Modulation of Measles Virus NTAIL Interactions through Fuzziness and Sequence Features of Disordered Binding Sites



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
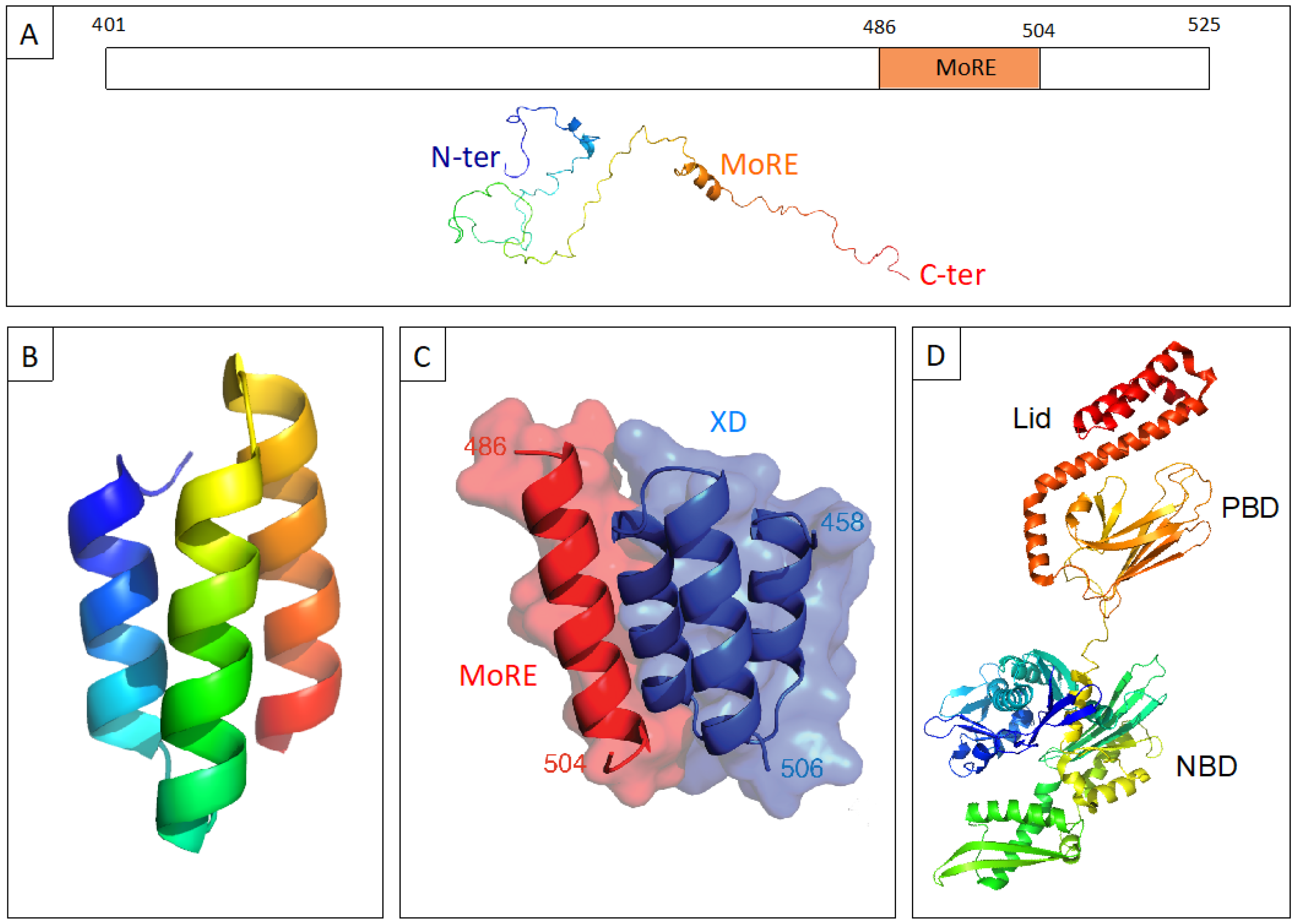
:1. Structural Properties and Molecular Partnership of NTAIL
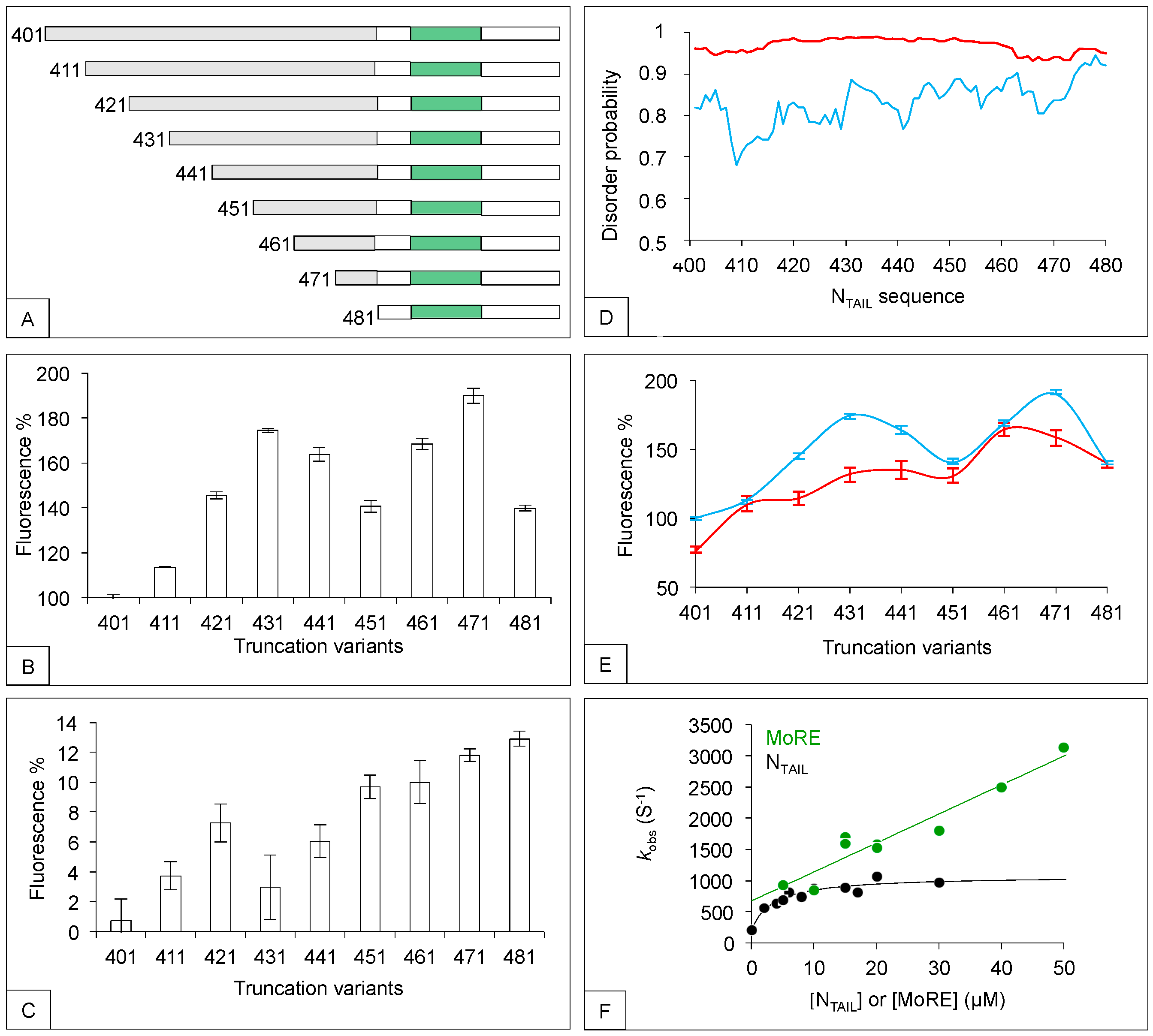
2. The N-Terminal Fuzzy Region of NTAIL down Regulates the Binding of the MoRE to Both XD and Hsp70
3. The Bindings of XD and Hsp70 to NTAIL MoRE Rely on Different Primary and Secondary Structure Requirements
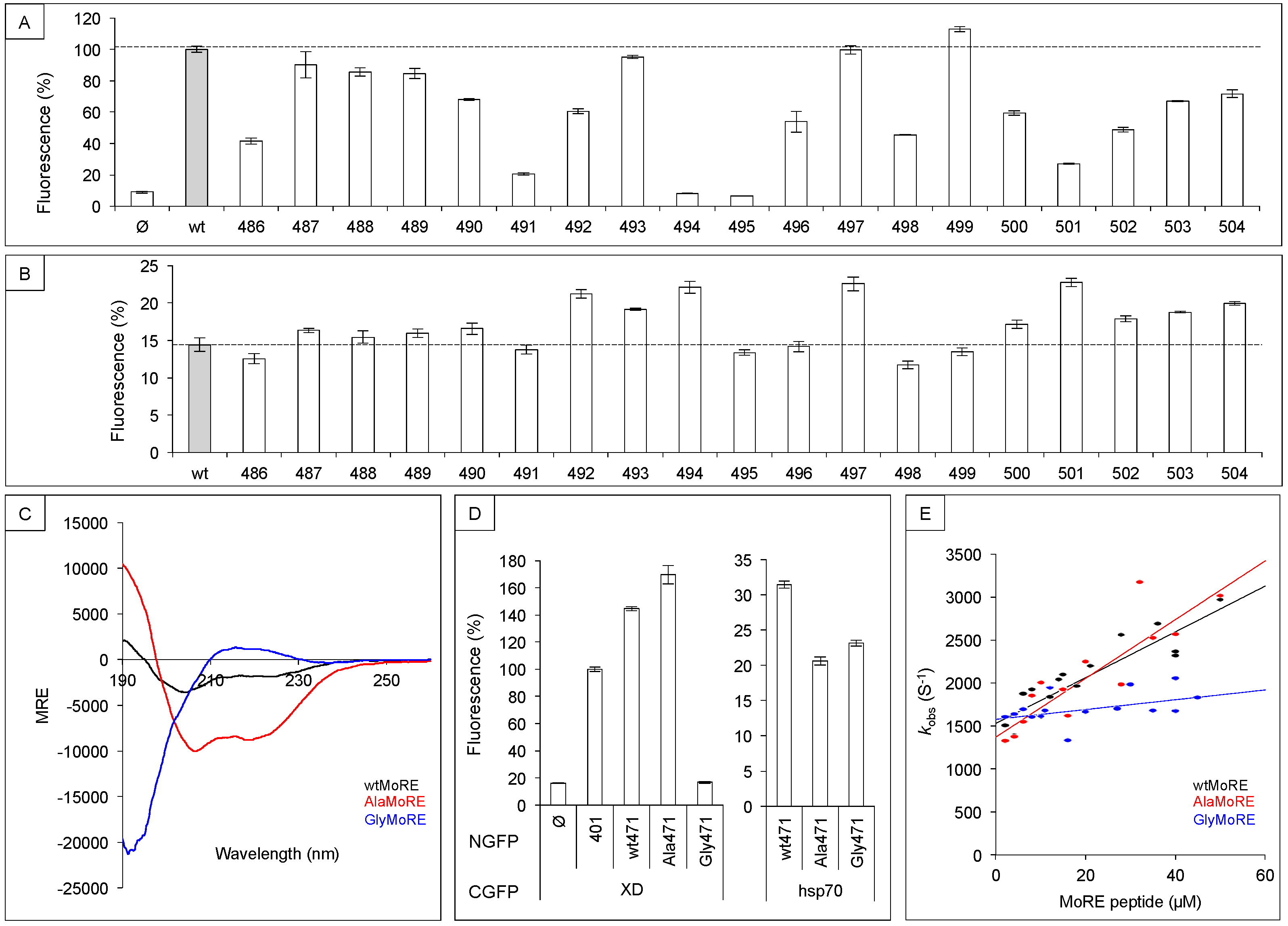
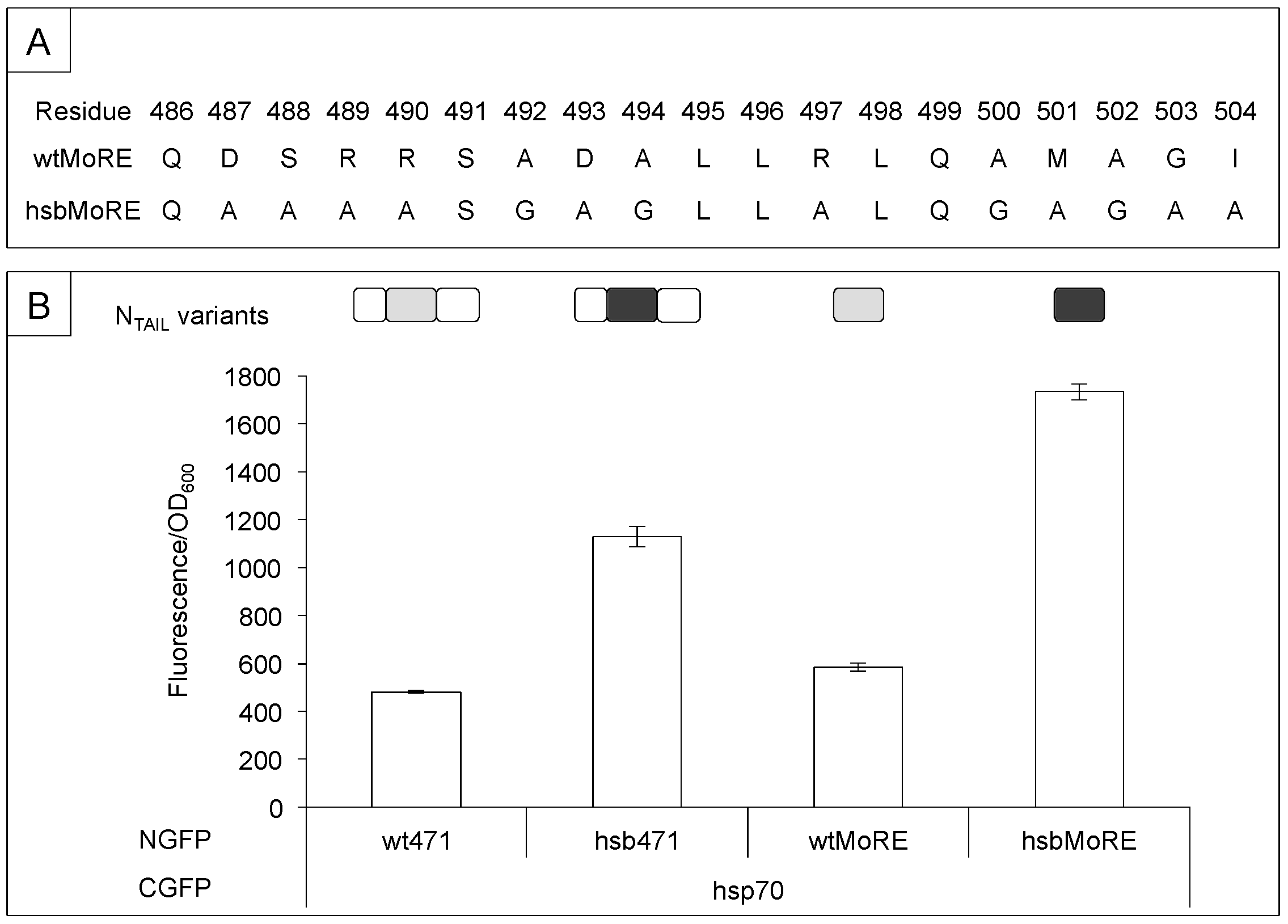
3.1. Sequence Requirements of NTAIL Molecular Recognition Element for XD and Hsp70 Binding
3.2. Secondary Structure Requirements of NTAIL Molecular Recognition Element for XD and Hsp70 Binding
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Longhi, S.; Receveur-Brechot, V.; Karlin, D.; Johansson, K.; Darbon, H.; Bhella, D.; Yeo, R.; Finet, S.; Canard, B. The C-terminal domain of the measles virus nucleoprotein is intrinsically disordered and folds upon binding to the C-terminal moiety of the phosphoprotein. J. Biol. Chem. 2003, 278, 18638–18648. [Google Scholar] [CrossRef] [PubMed]
- Heggeness, M.H.; Scheid, A.; Choppin, P.W. Conformation of the helical nucleocapsids of paramyxoviruses and vesicular stomatitis virus: Reversible coiling and uncoiling induced by changes in salt concentration. Proc. Natl. Acad. Sci. USA 1980, 77, 2631–2635. [Google Scholar] [CrossRef] [PubMed]
- Heggeness, M.H.; Scheid, A.; Choppin, P.W. The relationship of conformational changes in the Sendai virus nucleocapsid to proteolytic cleavage of the NP polypeptide. Virology 1981, 114, 555–562. [Google Scholar] [CrossRef]
- Karlin, D.; Longhi, S.; Canard, B. Substitution of two residues in the measles virus nucleoprotein results in an impaired self-association. Virology 2002, 302, 420–432. [Google Scholar] [CrossRef] [PubMed]
- Ringkjøbing Jensen, M.; Communie, G.; Ribeiro, E.D., Jr.; Martinez, N.; Desfosses, A.; Salmon, L.; Mollica, L.; Gabel, F.; Jamin, M.; Longhi, S.; et al. Intrinsic disorder in measles virus nucleocapsids. Proc. Natl. Acad. Sci. USA 2011, 108, 9839–9844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gutsche, I.; Desfosses, A.; Effantin, G.; Ling, W.L.; Haupt, M.; Ruigrok, R.W.; Sachse, C.; Schoehn, G. Near-atomic cryo-EM structure of the helical measles virus nucleocapsid. Science 2015, 348, 704–707. [Google Scholar] [CrossRef] [PubMed]
- Bourhis, J.; Johansson, K.; Receveur-Bréchot, V.; Oldfield, C.J.; Dunker, A.K.; Canard, B.; Longhi, S. The C-terminal domain of measles virus nucleoprotein belongs to the class of intrinsically disordered proteins that fold upon binding to their physiological partner. Virus Res. 2004, 99, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Bourhis, J.M.; Receveur-Bréchot, V.; Oglesbee, M.; Zhang, X.; Buccellato, M.; Darbon, H.; Canard, B.; Finet, S.; Longhi, S. The intrinsically disordered C-terminal domain of the measles virus nucleoprotein interacts with the C-terminal domain of the phosphoprotein via two distinct sites and remains predominantly unfolded. Protein Sci. 2005, 14, 1975–1992. [Google Scholar] [CrossRef] [Green Version]
- Johansson, K.; Bourhis, J.M.; Campanacci, V.; Cambillau, C.; Canard, B.; Longhi, S. Crystal structure of the measles virus phosphoprotein domain responsible for the induced folding of the C-terminal domain of the nucleoprotein. J. Biol. Chem. 2003, 278, 44567–44573. [Google Scholar] [CrossRef]
- Kingston, R.L.; Hamel, D.J.; Gay, L.S.; Dahlquist, F.W.; Matthews, B.W. Structural basis for the attachment of a paramyxoviral polymerase to its template. Proc. Natl. Acad. Sci. USA 2004, 101, 8301–8306. [Google Scholar] [CrossRef] [Green Version]
- Longhi, S.; Bloyet, L.M.; Gianni, S.; Gerlier, D. How order and disorder within paramyxoviral nucleoproteins and phosphoproteins orchestrate the molecular interplay of transcription and replication. Cell. Mol. Life Sci. 2017, 74, 3091–3118. [Google Scholar] [CrossRef] [PubMed]
- Dunker, A.K.; Cortese, M.S.; Romero, P.; Iakoucheva, L.M.; Uversky, V.N. Flexible nets. FEBS J. 2005, 272, 5129–5148. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N.; Oldfield, C.J.; Dunker, A.K. Showing your ID: Intrinsic disorder as an ID for recognition, regulation and cell signaling. J. Mol. Recognit. 2005, 18, 343–384. [Google Scholar] [CrossRef] [PubMed]
- Haynes, C.; Oldfield, C.J.; Ji, F.; Klitgord, N.; Cusick, M.E.; Radivojac, P.; Uversky, V.N.; Vidal, M.; Iakoucheva, L.M. Intrinsic disorder is a common feature of hub proteins from four eukaryotic interactomes. PLoS Comput. Biol. 2006, 2, e100. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, M.; Takeda, M.; Shirogane, Y.; Nakatsu, Y.; Nakamura, T.; Yanagi, Y. The matrix protein of measles virus regulates viral RNA synthesis and assembly by interacting with the nucleocapsid protein. J. Virol. 2009, 83, 10374–10383. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Glendening, C.; Linke, H.; Parks, C.L.; Brooks, C.; Udem, S.A.; Oglesbee, M. Identification and characterization of a regulatory domain on the carboxyl terminus of the measles virus nucleocapsid protein. J. Virol. 2002, 76, 8737–8746. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Bourhis, J.M.; Longhi, S.; Carsillo, T.; Buccellato, M.; Morin, B.; Canard, B.; Oglesbee, M. Hsp72 recognizes a P binding motif in the measles virus N protein C-terminus. Virology 2005, 337, 162–174. [Google Scholar] [CrossRef] [Green Version]
- Couturier, M.; Buccellato, M.; Costanzo, S.; Bourhis, J.M.; Shu, Y.; Nicaise, M.; Desmadril, M.; Flaudrops, C.; Longhi, S.; Oglesbee, M. High Affinity Binding between Hsp70 and the C-Terminal Domain of the Measles Virus Nucleoprotein Requires an Hsp40 Co-Chaperone. J. Mol. Recognit. 2010, 23, 301–315. [Google Scholar] [CrossRef]
- Sato, H.; Masuda, M.; Miura, R.; Yoneda, M.; Kai, C. Morbillivirus nucleoprotein possesses a novel nuclear localization signal and a CRM1-independent nuclear export signal. Virology 2006, 352, 121–130. [Google Scholar] [CrossRef] [Green Version]
- TenOever, B.R.; Servant, M.J.; Grandvaux, N.; Lin, R.; Hiscott, J. Recognition of the Measles Virus Nucleocapsid as a Mechanism of IRF-3 Activation. J. Virol. 2002, 76, 3659–3669. [Google Scholar] [CrossRef] [Green Version]
- Colombo, M.; Bourhis, J.M.; Chamontin, C.; Soriano, C.; Villet, S.; Costanzo, S.; Couturier, M.; Belle, V.; Fournel, A.; Darbon, H.; et al. The interaction between the measles virus nucleoprotein and the Interferon Regulator Factor 3 relies on a specific cellular environment. Virol. J. 2009, 6, 59. [Google Scholar] [CrossRef] [PubMed]
- Laine, D.; Bourhis, J.; Longhi, S.; Flacher, M.; Cassard, L.; Canard, B.; Sautès-Fridman, C.; Rabourdin-Combe, C.; Valentin, H. Measles virus nucleoprotein induces cell proliferation arrest and apoptosis through NTAIL/NR and NCORE/FcgRIIB1 interactions, respectively. J. Gen. Virol. 2005, 86, 1771–1784. [Google Scholar] [CrossRef] [PubMed]
- Laine, D.; Trescol-Biémont, M.; Longhi, S.; Libeau, G.; Marie, J.; Vidalain, P.; Azocar, O.; Diallo, A.; Canard, B.; Rabourdin-Combe, C.; et al. Measles virus nucleoprotein binds to a novel cell surface receptor distinct from FcgRII via its C-terminal domain: Role in MV-induced immunosuppression. J. Virol. 2003, 77, 11332–11346. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, A.; Yoneda, M.; Ikeda, F.; Sugai, A.; Sato, H.; Kai, C. Peroxiredoxin 1 is required for efficient transcription and replication of measles virus. J. Virol. 2011, 85, 2247–2253. [Google Scholar] [CrossRef] [PubMed]
- De, B.P.; Banerjee, A.K. Involvement of actin microfilaments in the transcription/replication of human parainfluenza virus type 3: Possible role of actin in other viruses. Microsc. Res. Tech. 1999, 47, 114–123. [Google Scholar] [CrossRef]
- Moyer, S.A.; Baker, S.C.; Horikami, S.M. Host cell proteins required for measles virus reproduction. J. Gen. Virol. 1990, 71, 775–783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Habchi, J.; Blangy, S.; Mamelli, L.; Ringkjobing Jensen, M.; Blackledge, M.; Darbon, H.; Oglesbee, M.; Shu, Y.; Longhi, S. Characterization of the interactions between the nucleoprotein and the phosphoprotein of Henipaviruses. J. Biol. Chem. 2011, 286, 13583–13602. [Google Scholar] [CrossRef] [PubMed]
- Dosnon, M.; Bonetti, D.; Morrone, A.; Erales, J.; di Silvio, E.; Longhi, S.; Gianni, S. Demonstration of a folding after binding mechanism in the recognition between the measles virus NTAIL and X domains. ACS Chem. Biol. 2015, 10, 795–802. [Google Scholar] [CrossRef]
- Gely, S.; Lowry, D.F.; Bernard, C.; Ringkjobing-Jensen, M.; Blackledge, M.; Costanzo, S.; Darbon, H.; Daughdrill, G.W.; Longhi, S. Solution structure of the C-terminal X domain of the measles virus phosphoprotein and interaction with the intrinsically disordered C-terminal domain of the nucleoprotein. J. Mol. Recognit. 2010, 23, 435–447. [Google Scholar] [CrossRef]
- D’Urzo, A.; Konijnenberg, A.; Rossetti, G.; Habchi, J.; Li, J.; Carloni, P.; Sobott, F.; Longhi, S.; Grandori, R. Molecular Basis for Structural Heterogeneity of an Intrinsically Disordered Protein Bound to a Partner by Combined ESI-IM-MS and Modeling. J. Am. Soc. Mass Spectrom. 2015, 26, 472–481. [Google Scholar] [CrossRef]
- Bonetti, D.; Camilloni, C.; Visconti, L.; Longhi, S.; Brunori, M.; Vendruscolo, M.; Gianni, S. Identification and Structural Characterization of an Intermediate in the Folding of the Measles Virus X Domain. J. Biol. Chem. 2016, 291, 10886–10892. [Google Scholar] [CrossRef]
- Habchi, J.; Mamelli, L.; Darbon, H.; Longhi, S. Structural Disorder within Henipavirus Nucleoprotein and Phosphoprotein: From Predictions to Experimental Assessment. PLoS ONE 2010, 5, e11684. [Google Scholar] [CrossRef] [PubMed]
- Communie, G.; Habchi, J.; Yabukarski, F.; Blocquel, D.; Schneider, R.; Tarbouriech, N.; Papageorgiou, N.; Ruigrok, R.W.; Jamin, M.; Ringkjøbing-Jensen, M.; et al. Atomic resolution description of the interaction between the nucleoprotein and phosphoprotein of Hendra virus. PLoS Pathog. 2013, 9, e1003631. [Google Scholar] [CrossRef] [PubMed]
- Fuxreiter, M.; Simon, I.; Friedrich, P.; Tompa, P. Preformed structural elements feature in partner recognition by intrinsically unstructured proteins. J. Mol. Biol. 2004, 338, 1015–1026. [Google Scholar] [CrossRef] [PubMed]
- Morin, B.; Bourhis, J.M.; Belle, V.; Woudstra, M.; Carrière, F.; Guigliarelli, B.; Fournel, A.; Longhi, S. Assessing induced folding of an intrinsically disordered protein by site-directed spin-labeling EPR spectroscopy. J. Phys. Chem. B 2006, 110, 20596–20608. [Google Scholar] [CrossRef]
- Belle, V.; Rouger, S.; Costanzo, S.; Liquiere, E.; Strancar, J.; Guigliarelli, B.; Fournel, A.; Longhi, S. Mapping alpha-helical induced folding within the intrinsically disordered C-terminal domain of the measles virus nucleoprotein by site-directed spin-labeling EPR spectroscopy. Proteins Struct. Funct. Bioinform. 2008, 73, 973–988. [Google Scholar] [CrossRef] [PubMed]
- Martinho, M.; Habchi, J.; El Habre, Z.; Nesme, L.; Guigliarelli, B.; Belle, V.; Longhi, S. Assessing induced folding within the intrinsically disordered C-terminal domain of the Henipavirus nucleoproteins by site directed spin labeling EPR spectroscopy. J. Biomol. Struct. Dyn. 2013, 31, 453–471. [Google Scholar] [CrossRef] [PubMed]
- Baronti, L.; Erales, J.; Habchi, J.; Felli, I.C.; Pierattelli, R.; Longhi, S. Dynamics of the intrinsically disordered C-terminal domain of the Nipah virus nucleoprotein and interaction with the X domain of the phosphoprotein as unveiled by NMR spectroscopy. ChemBioChem 2015, 16, 268–276. [Google Scholar] [CrossRef]
- Wang, Y.; Chu, X.; Longhi, S.; Roche, P.; Han, W.; Wang, E.; Wang, J. Multiscaled exploration of coupled folding and binding of an intrinsically disordered molecular recognition element in measles virus nucleoprotein. Proc. Natl. Acad. Sci. USA 2013, 110, e3743–e3752. [Google Scholar] [CrossRef]
- Shang, X.; Chu, W.; Chu, X.; Xu, L.; Longhi, S.; Wang, J. Exploration of nucleoprotein alpha-MoRE and XD interactions of Nipah and Hendra viruses. J. Mol. Model. 2018, 24, 113. [Google Scholar] [CrossRef]
- Bonetti, D.; Troilo, F.; Toto, A.; Brunori, M.; Longhi, S.; Gianni, S. Analyzing the folding and binding steps of an intrinsically disordered protein by protein engineering. Biochemistry 2017, 56, 3780–3786. [Google Scholar] [CrossRef] [PubMed]
- Belle, V.; Rouger, S.; Costanzo, S.; Longhi, S.; Fournel, A. Site-directed spin labeling EPR spectroscopy. In Instrumental Analysis of Intrinsically Disordered Proteins: Assessing Structure and Conformation; Uversky, V.N., Longhi, S., Eds.; John Wiley and Sons: Hoboken, NJ, USA, 2010. [Google Scholar]
- Blocquel, D.; Habchi, J.; Gruet, A.; Blangy, S.; Longhi, S. Compaction and binding properties of the intrinsically disordered C-terminal domain of Henipavirus nucleoprotein as unveiled by deletion studies. Mol. Biosyst. 2012, 8, 392–410. [Google Scholar] [CrossRef] [PubMed]
- Troilo, F.; Bignon, C.; Gianni, S.; Fuxreiter, M.; Longhi, S. Experimental characterization of fuzzy protein assemblies: Interactions of paramyxoviral NTAIL domains with their functional partners. Methods Enzymol. 2018, 611, 137–192. [Google Scholar] [PubMed]
- Tompa, P.; Fuxreiter, M. Fuzzy complexes: Polymorphism and structural disorder in protein-protein interactions. Trends Biochem. Sci. 2008, 33, 2–8. [Google Scholar] [CrossRef] [PubMed]
- Fuxreiter, M. Fuzziness: Linking regulation to protein dynamics. Mol. Biosyst. 2012, 8, 168–177. [Google Scholar] [CrossRef] [PubMed]
- Fuxreiter, M. Fuzziness in protein interactions—A historical perspective. J. Mol. Biol. 2018, 430, 2278–2287. [Google Scholar] [CrossRef]
- Fuxreiter, M. Fold or not to fold upon binding—Does it really matter? Curr. Opin. Struct. Biol. 2018, 54, 19–25. [Google Scholar] [CrossRef]
- Ozenne, V.; Bauer, F.; Salmon, L.; Huang, J.R.; Jensen, M.R.; Segard, S.; Bernado, P.; Charavay, C.; Blackledge, M. Flexible-meccano: A tool for the generation of explicit ensemble descriptions of intrinsically disordered proteins and their associated experimental observables. Bioinformatics 2012, 28, 1463–1470. [Google Scholar] [CrossRef]
- Carsillo, T.; Zhang, X.; Vasconcelos, D.; Niewiesk, S.; Oglesbee, M. A single codon in the nucleocapsid protein C terminus contributes to in vitro and in vivo fitness of Edmonston measles virus. J. Virol. 2006, 80, 2904–2912. [Google Scholar] [CrossRef]
- Oglesbee, M. Nucleocapsid protein interactions with the major inducible heat shock protein. In Measles Virus Nucleoprotein; Longhi, S., Ed.; Nova Publishers Inc.: Hauppage, NY, USA, 2007; pp. 53–98. [Google Scholar]
- Oglesbee, M.J.; Kenney, H.; Kenney, T.; Krakowka, S. Enhanced production of morbillivirus gene-specific RNAs following induction of the cellular stress response in stable persistent infection. Virology 1993, 192, 556–567. [Google Scholar] [CrossRef]
- Oglesbee, M.J.; Liu, Z.; Kenney, H.; Brooks, C.L. The highly inducible member of the 70 kDa family of heat shock proteins increases canine distemper virus polymerase activity. J. Gen. Virol. 1996, 77, 2125–2135. [Google Scholar] [CrossRef] [PubMed]
- Vasconcelos, D.; Norrby, E.; Oglesbee, M. The cellular stress response increases measles virus-induced cytopathic effect. J. Gen. Virol. 1998, 79, 1769–1773. [Google Scholar] [CrossRef] [PubMed]
- Vasconcelos, D.Y.; Cai, X.H.; Oglesbee, M.J. Constitutive overexpression of the major inducible 70 kDa heat shock protein mediates large plaque formation by measles virus. J. Gen. Virol. 1998, 79, 2239–2247. [Google Scholar] [CrossRef]
- Longhi, S.; Oglesbee, M. Structural disorder within the measles virus nucleoprotein and phosphoprotein. Protein Peptide Lett. 2010, 17, 961–978. [Google Scholar] [CrossRef]
- Wilson, C.G.; Magliery, T.J.; Regan, L. Detecting protein-protein interactions with GFP-fragment reassembly. Nat. Methods 2004, 1, 255–262. [Google Scholar] [CrossRef] [PubMed]
- Gruet, A.; Dosnon, M.; Vassena, A.; Lombard, V.; Gerlier, D.; Bignon, C.; Longhi, S. Dissecting partner recognition by an intrinsically disordered protein using descriptive random mutagenesis. J. Mol. Biol. 2013, 425, 3495–3509. [Google Scholar] [CrossRef]
- Jackrel, M.E.; Cortajarena, A.L.; Liu, T.Y.; Regan, L. Screening libraries to identify proteins with desired binding activities using a split-GFP reassembly assay. ACS Chem. Biol. 2010, 5, 553–562. [Google Scholar] [CrossRef]
- Magliery, T.J.; Wilson, C.G.; Pan, W.; Mishler, D.; Ghosh, I.; Hamilton, A.D.; Regan, L. Detecting protein-protein interactions with a green fluorescent protein fragment reassembly trap: Scope and mechanism. J. Am. Chem. Soc. 2005, 127, 146–157. [Google Scholar] [CrossRef]
- Gruet, A.; Dosnon, M.; Blocquel, D.; Brunel, J.; Gerlier, D.; Das, R.K.; Bonetti, D.; Gianni, S.; Fuxreiter, M.; Longhi, S.; et al. Fuzzy regions in an intrinsically disordered protein impair protein-protein interactions. FEBS J. 2016, 283, 576–594. [Google Scholar] [CrossRef] [Green Version]
- Cassonnet, P.; Rolloy, C.; Neveu, G.; Vidalain, P.O.; Chantier, T.; Pellet, J.; Jones, L.; Muller, M.; Demeret, C.; Gaud, G.; et al. Benchmarking a luciferase complementation assay for detecting protein complexes. Nat. Methods 2011, 8, 990–992. [Google Scholar] [CrossRef]
- Keul, N.D.; Oruganty, K.; Schaper Bergman, E.T.; Beattie, N.R.; McDonald, W.E.; Kadirvelraj, R.; Gross, M.L.; Phillips, R.S.; Harvey, S.C.; Wood, Z.A. The entropic force generated by intrinsically disordered segments tunes protein function. Nature 2018. [Google Scholar] [CrossRef] [PubMed]
- Dosztanyi, Z.; Csizmok, V.; Tompa, P.; Simon, I. IUPred: Web server for the prediction of intrinsically unstructured regions of proteins based on estimated energy content. Bioinformatics 2005, 21, 3433–3434. [Google Scholar] [CrossRef]
- Kim, M.Y.; Shu, Y.; Carsillo, T.; Zhang, J.; Yu, L.; Peterson, C.; Longhi, S.; Girod, S.; Niewiesk, S.; Oglesbee, M. Hsp70 and a novel axis of type I interferon-dependent antiviral immunity in the measles virus-infected brain. J. Virol. 2013, 87, 998–1009. [Google Scholar] [CrossRef] [PubMed]
- Bignon, C.; Troilo, F.; Gianni, S.; Longhi, S. Partner-mediated polymorphism of an intrinsically disordered protein. J. Mol. Biol. 2018, 430, 2493–2507. [Google Scholar] [CrossRef] [PubMed]
- Reichmann, D.; Xu, Y.; Cremers, C.M.; Ilbert, M.; Mittelman, R.; Fitzgerald, M.C.; Jakob, U. Order out of disorder: Working cycle of an intrinsically unfolded chaperone. Cell 2012, 148, 947–957. [Google Scholar] [CrossRef] [PubMed]
- O’Neil, K.T.; DeGrado, W.F. A thermodynamic scale for the helix-forming tendencies of the commonly occurring amino acids. Science 1990, 250, 646–651. [Google Scholar] [CrossRef] [PubMed]
- Kolakofsky, D.; Le Mercier, P.; Iseni, F.; Garcin, D. Viral DNA polymerase scanning and the gymnastics of Sendai virus RNA synthesis. Virology 2004, 318, 463–473. [Google Scholar] [CrossRef]
- Thakkar, V.D.; Cox, R.M.; Sawatsky, B.; da Fontoura Budaszewski, R.; Sourimant, J.; Wabbel, K.; Makhsous, N.; Greninger, A.L.; von Messling, V.; Plemper, R.K. The Unstructured Paramyxovirus Nucleocapsid Protein Tail Domain Modulates Viral Pathogenesis through Regulation of Transcriptase Activity. J. Virol. 2018, 92. [Google Scholar] [CrossRef]
- Brunel, J.; Chopy, D.; Dosnon, M.; Bloyet, L.M.; Devaux, P.; Urzua, E.; Cattaneo, R.; Longhi, S.; Gerlier, D. Sequence of events in measles virus replication: Role of phosphoprotein-nucleocapsid interactions. J. Virol. 2014, 88, 10851–10863. [Google Scholar] [CrossRef]
- Bloyet, L.; Brunel, J.; Dosnon, M.; Hamon, V.; Erales, J.; Gruet, A.; Lazert, C.; Bignon, C.; Roche, P.; Longhi, S.; et al. Modulation of re-initiation of measles virus transcription at intergenic regions by PXD to NTAIL binding strength. PLoS Pathog. 2016, 12, e1006058. [Google Scholar] [CrossRef]
- Shu, Y.; Habchi, J.; Costanzo, S.; Padilla, A.; Brunel, J.; Gerlier, D.; Oglesbee, M.; Longhi, S. Plasticity in structural and functional interactions between the phosphoprotein and nucleoprotein of measles virus. J. Biol. Chem. 2012, 287, 11951–11967. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Bu, L.; Wang, C.; Zhang, Y.; Zhou, H.; Zhang, X.; Guo, W.; Long, C.; Guo, D.; Sun, X. The Hsp70 inhibitor 2-phenylethynesulfonamide inhibits replication and carcinogenicity of Epstein-Barr virus by inhibiting the molecular chaperone function of Hsp70. Cell Death Dis. 2018, 9, 734. [Google Scholar] [CrossRef] [PubMed]
- Leu, J.I.; Pimkina, J.; Frank, A.; Murphy, M.E.; George, D.L. A small molecule inhibitor of inducible heat shock protein 70. Mol. Cell 2009, 36, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Granato, M.; Lacconi, V.; Peddis, M.; Lotti, L.V.; Di Renzo, L.; Gonnella, R.; Santarelli, R.; Trivedi, P.; Frati, L.; D’Orazi, G.; et al. HSP70 inhibition by 2-phenylethynesulfonamide induces lysosomal cathepsin D release and immunogenic cell death in primary effusion lymphoma. Cell Death Dis. 2013, 4, e730. [Google Scholar] [CrossRef] [PubMed]




© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bignon, C.; Troilo, F.; Gianni, S.; Longhi, S. Modulation of Measles Virus NTAIL Interactions through Fuzziness and Sequence Features of Disordered Binding Sites. Biomolecules 2019, 9, 8. https://doi.org/10.3390/biom9010008
Bignon C, Troilo F, Gianni S, Longhi S. Modulation of Measles Virus NTAIL Interactions through Fuzziness and Sequence Features of Disordered Binding Sites. Biomolecules. 2019; 9(1):8. https://doi.org/10.3390/biom9010008
Chicago/Turabian StyleBignon, Christophe, Francesca Troilo, Stefano Gianni, and Sonia Longhi. 2019. "Modulation of Measles Virus NTAIL Interactions through Fuzziness and Sequence Features of Disordered Binding Sites" Biomolecules 9, no. 1: 8. https://doi.org/10.3390/biom9010008