- Lindorff-Larsen, K.; Piana, S.; Dror, R.O.; Shaw, D.E. How Fast-Folding Proteins Fold. Science 2011, 334, 517–520. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.; Vaidehi, N.; Rodriguez, G. A Fast Recursive Algorithm for Molecular-Dynamics Simulation. J. Comput. Phys. 1993, 106, 258–268. [Google Scholar] [CrossRef]
- Vaidehi, N.; Jain, A.; Goddard, W.A. Constant temperature constrained molecular dynamics: The Newton-Euler inverse mass operator method. J. Phys. Chem. 1996, 100, 10508–10517. [Google Scholar] [CrossRef]
- Vaidehi, N.; Jain, A. Internal Coordinate Molecular Dynamics: A Foundation for Multiscale Dynamics. J. Phys. Chem. B 2015, 119, 1233–1242. [Google Scholar] [CrossRef] [PubMed]
- Gangupomu, V.K.; Wagner, J.R.; Park, I.H.; Jain, A.; Vaidehi, N. Mapping Conformational Dynamics of Proteins Using Torsional Dynamics Simulations. Biophys. J. 2013, 104, 1999–2008. [Google Scholar] [CrossRef]
- Kandel, S.; Salomon-Ferrer, R.; Larsen, A.B.; Jain, A.; Vaidehi, N. Overcoming potential energy distortions in constrained internal coordinate molecular dynamics simulations. J. Chem. Phys. 2016, 144, 044112. [Google Scholar] [CrossRef]
- Bui, J.M.; McCammon, J.A. Protein complex formation by acetylcholinesterase and the neurotoxin fasciculin-2 appears to involve an induced-fit mechanism. Proc. Natl. Acad. Sci. USA 2006, 103, 15451–15456. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.W.; Jasnow, D.; Zuckerman, D.M. Efficient and verified simulation of a path ensemble for conformational change in a united-residue model of calmodulin. Proc. Natl. Acad. Sci. USA 2007, 104, 18043–18048. [Google Scholar] [CrossRef]
- Elson, E.L. Fluorescence Correlation Spectroscopy: Past, Present, Future. Biophys. J. 2011, 101, 2855–2870. [Google Scholar] [CrossRef] [PubMed]
- Zerze, G.H.; Mittal, J.; Best, R.B. Diffusive Dynamics of Contact Formation in Disordered Polypeptides. Phys. Rev. Lett. 2016, 116, 068102. [Google Scholar] [CrossRef]
- Soranno, A.; Holla, A.; Dingfelder, F.; Nettels, D.; Makarov, D.E.; Schuler, B. Integrated view of internal friction in unfolded proteins from single-molecule FRET, contact quenching, theory, and simulations. Proc. Natl. Acad. Sci. USA 2017, 114, E1833–E1839. [Google Scholar] [CrossRef]
- Rezaei-Ghaleh, N.; Parigi, G.; Soranno, A.; Holla, A.; Becker, S.; Schuler, B.; Luchinat, C.; Zweckstetter, M. Local and Global Dynamics in Intrinsically Disordered Synuclein. Angew. Chem. Int. Edit. 2018, 57, 15262–15266. [Google Scholar] [CrossRef]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef]
- Best, R.B.; Hummer, G. Optimized molecular dynamics force fields applied to the helix-coil transition of polypeptides. J. Phys. Chem. B 2009, 113, 9004–9015. [Google Scholar] [CrossRef]
- Lindorff-Larsen, K.; Piana, S.; Palmo, K.; Maragakis, P.; Klepeis, J.L.; Dror, R.O.; Shaw, D.E. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins 2010, 78, 1950–1958. [Google Scholar] [CrossRef]
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, M.; de Groot, B.L.; Grubmuller, H.; MacKerell, A.D., Jr. CHARMM36m: an improved force field for folded and intrinsically disordered proteins. Nat. Methods 2017, 14, 71–73. [Google Scholar] [CrossRef]
- Palazzesi, F.; Prakash, M.K.; Bonomi, M.; Barducci, A. Accuracy of current all-atom force-fields in modeling protein disordered states. J. Chem. Theory Comput. 2015, 11, 2–7. [Google Scholar] [CrossRef]
- Rauscher, S.; Gapsys, V.; Gajda, M.J.; Zweckstetter, M.; de Groot, B.L.; Grubmuller, H. Structural Ensembles of Intrinsically Disordered Proteins Depend Strongly on Force Field: A Comparison to Experiment. J. Chem. Theory Comput. 2015, 11, 5513–5524. [Google Scholar] [CrossRef]
- Piana, S.; Lindorff-Larsen, K.; Shaw, D.E. How robust are protein folding simulations with respect to force field parameterization? Biophys. J. 2011, 100, L47–L49. [Google Scholar] [CrossRef] [PubMed]
- Best, R.B.; Zheng, W.; Mittal, J. Balanced Protein-Water Interactions Improve Properties of Disordered Proteins and Non-Specific Protein Association. J. Chem. Theory. Comput. 2014, 10, 5113–5124. [Google Scholar] [CrossRef]
- Jiang, F.; Zhou, C.Y.; Wu, Y.D. Residue-specific force field based on the protein coil library. RSFF1: modification of OPLS-AA/L. J. Phys. Chem. B 2014, 118, 6983–6998. [Google Scholar] [CrossRef]
- Zhou, C.Y.; Jiang, F.; Wu, Y.D. Residue-specific force field based on protein coil library. RSFF2: modification of AMBER ff99SB. J. Phys. Chem. B 2015, 119, 1035–1047. [Google Scholar] [CrossRef]
- Mercadante, D.; Milles, S.; Fuertes, G.; Svergun, D.I.; Lemke, E.A.; Grater, F. Kirkwood-Buff Approach Rescues Overcollapse of a Disordered Protein in Canonical Protein Force Fields. J. Phys. Chem. B 2015, 119, 7975–7984. [Google Scholar] [CrossRef]
- Yoo, J.; Aksimentiev, A. New tricks for old dogs: improving the accuracy of biomolecular force fields by pair-specific corrections to non-bonded interactions. Phys. Chem. Chem. Phys. 2018, 20, 8432–8449. [Google Scholar] [CrossRef]
- Song, D.; Wang, W.; Ye, W.; Ji, D.; Luo, R.; Chen, H.F. ff14IDPs force field improving the conformation sampling of intrinsically disordered proteins. Chem. Biol. Drug. Des. 2017, 89, 5–15. [Google Scholar] [CrossRef]
- Still, W.C.; Tempczyk, A.; Hawley, R.C.; Hendrickson, T. Semianalytical Treatment of Solvation for Molecular Mechanics and Dynamics. J. Am. Chem. Soc. 1990, 112, 6127–6129. [Google Scholar] [CrossRef]
- Kleinjung, J.; Fraternali, F. Design and application of implicit solvent models in biomolecular simulations. Curr. Opin. Struc. Biol. 2014, 25, 126–134. [Google Scholar] [CrossRef]
- Zhou, R.H. Free energy landscape of protein folding in water: Explicit vs. implicit solvent. Proteins-Structure Function Genetics 2003, 53, 148–161. [Google Scholar] [CrossRef]
- Awile, O.; Krisko, A.; Sbalzarini, I.F.; Zagrovic, B. Intrinsically Disordered Regions May Lower the Hydration Free Energy in Proteins: A Case Study of Nudix Hydrolase in the Bacterium Deinococcus radiodurans. PLoS Comput. Biol. 2010, 6, e1000854. [Google Scholar] [CrossRef]
- Vitalis, A.; Pappu, R.V. ABSINTH: A New Continuum Solvation Model for Simulations of Polypeptides in Aqueous Solutions. J. Comput. Chem. 2009, 30, 673–699. [Google Scholar] [CrossRef]
- Lee, K.H.; Chen, J.H. Optimization of the GBMV2 implicit solvent force field for accurate simulation of protein conformational equilibria. J. Comput. Chem. 2017, 38, 1332–1341. [Google Scholar] [CrossRef]
- Lee, M.S.; Feig, M.; Salsbury, F.R.; Brooks, C.L. New analytic approximation to the standard molecular volume definition and its application to generalized born calculations. J. Comput. Chem. 2003, 24, 1348–1356. [Google Scholar] [CrossRef]
- Choi, J.M.; Pappu, R.V. Improvements to the ABSINTH Force Field for Proteins Based on Experimentally Derived Amino Acid Specific Backbone Conformational Statistics. J. Chem. Theory Comput. 2019, 15, 1367–1382. [Google Scholar] [CrossRef]
- Lazaridis, T.; Karplus, M. Effective energy function for proteins in solution. Proteins 1999, 35, 133–152. [Google Scholar] [CrossRef]
- Das, R.K.; Pappu, R.V. Conformations of intrinsically disordered proteins are influenced by linear sequence distributions of oppositely charged residues. Proc. Natl. Acad. Sci. USA 2013, 110, 13392–13397. [Google Scholar] [CrossRef]
- Wuttke, R.; Hofmann, H.; Nettels, D.; Borgia, M.B.; Mittal, J.; Best, R.B.; Schuler, B. Temperature-dependent solvation modulates the dimensions of disordered proteins. Proc. Natl. Acad. Sci. USA 2014, 111, 5213–5218. [Google Scholar] [CrossRef]
- Davtyan, A.; Schafer, N.P.; Zheng, W.H.; Clementi, C.; Wolynes, P.G.; Papoian, G.A. AWSEM-MD: Protein Structure Prediction Using Coarse-Grained Physical Potentials and Bioinformatically Based Local Structure Biasing. J. Phys. Chem. B 2012, 116, 8494–8503. [Google Scholar] [CrossRef]
- Davtyan, A.; Zheng, W.H.; Schafer, N.; Wolynes, P.; Papoian, G. AWSEM-MD: Coarse-Grained Protein Structure Prediction using Physical Potentials and Bioinformatically Based Local Structure Biasing. Biophys. J. 2012, 102, 619a. [Google Scholar] [CrossRef]
- Wu, H.; Wolynes, P.G.; Papoian, G.A. AWSEM-IDP: A Coarse-Grained Force Field for Intrinsically Disordered Proteins. J. Phys. Chem. B 2018, 122, 11115–11125. [Google Scholar] [CrossRef]
- Lin, X.C.; Roy, S.; Jolly, M.K.; Bocci, F.; Schafer, N.P.; Tsai, M.Y.; Chen, Y.H.; He, Y.N.; Grishaev, A.; Weninger, K.; et al. PAGE4 and Conformational Switching: Insights from Molecular Dynamics Simulations and Implications for Prostate Cancer. J. Mol. Biol. 2018, 430, 2422–2438. [Google Scholar] [CrossRef]
- Bereau, T.; Deserno, M. Generic coarse-grained model for protein folding and aggregation. J. Chem. Phys. 2009, 130, 235106. [Google Scholar] [CrossRef]
- Rutter, G.O.; Brown, A.H.; Quigley, D.; Walsh, T.R.; Allen, M.P. Testing the transferability of a coarse-grained model to intrinsically disordered proteins. Phys. Chem. Chem. Phys. 2015, 17, 31741–31749. [Google Scholar] [CrossRef]
- Rutter, G.O.; Brown, A.H.; Quigley, D.; Walsh, T.R.; Allen, M.P. Emergence of order in self-assembly of the intrinsically disordered biomineralisation peptide n16N. Mol. Simulat. 2018, 44, 463–469. [Google Scholar] [CrossRef]
- Salmon, L.; Nodet, G.; Ozenne, V.; Yin, G.W.; Jensen, M.R.; Zweckstetter, M.; Blackledge, M. NMR Characterization of Long-Range Order in Intrinsically Disordered Proteins. J. Am. Chem. Soc. 2010, 132, 8407–8418. [Google Scholar] [CrossRef]
- Hsu, W.L.; Oldfield, C.J.; Xue, B.; Meng, J.; Huang, F.; Romero, P.; Uversky, V.N.; Dunker, A.K. Exploring the binding diversity of intrinsically disordered proteins involved in one-to-many binding. Protein Sci. 2013, 22, 258–273. [Google Scholar] [CrossRef]
- Sharma, R.; Raduly, Z.; Miskei, M.; Fuxreiter, M. Fuzzy complexes: Specific binding without complete folding. Febs. Lett. 2015, 589, 2533–2542. [Google Scholar] [CrossRef]
- Arbesu, M.; Iruela, G.; Fuentes, H.; Teixeira, J.M.C.; Pons, M. Intramolecular Fuzzy Interactions Involving Intrinsically Disordered Domains. Front. Mol. Biosci. 2018, 5. [Google Scholar] [CrossRef]
- Jakob, U.; Kriwacki, R.; Uversky, V.N. Conditionally and Transiently Disordered Proteins: Awakening Cryptic Disorder to Regulate Protein Function. Chem. Rev. 2014, 114, 6779–6805. [Google Scholar] [CrossRef]
- Sigalov, A.B. Membrane binding of intrinsically disordered proteins: Critical importance of an appropriate membrane model. Self. Nonself. 2010, 1, 129–132. [Google Scholar] [CrossRef]
- Milles, S.; Mercadante, D.; Aramburu, I.V.; Jensen, M.R.; Banterle, N.; Koehler, C.; Tyagi, S.; Clarke, J.; Shammas, S.L.; Blackledge, M.; et al. Plasticity of an Ultrafast Interaction between Nucleoporins and Nuclear Transport Receptors. Cell 2015, 163, 734–745. [Google Scholar] [CrossRef]
- Arai, M.; Sugase, K.; Dyson, H.J.; Wright, P.E. Conformational propensities of intrinsically disordered proteins influence the mechanism of binding and folding. Proc. Natl. Acad. Sci. USA 2015, 112, 9614–9619. [Google Scholar] [CrossRef]
- Wang, Y.; Chu, X.K.; Longhi, S.; Roche, P.; Han, W.; Wang, E.K.; Wang, J. Multiscaled exploration of coupled folding and binding of an intrinsically disordered molecular recognition element in measles virus nucleoprotein. Proc. Natl. Acad. Sci. USA 2013, 110, E3743–E3752. [Google Scholar] [CrossRef]
- Ganguly, D.; Chen, J.H. Topology-based modeling of intrinsically disordered proteins: Balancing intrinsic folding and intermolecular interactions. Proteins 2011, 79, 1251–1266. [Google Scholar] [CrossRef]
- Turjanski, A.G.; Gutkind, J.S.; Best, R.B.; Hummer, G. Binding-induced folding of a natively unstructured transcription factor. PLoS Comput. Biol. 2008, 4, e1000060. [Google Scholar] [CrossRef]
- Lu, Q.; Lu, H.P.; Wang, J. Exploring the mechanism of flexible biomolecular recognition with single molecule dynamics. Phys. Rev. Lett. 2007, 98, 263602. [Google Scholar] [CrossRef]
- Ganguly, D.; Zhang, W.H.; Chen, J.H. Electrostatically Accelerated Encounter and Folding for Facile Recognition of Intrinsically Disordered Proteins. PLoS Comput. Biol. 2013, 9, e1003363. [Google Scholar] [CrossRef]
- Knott, M.; Best, R.B. Discriminating binding mechanisms of an intrinsically disordered protein via a multi-state coarse-grained model. J. Chem. Phys. 2014, 140, 175102. [Google Scholar] [CrossRef] [PubMed]
- Clementi, C.; Nymeyer, H.; Onuchic, J.N. Topological and energetic factors: What determines the structural details of the transition state ensemble and "en-route" intermediates for protein folding? An investigation for small globular proteins. J. Mol. Biol. 2000, 298, 937–953. [Google Scholar] [CrossRef]
- Bryngelson, J.D.; Wolynes, P.G. Spin-Glasses and the Statistical-Mechanics of Protein Folding. Proc. Natl. Acad. Sci. USA 1987, 84, 7524–7528. [Google Scholar] [CrossRef]
- Bryngelson, J.D.; Onuchic, J.N.; Socci, N.D.; Wolynes, P.G. Funnels, Pathways, and the Energy Landscape of Protein-Folding - a Synthesis. Proteins 1995, 21, 167–195. [Google Scholar] [CrossRef]
- Uversky, V.N. Intrinsically disordered proteins in overcrowded milieu: Membrane-less organelles, phase separation, and intrinsic disorder. Curr. Opin. Struc. Biol. 2017, 44, 18–30. [Google Scholar] [CrossRef] [PubMed]
- Cuevas-Velazquez, C.L.; Dinneny, J.R. Organization out of disorder: liquid-liquid phase separation in plants. Curr. Opin. Plant. Biol. 2018, 45, 68–74. [Google Scholar] [CrossRef]
- Darling, A.L.; Liu, Y.; Oldfield, C.J.; Uversky, V.N. Intrinsically Disordered Proteome of Human Membrane-Less Organelles. Proteomics 2018, 18, 1700193. [Google Scholar] [CrossRef]
- Ambadipudi, S.; Biernat, J.; Riedel, D.; Mandelkow, E.; Zweckstetter, M. Liquid-liquid phase separation of the microtubule-binding repeats of the Alzheimer-related protein Tau. Nat. Commun. 2017, 8, 275. [Google Scholar] [CrossRef]
- Dignon, G.L.; Zheng, W.W.; Kim, Y.C.; Best, R.B.; Mittal, J. Sequence determinants of protein phase behavior from a coarse-grained model. PLoS Comput. Biol. 2018, 14, e1005941. [Google Scholar] [CrossRef]
- Boeynaems, S.; Holehouse, A.S.; Weinhardt, V.; Kovacs, D.; Van Lindt, J.; Larabell, C.; Van Den Bosch, L.; Das, R.; Tompa, P.S.; Pappu, R.V.; et al. Spontaneous driving forces give rise to protein-RNA condensates with coexisting phases and complex material properties. Proc. Natl. Acad. Sci. USA 2019. [Google Scholar] [CrossRef]
- Lin, Y.H.; Forman-Kay, J.D.; Chan, H.S. Theories for Sequence-Dependent Phase Behaviors of Biomolecular Condensates. Biochemistry 2018, 57, 2499–2508. [Google Scholar] [CrossRef]
- Ruff, K.M.; Pappu, R.V.; Holehouse, A.S. Conformational preferences and phase behavior of intrinsically disordered low complexity sequences: insights from multiscale simulations. Curr. Opin. Struct. Biol. 2018, 56, 1–10. [Google Scholar] [CrossRef]
- Kremer, K.; Binder, K. Monte-Carlo Simulations of Lattice Models for Macromolecules. Comput. Phys. Rep. 1988, 7, 259–310. [Google Scholar] [CrossRef]
- Bhattacharya, S.; Coasne, B.; Hung, F.R.; Gubbins, K.E. Modeling Micelle-Templated Mesoporous Material SBA-15: Atomistic Model and Gas Adsorption Studies. Langmuir 2009, 25, 5802–5813. [Google Scholar] [CrossRef]
- McCarty, J.; Delaney, K.T.; Danielsen, S.P.O.; Fredrickson, G.H.; Shea, J.E. Complete Phase Diagram for Liquid-Liquid Phase Separation of Intrinsically Disordered Proteins. J. Phys. Chem. Lett. 2019, 10, 1644–1652. [Google Scholar] [CrossRef]
- Levine, Z.A.; Larini, L.; LaPointe, N.E.; Feinstein, S.C.; Shea, J.E. Regulation and aggregation of intrinsically disordered peptides. Proc. Natl. Acad. Sci. USA 2015, 112, 2758–2763. [Google Scholar] [CrossRef]
- Das, P.; Murray, B.; Belfort, G. Alzheimer’s protective A2T mutation changes the conformational landscape of the Aβ1–42 monomer differently than does the A2V mutation. Biophys. J. 2015, 108, 738–747. [Google Scholar] [CrossRef]
- Chakraborty, S.; Das, P. Emergence of Alternative Structures in Amyloid Beta 1-42 Monomeric Landscape by N-terminal Hexapeptide Amyloid Inhibitors. Sci. Rep.-UK 2017, 7, 9941. [Google Scholar] [CrossRef]
- Mondal, B.; Reddy, G. Cosolvent Effects on the Growth of Protein Aggregates Formed by a Single Domain Globular Protein and an Intrinsically Disordered Protein. J. Phys. Chem. B 2019, 123, 1950–1960. [Google Scholar] [CrossRef]
- Zheng, W.H.; Schafer, N.P.; Wolynes, P.G. Free energy landscapes for initiation and branching of protein aggregation. Proc. Natl. Acad. Sci. USA 2013, 110, 20515–20520. [Google Scholar] [CrossRef] [PubMed]
- Metallo, S.J. Intrinsically disordered proteins are potential drug targets. Curr. Opin. Chem. Biol. 2010, 14, 481–488. [Google Scholar] [CrossRef] [PubMed]
- Camargo, D.C.R.; Tripsianes, K.; Buday, K.; Franko, A.; Gobl, C.; Hartlmuller, C.; Sarkar, R.; Aichler, M.; Mettenleiter, G.; Schulz, M.; et al. The redox environment triggers conformational changes and aggregation of hIAPP in Type II Diabetes. Sci. Rep.-UK 2017, 7, 44041. [Google Scholar] [CrossRef]
- Follis, A.V.; Hammoudeh, D.I.; Wang, H.B.; Prochownik, E.V.; Metallo, S.J. Structural Rationale for the Coupled Binding and Unfolding of the c-Myc Oncoprotein by Small Molecules. Chem. Biol. 2008, 15, 1149–1155. [Google Scholar] [CrossRef]
- Hammoudeh, D.I.; Follis, A.V.; Prochownik, E.V.; Metallo, S.J. Multiple Independent Binding Sites for Small-Molecule Inhibitors on the Oncoprotein c-Myc. J. Am. Chem. Soc. 2009, 131, 7390–7401. [Google Scholar] [CrossRef]
- Neira, J.L.; Bintz, J.; Arruebo, M.; Rizzuti, B.; Bonacci, T.; Vega, S.; Lanas, A.; Velazquez-Campoy, A.; Iovanna, J.L.; Abian, O. Identification of a Drug Targeting an Intrinsically Disordered Protein Involved in Pancreatic Adenocarcinoma. Sci. Rep.-UK 2017, 7, 39732. [Google Scholar] [CrossRef] [PubMed]
- Price, D.L.; Koike, M.A.; Khan, A.; Wrasidlo, W.; Rockenstein, E.; Masliah, E.; Bonhaus, D. The small molecule alpha-synuclein misfolding inhibitor, NPT200-11, produces multiple benefits in an animal model of Parkinson’s disease. Sci. Rep.-UK 2018, 8, 16165. [Google Scholar] [CrossRef]
- Pujols, J.; Pena-Diaz, S.; Lazaro, D.F.; Peccati, F.; Pinheiro, F.; Gonzalez, D.; Carija, A.; Navarro, S.; Conde-Gimenez, M.; Garcia, J.; et al. Small molecule inhibits alpha-synuclein aggregation, disrupts amyloid fibrils, and prevents degeneration of dopaminergic neurons. Proc. Natl. Acad. Sci. USA 2018, 115, 10481–10486. [Google Scholar] [CrossRef]
- Ruan, H.; Sun, Q.; Zhang, W.L.; Liu, Y.; Lai, L.H. Targeting intrinsically disordered proteins at the edge of chaos. Drug Discov. Today 2019, 24, 217–227. [Google Scholar] [CrossRef]
- Joshi, P.; Vendruscolo, M. Druggability of Intrinsically Disordered Proteins. Adv. Exp. Med. Biol. 2015, 870, 383–400. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; LeGall, T.; Oldfield, C.J.; Mueller, J.P.; Van, Y.Y.J.; Romero, P.; Cortese, M.S.; Uversky, V.N.; Dunker, A.K. Rational drug design via intrinsically disordered protein. Trends Biotechnol. 2006, 24, 435–442. [Google Scholar] [CrossRef]
- Subramaniam, S.; Mehrotra, M.; Gupta, D. Virtual high throughput screening (vHTS)—A perspective. Bioinformation 2008, 3, 14–17. [Google Scholar] [CrossRef]
- Zhang, Y.; Cao, H.; Liu, Z. Binding cavities and druggability of intrinsically disordered proteins. Protein Sci. 2015, 24, 688–705. [Google Scholar] [CrossRef]
- Michel, J.; Cuchillo, R. The Impact of Small Molecule Binding on the Energy Landscape of the Intrinsically Disordered Protein C-Myc. PLoS ONE 2012, 7. [Google Scholar] [CrossRef]
- Venken, T.; Voet, A.; De Maeyer, M.; De Fabritiis, G.; Sadiq, S.K. Rapid Conformational Fluctuations of Disordered HIV-1 Fusion Peptide in Solution. J. Chem. Theory Comput. 2013, 9, 2870–2874. [Google Scholar] [CrossRef]
- Motlagh, H.N.; Wrabl, J.O.; Li, J.; Hilser, V.J. The ensemble nature of allostery. Nature 2014, 508, 331–339. [Google Scholar] [CrossRef]
- Motlagh, H.N.; Li, J.; Thompson, E.B.; Hilser, V.J. Interplay between allostery and intrinsic disorder in an ensemble. Biochem. Soc. T. 2012, 40, 975–980. [Google Scholar] [CrossRef] [PubMed]
- Ferreon, A.C.M.; Ferreon, J.C.; Wright, P.E.; Deniz, A.A. Modulation of allostery by protein intrinsic disorder. Nature 2013, 498, 390–394. [Google Scholar] [CrossRef] [PubMed]
- Bai, F.; Branch, R.W.; Nicolau, D.V.; Pilizota, T.; Steel, B.C.; Maini, P.K.; Berry, R.M. Conformational Spread as a Mechanism for Cooperativity in the Bacterial Flagellar Switch. Science 2010, 327, 685–689. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, S.; Vaidehi, N. Differences in Allosteric Communication Pipelines in the Inactive and Active States of a GPCR. Biophys. J. 2014, 107, 422–434. [Google Scholar] [CrossRef]
- Tautermann, C.S.; Binder, F.; Buttner, F.H.; Eickmeier, C.; Fiegen, D.; Gross, U.; Grundl, M.A.; Heilker, R.; Hobson, S.; Hoerer, S.; et al. Allosteric Activation of Striatal-Enriched Protein Tyrosine Phosphatase (STEP, PTPN5) by a Fragment-like Molecule. J. Med. Chem. 2019, 62, 306–316. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Kasam, V.; Tautermann, C.S.; Seeliger, D.; Vaidehi, N. Computational Method to Identify Druggable Binding Sites That Target Protein-Protein Interactions. J. Chem. Infor. Model. 2014, 54, 1391–1400. [Google Scholar] [CrossRef]
- Le Guilloux, V.; Schmidtke, P.; Tuffery, P. Fpocket: An open source platform for ligand pocket detection. BMC Bioinformatics 2009, 10. [Google Scholar] [CrossRef] [PubMed]
- Fisher, C.K.; Huang, A.; Stultz, C.M. Modeling Intrinsically Disordered Proteins with Bayesian Statistics. J. Am. Chem. Soc. 2010, 132, 14919–14927. [Google Scholar] [CrossRef] [PubMed]
- Gong, H.C.; Zhang, S.; Wang, J.D.; Gong, H.P.; Zeng, J.Y. Constructing Structure Ensembles of Intrinsically Disordered Proteins from Chemical Shift Data. J. Comput. Biol. 2016, 23, 300–310. [Google Scholar] [CrossRef] [PubMed]
- Sickmeier, M.; Hamilton, J.A.; LeGall, T.; Vacic, V.; Cortese, M.S.; Tantos, A.; Szabo, B.; Tompa, P.; Chen, J.; Uversky, V.N.; et al. DisProt: the database of disordered proteins. Nucleic Acids Res. 2007, 35, D786–D793. [Google Scholar] [CrossRef] [PubMed]
- Varadi, M.; Kosol, S.; Lebrun, P.; Valentini, E.; Blackledge, M.; Dunker, A.K.; Felli, I.C.; Forman-Kay, J.D.; Kriwacki, R.W.; Pierattelli, R.; et al. pE-DB: a database of structural ensembles of intrinsically disordered and of unfolded proteins. Nucleic Acids Res. 2014, 42, D326–D335. [Google Scholar] [CrossRef]
- Morcos, F.; Pagnani, A.; Lunt, B.; Bertolino, A.; Marks, D.S.; Sander, C.; Zecchina, R.; Onuchic, J.N.; Hwa, T.; Weigt, M. Direct-coupling analysis of residue coevolution captures native contacts across many protein families. Proc. Natl. Acad. Sci. USA 2011, 108, E1293–E1301. [Google Scholar] [CrossRef] [PubMed]
- Bai, F.; Morcos, F.; Cheng, R.R.; Jiang, H.L.; Onuchic, J.N. Elucidating the druggable interface of protein-protein interactions using fragment docking and coevolutionary analysis. Proc. Natl. Acad. Sci. USA 2016, 113, E8051–E8058. [Google Scholar] [CrossRef]
- Pancsa, R.; Zsolyomi, F.; Tompa, P. Co-Evolution of Intrinsically Disordered Proteins with Folded Partners Witnessed by Evolutionary Couplings. Int. J. Mol. Sci. 2018, 19, 3315. [Google Scholar] [CrossRef] [PubMed]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bhattacharya, S.; Lin, X. Recent Advances in Computational Protocols Addressing Intrinsically Disordered Proteins. Biomolecules 2019, 9, 146. https://doi.org/10.3390/biom9040146
Bhattacharya S, Lin X. Recent Advances in Computational Protocols Addressing Intrinsically Disordered Proteins. Biomolecules. 2019; 9(4):146. https://doi.org/10.3390/biom9040146
Chicago/Turabian StyleBhattacharya, Supriyo, and Xingcheng Lin. 2019. "Recent Advances in Computational Protocols Addressing Intrinsically Disordered Proteins" Biomolecules 9, no. 4: 146. https://doi.org/10.3390/biom9040146