Combining Optical Approaches with Human Inducible Pluripotent Stem Cells in G Protein-Coupled Receptor Drug Screening and Development

Abstract
:1. Introduction
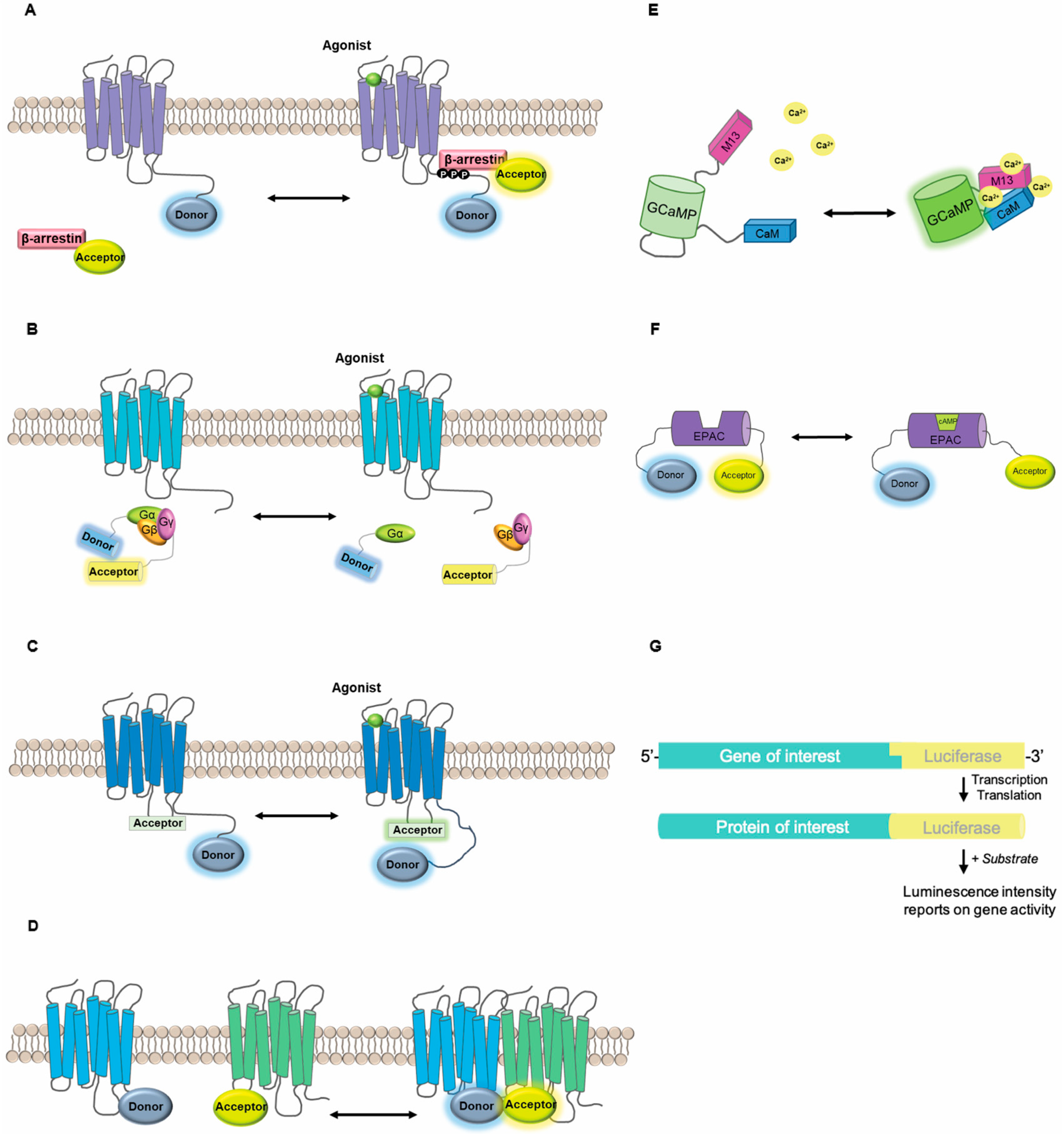
2. Genetically-Encoded Optical Biosensors
2.1. Design Considerations
2.2. Biosensor Applications
3. How Cell Context Effects Signaling: Evidence from In Vivo Studies
3.1. Single Cell Sequencing
3.2. Single Cell Signaling Assays
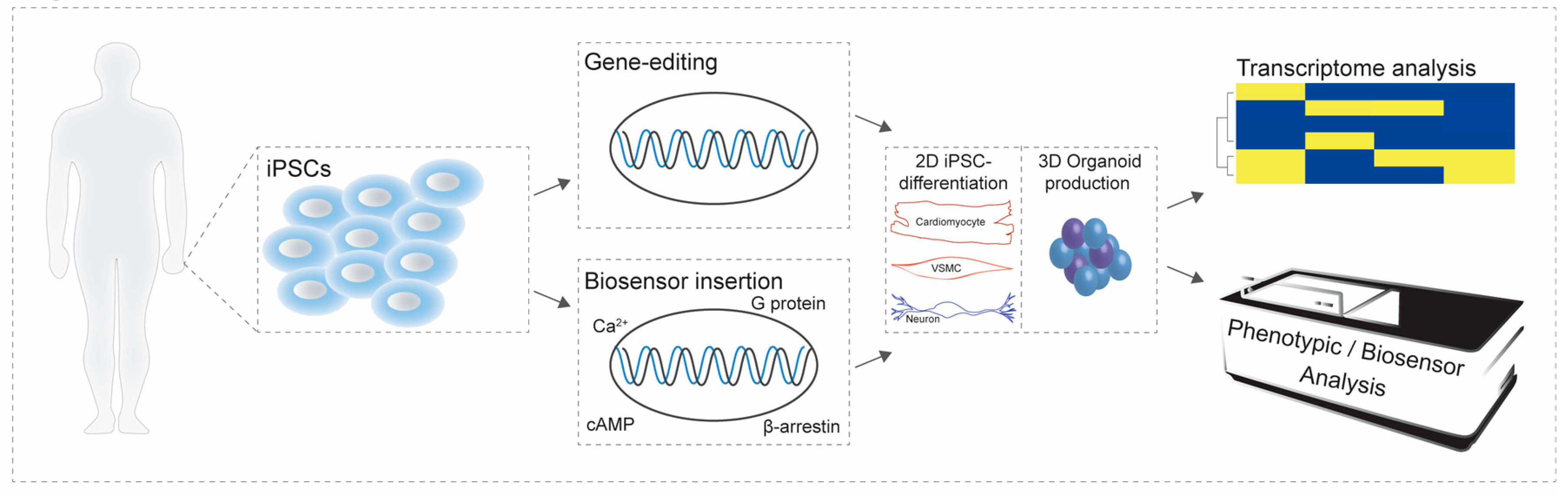
4. Incorporating Cell Context into Screening Approaches
4.1. Inducible Pluripotent Stem Cells and their Differentiated Derivatives: Gene Expression and Functional Validation
4.2. Application of Inducible Pluripotent Stem Cells for Screening Purposes
5. Considerations When Studying Human Disease in Inducible Pluripotent Stem Cell-Based Cellular Models
6. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Thomson, J.A.; Itskovitz-Eldor, J.; Shapiro, S.S.; Waknitz, M.A.; Swiergiel, J.J.; Marshall, V.S.; Jones, J.M. Embryonic stem cell lines derived from human blastocysts. Science 1998, 282, 1145–1147. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Vodyanik, M.A.; Smuga-Otto, K.; Antosiewicz-Bourget, J.; Frane, J.L.; Tian, S.; Nie, J.; Jonsdottir, G.A.; Ruotti, V.; Stewart, R.; et al. Induced pluripotent stem cell lines derived from human somatic cells. Science 2007, 318, 1917–1920. [Google Scholar] [CrossRef] [PubMed]
- Tiscornia, G.; Vivas, E.L.; Izpisua Belmonte, J.C. Diseases in a dish: Modeling human genetic disorders using induced pluripotent cells. Nat. Med. 2011, 17, 1570–1576. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Inoue, H.; Wu, J.C.; Yamanaka, S. Induced pluripotent stem cell technology: A decade of progress. Nat. Rev. Drug Discov. 2017, 16, 115–130. [Google Scholar] [CrossRef] [PubMed]
- Sayed, N.; Liu, C.; Wu, J.C. Translation of Human-Induced Pluripotent Stem Cells: From Clinical Trial in a Dish to Precision Medicine. J. Am. Coll. Cardiol. 2016, 67, 2161–2176. [Google Scholar] [CrossRef] [PubMed]
- Miranda, C.C.; Fernandes, T.G.; Diogo, M.M.; Cabral, J.M.S. Towards Multi-Organoid Systems for Drug Screening Applications. Bioengineering 2018, 5, 49. [Google Scholar] [CrossRef] [PubMed]
- Devarasetty, M.; Mazzocchi, A.R.; Skardal, A. Applications of Bioengineered 3D Tissue and Tumor Organoids in Drug Development and Precision Medicine: Current and Future. BioDrugs Clin. Immunother. Biopharm. Gene Ther. 2018, 32, 53–68. [Google Scholar] [CrossRef] [PubMed]
- Takata, N.; Eiraku, M. Stem cells and genome editing: Approaches to tissue regeneration and regenerative medicine. J. Hum. Genet. 2018, 63, 165–178. [Google Scholar] [CrossRef] [PubMed]
- Marinissen, M.J.; Gutkind, J.S. G-protein-coupled receptors and signaling networks: Emerging paradigms. Trends Pharmacol. Sci. 2001, 22, 368–376. [Google Scholar] [CrossRef]
- Ritter, S.L.; Hall, R.A. Fine-tuning of GPCR activity by receptor-interacting proteins. Nat. Rev. Mol. Cell Biol. 2009, 10, 819–830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goyet, E.; Bouquier, N.; Ollendorff, V.; Perroy, J. Fast and high resolution single-cell BRET imaging. Sci. Rep. 2016, 6, 28231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kauk, M.; Hoffmann, C. Intramolecular and Intermolecular FRET Sensors for GPCRs—Monitoring Conformational Changes and Beyond. Trends Pharmacol. Sci. 2018, 39, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Lohse, M.J.; Nuber, S.; Hoffmann, C. Fluorescence/bioluminescence resonance energy transfer techniques to study G-protein-coupled receptor activation and signaling. Pharmacol. Rev. 2012, 64, 299–336. [Google Scholar] [CrossRef] [PubMed]
- Hochreiter, B.; Garcia, A.P.; Schmid, J.A. Fluorescent proteins as genetically encoded FRET biosensors in life sciences. Sensors 2015, 15, 26281–26314. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.-W.; Wardill, T.J.; Sun, Y.; Pulver, S.R.; Renninger, S.L.; Baohan, A.; Schreiter, E.R.; Kerr, R.A.; Orger, M.B.; Jayaraman, V.; et al. Ultrasensitive fluorescent proteins for imaging neuronal activity. Nature 2013, 499, 295. [Google Scholar] [CrossRef] [PubMed]
- Hackley, C.R.; Mazzoni, E.O.; Blau, J. cAMPr: A single-wavelength fluorescent sensor for cyclic AMP. Sci. Signal. 2018, 11. [Google Scholar] [CrossRef] [PubMed]
- Harada, K.; Ito, M.; Wang, X.; Tanaka, M.; Wongso, D.; Konno, A.; Hirai, H.; Hirase, H.; Tsuboi, T.; Kitaguchi, T. Red fluorescent protein-based cAMP indicator applicable to optogenetics and in vivo imaging. Sci. Rep. 2017, 7, 7351. [Google Scholar] [CrossRef] [PubMed]
- Ross, B.L.; Tenner, B.; Markwardt, M.L.; Zviman, A.; Shi, G.; Kerr, J.P.; Snell, N.E.; McFarland, J.J.; Mauban, J.R.; Ward, C.W.; et al. Single-color, ratiometric biosensors for detecting signaling activities in live cells. eLife 2018, 7, e35458. [Google Scholar] [CrossRef] [PubMed]
- Gealageas, R.; Malikova, N.P.; Picaud, S.; Borgdorff, A.J.; Burakova, L.P.; Brulet, P.; Vysotski, E.S.; Dodd, R.H. Bioluminescent properties of obelin and aequorin with novel coelenterazine analogues. Anal. Bioanal. Chem. 2014, 406, 2695–2707. [Google Scholar] [CrossRef] [PubMed]
- Ayoub, M.A. Resonance Energy Transfer-Based Approaches to Study GPCRs. Methods Cell Biol. 2016, 132, 255–292. [Google Scholar] [PubMed]
- Bourque, K.; Pétrin, D.; Sleno, R.; Devost, D.; Zhang, A.; Hébert, T.E. Distinct Conformational Dynamics of Three G Protein-Coupled Receptors Measured Using FlAsH-BRET Biosensors. Front. Endocrinol. 2017, 8, 61. [Google Scholar] [CrossRef] [PubMed]
- Sleno, R.; Pétrin, D.; Devost, D.; Goupil, E.; Zhang, A.; Hébert, T.E. Designing BRET-based conformational biosensors for G protein-coupled receptors. Methods 2016, 92, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Nuber, S.; Zabel, U.; Lorenz, K.; Nuber, A.; Milligan, G.; Tobin, A.B.; Lohse, M.J.; Hoffmann, C. β-Arrestin biosensors reveal a rapid, receptor-dependent activation/deactivation cycle. Nature 2016, 531, 661–664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klarenbeek, J.; Goedhart, J.; van Batenburg, A.; Groenewald, D.; Jalink, K. Fourth-generation EPAC-based FRET sensors for cAMP feature exceptional brightness, photostability and dynamic range: Characterization of dedicated sensors for FLIM, for ratiometry and with high affinity. PLoS ONE 2015, 10, e0122513. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, N.; Aoki, K.; Yamada, M.; Yukinaga, H.; Fujita, Y.; Kamioka, Y.; Matsuda, M. Development of an optimized backbone of FRET biosensors for kinases and GTPases. Mol. Biol. Cell 2011, 22, 4647–4656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lossi, L.; Cocito, C.; Alasia, S.; Merighi, A. Ex vivo imaging of active caspase 3 by a FRET-based molecular probe demonstrates the cellular dynamics and localization of the protease in cerebellar granule cells and its regulation by the apoptosis-inhibiting protein survivin. Mol. Neurodegener. 2016, 11, 34. [Google Scholar] [CrossRef] [PubMed]
- Hébert, T.E.; Galés, C.; Rebois, R.V. Detecting and imaging protein-protein interactions during G protein-mediated signal transduction in vivo and in situ by using fluorescence-based techniques. Cell Biochem. Biophys. 2006, 45, 85–109. [Google Scholar] [CrossRef]
- van Unen, J.; Stumpf, A.D.; Schmid, B.; Reinhard, N.R.; Hordijk, P.L.; Hoffmann, C.; Gadella, T.W., Jr.; Goedhart, J. A New Generation of FRET Sensors for Robust Measurement of Gαi1, Gαi2 and Gαi3 Activation kinetics in single cells. PLoS ONE 2016, 11, e0146789. [Google Scholar] [CrossRef] [PubMed]
- Becker, W. Fluorescence lifetime imaging—techniques and applications. J. Microsc. 2012, 247, 119–136. [Google Scholar] [CrossRef] [PubMed]
- Miyawaki, A.; Niino, Y. Molecular spies for bioimaging--fluorescent protein-based probes. Mol. Cell 2015, 58, 632–643. [Google Scholar] [CrossRef] [PubMed]
- Couturier, C.; Deprez, B. Setting Up a Bioluminescence Resonance Energy Transfer High throughput Screening Assay to Search for Protein/Protein Interaction Inhibitors in Mammalian Cells. Front. Endocrinol. 2012, 3, 100. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Cumberbatch, D.; Centanni, S.; Shi, S.Q.; Winder, D.; Webb, D.; Johnson, C.H. Coupling optogenetic stimulation with NanoLuc-based luminescence (BRET) Ca(++) sensing. Nat. Commun. 2016, 7, 13268. [Google Scholar] [CrossRef] [PubMed]
- Marvin, J.S.; Borghuis, B.G.; Tian, L.; Cichon, J.; Harnett, M.T.; Akerboom, J.; Gordus, A.; Renninger, S.L.; Chen, T.W.; Bargmann, C.I.; et al. An optimized fluorescent probe for visualizing glutamate neurotransmission. Nat. Methods 2013, 10, 162–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patriarchi, T.; Cho, J.R.; Merten, K.; Howe, M.W.; Marley, A.; Xiong, W.H.; Folk, R.W.; Broussard, G.J.; Liang, R.; Jang, M.J.; et al. Ultrafast neuronal imaging of dopamine dynamics with designed genetically encoded sensors. Science 2018, 360. [Google Scholar] [CrossRef] [PubMed]
- Sun, F.; Zeng, J.; Jing, M.; Zhou, J.; Feng, J.; Owen, S.F.; Luo, Y.; Li, F.; Wang, H.; Yamaguchi, T.; et al. A Genetically Encoded Fluorescent Sensor Enables Rapid and Specific Detection of Dopamine in Flies, Fish, and Mice. Cell 2018, 174, 481–496. [Google Scholar] [CrossRef] [PubMed]
- Salahpour, A.; Espinoza, S.; Masri, B.; Lam, V.; Barak, L.S.; Gainetdinov, R.R. BRET biosensors to study GPCR biology, pharmacology, and signal transduction. Front. Endocrinol. 2012, 3, 105. [Google Scholar] [CrossRef] [PubMed]
- Devost, D.; Sleno, R.; Pétrin, D.; Zhang, A.; Shinjo, Y.; Okde, R.; Aoki, J.; Inoue, A.; Hébert, T.E. Conformational Profiling of the AT1 Angiotensin II Receptor Reflects Biased Agonism, G Protein Coupling, and Cellular Context. J. Biol. Chem. 2017, 292, 5443–5456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calebiro, D.; Nikolaev, V.O.; Persani, L.; Lohse, M.J. Signaling by internalized G-protein-coupled receptors. Trends Pharmacol. Sci. 2010, 31, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Salahpour, A.; Angers, S.; Bouvier, M. Functional significance of oligomerization of G-protein-coupled receptors. Trends Endocrinol. Metabol. TEM 2000, 11, 163–168. [Google Scholar] [CrossRef]
- Namkung, Y.; Radresa, O.; Armando, S.; Devost, D.; Beautrait, A.; Le Gouill, C.; Laporte, S.A. Quantifying biased signaling in GPCRs using BRET-based biosensors. Methods 2016, 92, 5–10. [Google Scholar] [CrossRef] [PubMed]
- Breton, B.; Sauvageau, E.; Zhou, J.; Bonin, H.; Le Gouill, C.; Bouvier, M. Multiplexing of multicolor bioluminescence resonance energy transfer. Biophys. J. 2010, 99, 4037–4046. [Google Scholar] [CrossRef] [PubMed]
- Vilardaga, J.P.; Bunemann, M.; Krasel, C.; Castro, M.; Lohse, M.J. Measurement of the millisecond activation switch of G protein-coupled receptors in living cells. Nat. Biotechnol. 2003, 21, 807–812. [Google Scholar] [CrossRef] [PubMed]
- Costa-Neto, C.M.; Parreiras, E.S.L.T.; Bouvier, M. A Pluridimensional View of Biased Agonism. Mol. Pharmacol. 2016, 90, 587–595. [Google Scholar] [CrossRef] [PubMed]
- Kenakin, T. The Effective Application of Biased Signaling to New Drug Discovery. Mol. Pharmacol. 2015, 88, 1055–1061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaur, H.; Carvalho, J.; Looso, M.; Singh, P.; Chennupati, R.; Preussner, J.; Günther, S.; Albarrán-Juárez, J.; Tischner, D.; Classen, S.; et al. Single-cell profiling reveals heterogeneity and functional patterning of GPCR expression in the vascular system. Nat. Commun. 2017, 8, 15700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tischner, D.; Grimm, M.; Kaur, H.; Staudenraus, D.; Carvalho, J.; Looso, M.; Gunther, S.; Wanke, F.; Moos, S.; Siller, N.; et al. Single-cell profiling reveals GPCR heterogeneity and functional patterning during neuroinflammation. JCI Insight 2017, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hohenhaus, D.M.; Schaale, K.; Le Cao, K.A.; Seow, V.; Iyer, A.; Fairlie, D.P.; Sweet, M.J. An mRNA atlas of G protein-coupled receptor expression during primary human monocyte/macrophage differentiation and lipopolysaccharide-mediated activation identifies targetable candidate regulators of inflammation. Immunobiology 2013, 218, 1345–1353. [Google Scholar] [CrossRef] [PubMed]
- Camp, J.G.; Wollny, D.; Treutlein, B. Single-cell genomics to guide human stem cell and tissue engineering. Nat. Methods 2018, 15, 661–667. [Google Scholar] [CrossRef] [PubMed]
- Pijuan-Sala, B.; Guibentif, C.; Gottgens, B. Single-cell transcriptional profiling: A window into embryonic cell-type specification. Nat. Rev. Mol. Cell Biol. 2018, 19, 399–412. [Google Scholar] [CrossRef] [PubMed]
- Marcott, P.F.; Gong, S.; Donthamsetti, P.; Grinnell, S.G.; Nelson, M.N.; Newman, A.H.; Birnbaumer, L.; Martemyanov, K.A.; Javitch, J.A.; Ford, C.P. Regional Heterogeneity of D2-Receptor Signaling in the Dorsal Striatum and Nucleus Accumbens. Neuron 2018, 98, 575–587. [Google Scholar] [CrossRef] [PubMed]
- Halls, M.L.; Yeatman, H.R.; Nowell, C.J.; Thompson, G.L.; Gondin, A.B.; Civciristov, S.; Bunnett, N.W.; Lambert, N.A.; Poole, D.P.; Canals, M. Plasma membrane localization of the μ-opioid receptor controls spatiotemporal signaling. Sci. Signal. 2016, 9, ra16. [Google Scholar] [CrossRef] [PubMed]
- Lein, E.S.; Hawrylycz, M.J.; Ao, N.; Ayres, M.; Bensinger, A.; Bernard, A.; Boe, A.F.; Boguski, M.S.; Brockway, K.S.; Byrnes, E.J.; et al. Genome-wide atlas of gene expression in the adult mouse brain. Nature 2007, 445, 168–176. [Google Scholar] [CrossRef] [PubMed]
- Zeisel, A.; Muñoz-Manchado, A.B.; Codeluppi, S.; Lönnerberg, P.; La Manno, G.; Juréus, A.; Marques, S.; Munguba, H.; He, L.; Betsholtz, C.; et al. Brain structure. Cell types in the mouse cortex and hippocampus revealed by single-cell RNA-seq. Science 2015, 347, 1138–1142. [Google Scholar] [CrossRef] [PubMed]
- Tasic, B.; Menon, V.; Nguyen, T.; Kim, T.; Jarsky, T.; Yao, Z.; Levi, B.; Gray, L.T.; Sorensen, S.A.; Dolbeare, T.; et al. Adult mouse cortical cell taxonomy revealed by single cell transcriptomics. Nat. Neurosci. 2016, 19, 335–346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xin, Y.; Kim, J.; Ni, M.; Wei, Y.; Okamoto, H.; Lee, J.; Adler, C.; Cavino, K.; Murphy, A.J.; Yancopoulos, G.D. Use of the Fluidigm C1 platform for RNA sequencing of single mouse pancreatic islet cells. Proc. Natl. Acad. Sci. USA 2016, 113, 3293–3298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macosko, E.Z.; Basu, A.; Satija, R.; Nemesh, J.; Shekhar, K.; Goldman, M.; Tirosh, I.; Bialas, A.R.; Kamitaki, N.; Martersteck, E.M.; et al. Highly Parallel Genome-wide Expression Profiling of Individual Cells Using Nanoliter Droplets. Cell 2015, 161, 1202–1214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Treutlein, B.; Brownfield, D.G.; Wu, A.R.; Neff, N.F.; Mantalas, G.L.; Espinoza, F.H.; Desai, T.J.; Krasnow, M.A.; Quake, S.R. Reconstructing lineage hierarchies of the distal lung epithelium using single-cell RNA-seq. Nature 2014, 509, 371–375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shalek, A.K.; Satija, R.; Shuga, J.; Trombetta, J.J.; Gennert, D.; Lu, D.; Chen, P.; Gertner, R.S.; Gaublomme, J.T.; Yosef, N.; et al. Single-cell RNA-seq reveals dynamic paracrine control of cellular variation. Nature 2014, 510, 363. [Google Scholar] [CrossRef] [PubMed]
- Handel, A.E.; Chintawar, S.; Lalic, T.; Whiteley, E.; Vowles, J.; Giustacchini, A.; Argoud, K.; Sopp, P.; Nakanishi, M.; Bowden, R.; et al. Assessing similarity to primary tissue and cortical layer identity in induced pluripotent stem cell-derived cortical neurons through single-cell transcriptomics. Hum. Mol. Genet. 2016, 25, 989–1000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paik, D.T.; Tian, L.; Lee, J.; Sayed, N.; Chen, I.Y.; Rhee, S.; Rhee, J.-W.; Kim, Y.; Wirka, R.C.; Buikema, J.W.; et al. Large-Scale Single-Cell RNA-Seq Reveals Molecular Signatures of Heterogeneous Populations of Human Induced Pluripotent Stem Cell-Derived Endothelial Cells. Circ. Res. 2018, 123, 443–450. [Google Scholar] [CrossRef] [PubMed]
- Friedman, C.E.; Nguyen, Q.; Lukowski, S.W.; Helfer, A.; Chiu, H.S.; Miklas, J.; Levy, S.; Suo, S.; Han, J.J.; Osteil, P.; et al. Single-Cell Transcriptomic Analysis of Cardiac Differentiation from Human PSCs Reveals HOPX-Dependent Cardiomyocyte Maturation. Cell Stem Cell 2018, 23, 586–598. [Google Scholar] [CrossRef] [PubMed]
- Tu, C.; Chao, B.S.; Wu, J.C. Strategies for Improving the Maturity of Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes. Circ. Res. 2018, 123, 512–514. [Google Scholar] [CrossRef] [PubMed]
- Engle, S.J.; Blaha, L.; Kleiman, R.J. Best Practices for Translational Disease Modeling Using Human iPSC-Derived Neurons. Neuron 2018, 100, 783–797. [Google Scholar] [CrossRef] [PubMed]
- Hirata, E.; Kiyokawa, E. Future Perspective of Single-Molecule FRET Biosensors and Intravital FRET Microscopy. Biophys. J. 2016, 111, 1103–1111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yanagawa, M.; Hiroshima, M.; Togashi, Y.; Abe, M.; Yamashita, T.; Shichida, Y.; Murata, M.; Ueda, M.; Sako, Y. Single-molecule diffusion-based estimation of ligand effects on G protein-coupled receptors. Sci. Signal. 2018, 11. [Google Scholar] [CrossRef] [PubMed]
- Partridge, J.G. Utilizing GCaMP transgenic mice to monitor endogenous Gq/11-coupled receptors. Front. Pharmacol. 2015, 6, 42. [Google Scholar] [CrossRef] [PubMed]
- Muntean, B.S.; Zucca, S.; MacMullen, C.M.; Dao, M.T.; Johnston, C.; Iwamoto, H.; Blakely, R.D.; Davis, R.L.; Martemyanov, K.A. Interrogating the Spatiotemporal Landscape of Neuromodulatory GPCR Signaling by Real-Time Imaging of cAMP in Intact Neurons and Circuits. Cell Rep. 2018, 24, 1081–1084. [Google Scholar] [CrossRef] [PubMed]
- Hiratsuka, T.; Sano, T.; Kato, H.; Komatsu, N.; Imajo, M.; Kamioka, Y.; Sumiyama, K.; Banno, F.; Miyata, T.; Matsuda, M. Live imaging of extracellular signal-regulated kinase and protein kinase A activities during thrombus formation in mice expressing biosensors based on Forster resonance energy transfer. J. Thromb. Haemost. JTH 2017, 15, 1487–1499. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, F.; Kamioka, Y.; Yano, T.; Matsuda, M. In Vivo FRET Imaging of Tumor Endothelial Cells Highlights a Role of Low PKA Activity in Vascular Hyperpermeability. Cancer Res. 2016, 76, 5266–5276. [Google Scholar] [CrossRef] [PubMed]
- Nobis, M.; Herrmann, D.; Warren, S.C.; Kadir, S.; Leung, W.; Killen, M.; Magenau, A.; Stevenson, D.; Lucas, M.C.; Reischmann, N.; et al. A RhoA-FRET Biosensor Mouse for Intravital Imaging in Normal Tissue Homeostasis and Disease Contexts. Cell Rep. 2017, 21, 274–288. [Google Scholar] [CrossRef] [PubMed]
- Strzelecka, P.M.; Ranzoni, A.M.; Cvejic, A. Dissecting human disease with single-cell omics: Application in model systems and in the clinic. Dis. Models Mech. 2018, 11. [Google Scholar] [CrossRef] [PubMed]
- Atwood, B.K.; Lopez, J.; Wager-Miller, J.; Mackie, K.; Straiker, A. Expression of G protein-coupled receptors and related proteins in HEK293, AtT20, BV2, and N18 cell lines as revealed by microarray analysis. BMC Genom. 2011, 12, 14. [Google Scholar] [CrossRef] [PubMed]
- Mary, S.; Damian, M.; Louet, M.; Floquet, N.; Fehrentz, J.A.; Marie, J.; Martinez, J.; Baneres, J.L. Ligands and signaling proteins govern the conformational landscape explored by a G protein-coupled receptor. Proc. Natl. Acad. Sci. USA 2012, 109, 8304–8309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barki-Harrington, L.; Luttrell, L.M.; Rockman, H.A. Dual inhibition of beta-adrenergic and angiotensin II receptors by a single antagonist: A functional role for receptor-receptor interaction in vivo. Circulation 2003, 108, 1611–1618. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Lan, H.; El-Battrawy, I.; Li, X.; Buljubasic, F.; Sattler, K.; Yucel, G.; Lang, S.; Tiburcy, M.; Zimmermann, W.H.; et al. Ion Channel Expression and Characterization in Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes. Stem Cells Int. 2018, 2018, 6067096. [Google Scholar] [CrossRef] [PubMed]
- Liang, P.; Lan, F.; Lee, A.S.; Gong, T.; Sanchez-Freire, V.; Wang, Y.; Diecke, S.; Sallam, K.; Knowles, J.W.; Wang, P.J.; et al. Drug screening using a library of human induced pluripotent stem cell-derived cardiomyocytes reveals disease-specific patterns of cardiotoxicity. Circulation 2013, 127, 1677–1691. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.S.; Leonardo, E.D.; Dranovsky, A. iPSC-derived neurons as a tool for probing molecular pharmacology of antipsychotic action. bioRxiv 2018. [Google Scholar] [CrossRef]
- Lian, X.; Zhang, J.; Azarin, S.M.; Zhu, K.; Hazeltine, L.B.; Bao, X.; Hsiao, C.; Kamp, T.J.; Palecek, S.P. Directed cardiomyocyte differentiation from human pluripotent stem cells by modulating Wnt/β-catenin signaling under fully defined conditions. Nat. Protoc. 2013, 8, 162–175. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.X.; Di Giorgio, F.P.; Yao, J.; Marchetto, M.C.; Brennand, K.; Wright, R.; Mei, A.; McHenry, L.; Lisuk, D.; Grasmick, J.M.; et al. Modeling hippocampal neurogenesis using human pluripotent stem cells. Stem Cell Rep. 2014, 2, 295–310. [Google Scholar] [CrossRef] [PubMed]
- Haile, Y.; Nakhaei-Nejad, M.; Boakye, P.A.; Baker, G.; Smith, P.A.; Murray, A.G.; Giuliani, F.; Jahroudi, N. Reprogramming of HUVECs into induced pluripotent stem cells (HiPSCs), generation and characterization of HiPSC-derived neurons and astrocytes. PLoS ONE 2015, 10, e0119617. [Google Scholar] [CrossRef] [PubMed]
- Bedut, S.; Seminatore-Nole, C.; Lamamy, V.; Caignard, S.; Boutin, J.A.; Nosjean, O.; Stephan, J.P.; Coge, F. High-throughput drug profiling with voltage- and calcium-sensitive fluorescent probes in human iPSC-derived cardiomyocytes. Am. J. Physiol. Heart Circ. Physiol. 2016, 311, H44–H53. [Google Scholar] [CrossRef] [PubMed]
- McPheeters, M.T.; Wang, Y.T.; Werdich, A.A.; Jenkins, M.W.; Laurita, K.R. An infrared optical pacing system for screening cardiac electrophysiology in human cardiomyocytes. PLoS ONE 2017, 12, e0183761. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.Z.; Schnitzer, M.J. Genetically encoded indicators of neuronal activity. Nat. Neurosci. 2016, 19, 1142–1153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Y.; Liu, H.; Klein, M.; Ostrominski, J.; Hong, S.G.; Yada, R.C.; Chen, G.; Navarengom, K.; Schwartzbeck, R.; San, H.; et al. Efficient differentiation of cardiomyocytes and generation of calcium-sensor reporter lines from nonhuman primate iPSCs. Sci. Rep. 2018, 8, 5907. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.; Ramirez, C.N.; Kim, H.; Zeltner, N.; Liu, B.; Radu, C.; Bhinder, B.; Kim, Y.J.; Choi, I.Y.; Mukherjee-Clavin, B.; et al. Large-scale screening using familial dysautonomia induced pluripotent stem cells identifies compounds that rescue IKBKAP expression. Nat. Biotechnol. 2012, 30, 1244–1248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakai, T.; Naito, A.T.; Kuramoto, Y.; Ito, M.; Okada, K.; Higo, T.; Nakagawa, A.; Shibamoto, M.; Yamaguchi, T.; Sumida, T.; et al. Phenotypic Screening Using Patient-Derived Induced Pluripotent Stem Cells Identified Pyr3 as a Candidate Compound for the Treatment of Infantile Hypertrophic Cardiomyopathy. Int. Heart J. 2018, 59, 1096–1105. [Google Scholar] [CrossRef] [PubMed]
- Stacey, P.; Wassermann, A.M.; Kammonen, L.; Impey, E.; Wilbrey, A.; Cawkill, D. Plate-Based Phenotypic Screening for Pain Using Human iPSC-Derived Sensory Neurons. SLAS Discov. Adv. Life Sci. R D 2018, 23, 585–596. [Google Scholar] [CrossRef] [PubMed]
- Brownjohn, P.W.; Smith, J.; Portelius, E.; Serneels, L.; Kvartsberg, H.; De Strooper, B.; Blennow, K.; Zetterberg, H.; Livesey, F.J. Phenotypic Screening Identifies Modulators of Amyloid Precursor Protein Processing in Human Stem Cell Models of Alzheimer’s Disease. Stem Cell Rep. 2017, 8, 870–882. [Google Scholar] [CrossRef] [PubMed]
- Luca, A.C.; Mersch, S.; Deenen, R.; Schmidt, S.; Messner, I.; Schafer, K.L.; Baldus, S.E.; Huckenbeck, W.; Piekorz, R.P.; Knoefel, W.T.; et al. Impact of the 3D microenvironment on phenotype, gene expression, and EGFR inhibition of colorectal cancer cell lines. PLoS ONE 2013, 8, e59689. [Google Scholar] [CrossRef] [PubMed]
- Broutier, L.; Mastrogiovanni, G.; Verstegen, M.M.; Francies, H.E.; Gavarro, L.M.; Bradshaw, C.R.; Allen, G.E.; Arnes-Benito, R.; Sidorova, O.; Gaspersz, M.P.; et al. Human primary liver cancer-derived organoid cultures for disease modeling and drug screening. Nat. Med. 2017, 23, 1424–1435. [Google Scholar] [CrossRef] [PubMed]
- Sampaziotis, F.; de Brito, M.C.; Madrigal, P.; Bertero, A.; Saeb-Parsy, K.; Soares, F.A.C.; Schrumpf, E.; Melum, E.; Karlsen, T.H.; Bradley, J.A.; et al. Cholangiocytes derived from human induced pluripotent stem cells for disease modeling and drug validation. Nat. Biotechnol. 2015, 33, 845–852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quadrato, G.; Nguyen, T.; Macosko, E.Z.; Sherwood, J.L.; Min Yang, S.; Berger, D.R.; Maria, N.; Scholvin, J.; Goldman, M.; Kinney, J.P.; et al. Cell diversity and network dynamics in photosensitive human brain organoids. Nature 2017, 545, 48–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raja, W.K.; Mungenast, A.E.; Lin, Y.T.; Ko, T.; Abdurrob, F.; Seo, J.; Tsai, L.H. Self-Organizing 3D Human Neural Tissue Derived from Induced Pluripotent Stem Cells Recapitulate Alzheimer’s Disease Phenotypes. PLoS ONE 2016, 11, e0161969. [Google Scholar] [CrossRef] [PubMed]
- Matsa, E.; Burridge, P.W.; Yu, K.H.; Ahrens, J.H.; Termglinchan, V.; Wu, H.; Liu, C.; Shukla, P.; Sayed, N.; Churko, J.M.; et al. Transcriptome Profiling of Patient-Specific Human iPSC-Cardiomyocytes Predicts Individual Drug Safety and Efficacy Responses. In Vitro Cell Stem Cell 2016, 19, 311–325. [Google Scholar] [CrossRef] [PubMed]
- Rouhani, F.; Kumasaka, N.; de Brito, M.C.; Bradley, A.; Vallier, L.; Gaffney, D. Genetic background drives transcriptional variation in human induced pluripotent stem cells. PLoS Genet. 2014, 10, e1004432. [Google Scholar] [CrossRef] [PubMed]
- Papp, B.; Plath, K. Epigenetics of reprogramming to induced pluripotency. Cell 2013, 152, 1324–1343. [Google Scholar] [CrossRef] [PubMed]
- Godini, R.; Lafta, H.Y.; Fallahi, H. Epigenetic modifications in the embryonic and induced pluripotent stem cells. Gene Exp. Patterns GEP 2018, 29, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Fermini, B.; Coyne, K.P.; Coyne, S.T. Challenges in designing and executing clinical trials in a dish studies. J. Pharmacol. Toxicol. Methods 2018, 94, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Shinnawi, R.; Huber, I.; Maizels, L.; Shaheen, N.; Gepstein, A.; Arbel, G.; Tijsen, A.J.; Gepstein, L. Monitoring Human-Induced Pluripotent Stem Cell-Derived Cardiomyocytes with Genetically Encoded Calcium and Voltage Fluorescent Reporters. Stem Cell Rep. 2015, 5, 582–596. [Google Scholar] [CrossRef] [PubMed]
- Leyton-Mange, J.S.; Mills, R.W.; Macri, V.S.; Jang, M.Y.; Butte, F.N.; Ellinor, P.T.; Milan, D.J. Rapid cellular phenotyping of human pluripotent stem cell-derived cardiomyocytes using a genetically encoded fluorescent voltage sensor. Stem Cell Rep. 2014, 2, 163–170. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| BRET | FRET | |
|---|---|---|
| Advantages |
|
|
| Disadvantages |
|
|
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bourque, K.; Jones-Tabah, J.; Mnasri, N.; Martin, R.D.; Hébert, T.E. Combining Optical Approaches with Human Inducible Pluripotent Stem Cells in G Protein-Coupled Receptor Drug Screening and Development. Biomolecules 2018, 8, 180. https://doi.org/10.3390/biom8040180
Bourque K, Jones-Tabah J, Mnasri N, Martin RD, Hébert TE. Combining Optical Approaches with Human Inducible Pluripotent Stem Cells in G Protein-Coupled Receptor Drug Screening and Development. Biomolecules. 2018; 8(4):180. https://doi.org/10.3390/biom8040180
Chicago/Turabian StyleBourque, Kyla, Jace Jones-Tabah, Nourhen Mnasri, Ryan D. Martin, and Terence E. Hébert. 2018. "Combining Optical Approaches with Human Inducible Pluripotent Stem Cells in G Protein-Coupled Receptor Drug Screening and Development" Biomolecules 8, no. 4: 180. https://doi.org/10.3390/biom8040180