Understanding the Positional Binding and Substrate Interaction of a Highly Thermostable GH10 Xylanase from Thermotoga maritima by Molecular Docking

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Amino Acid Sequence Comparison of TmxB and Other GH10 Xylanases
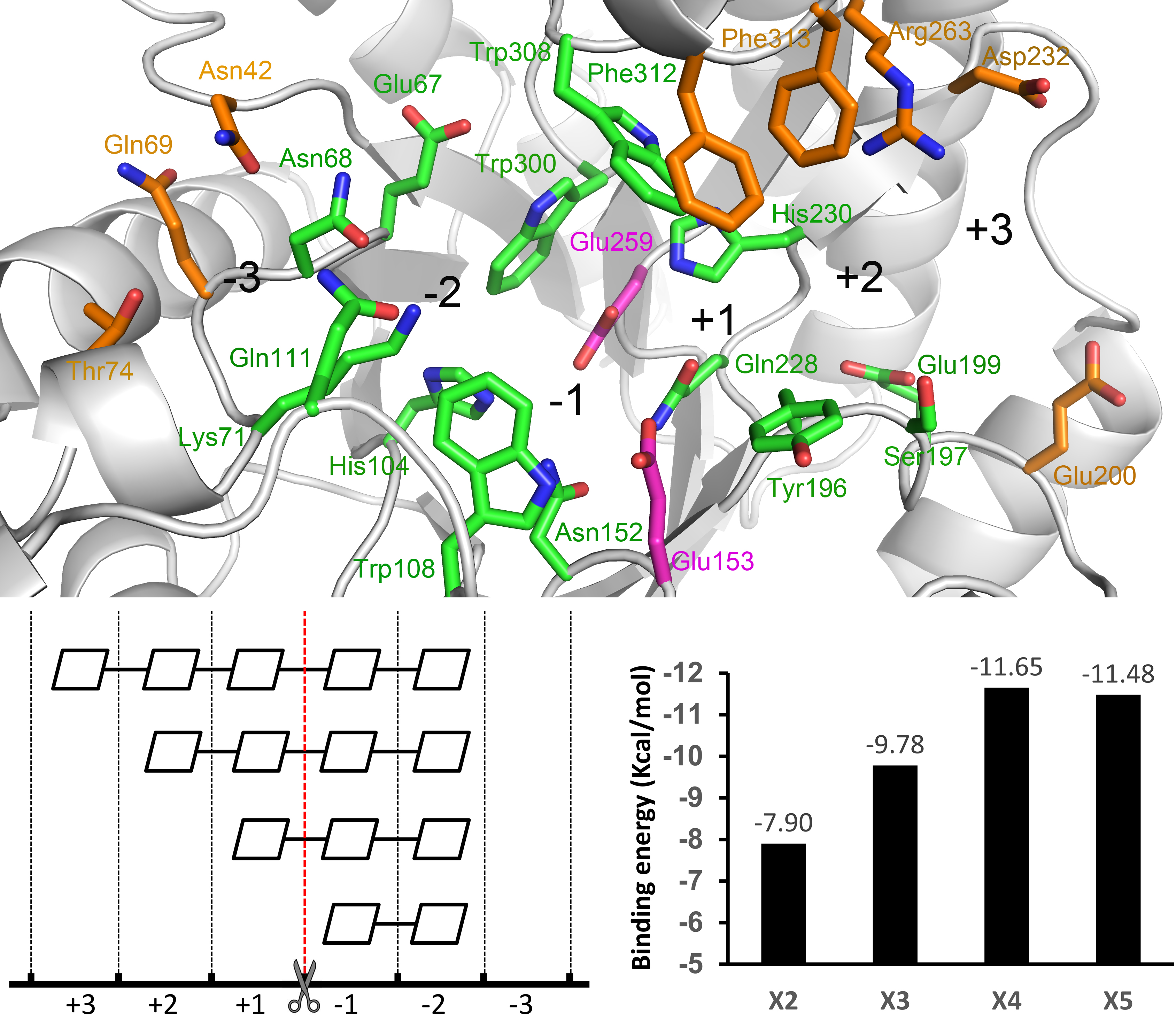
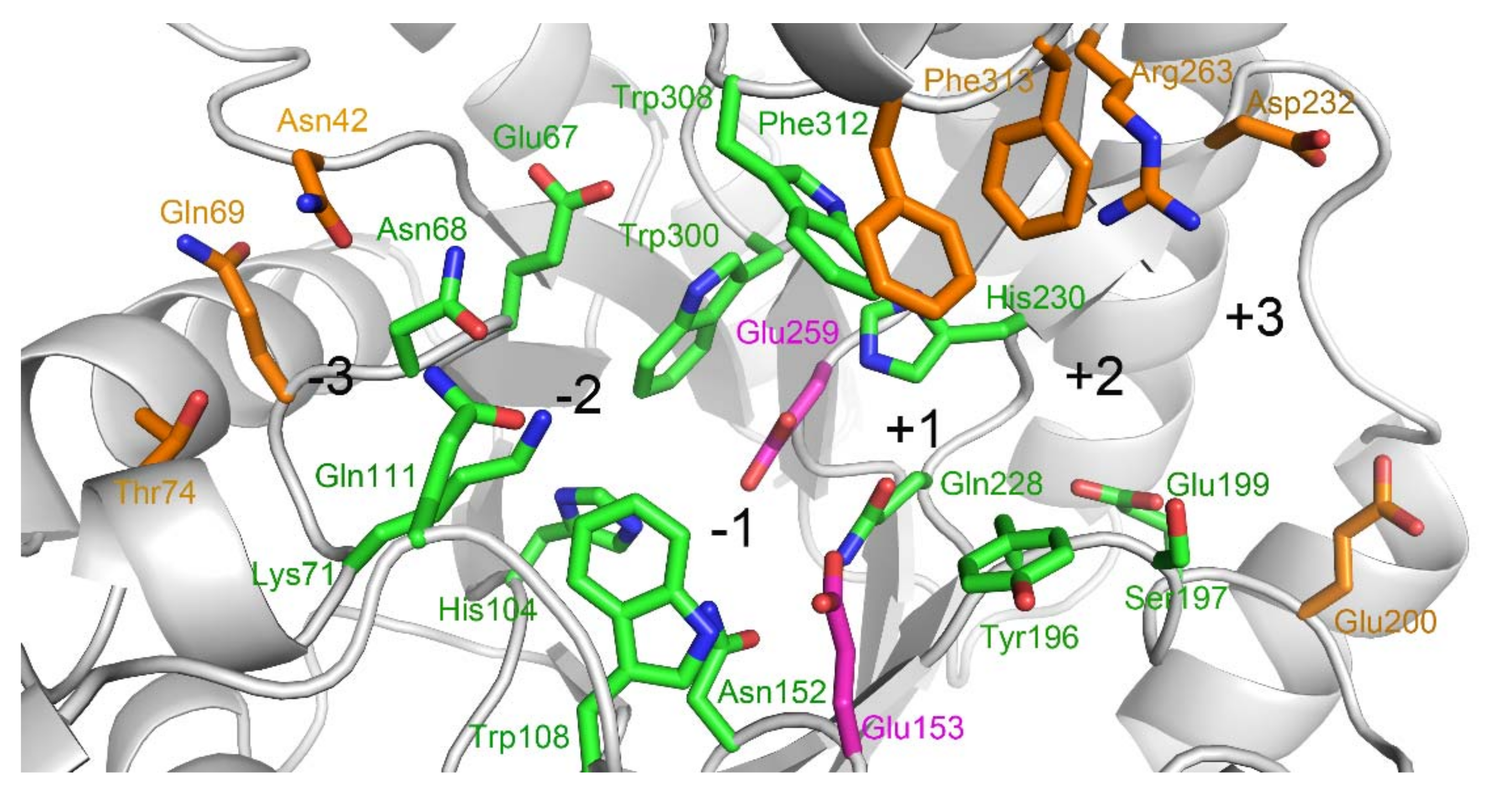
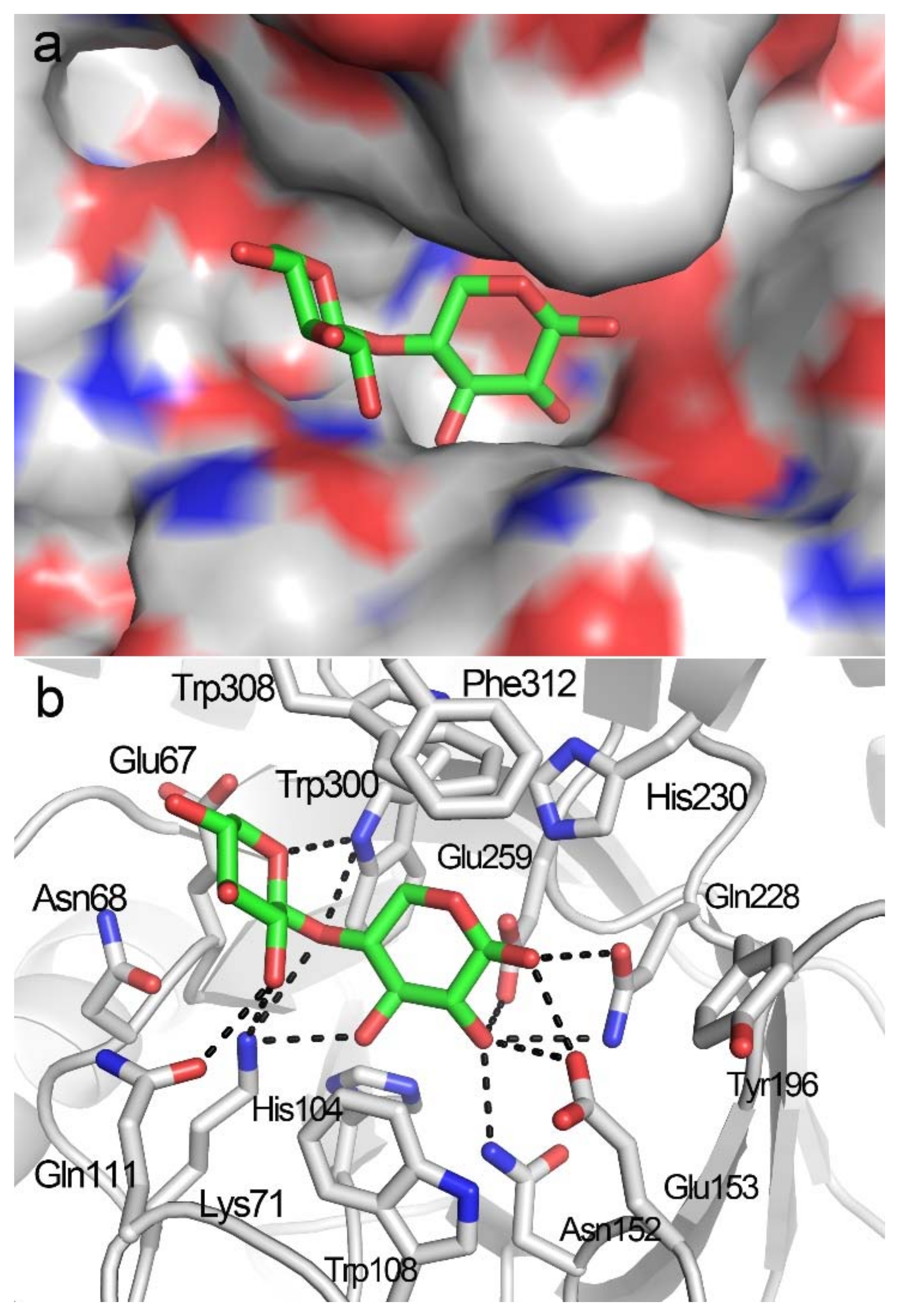
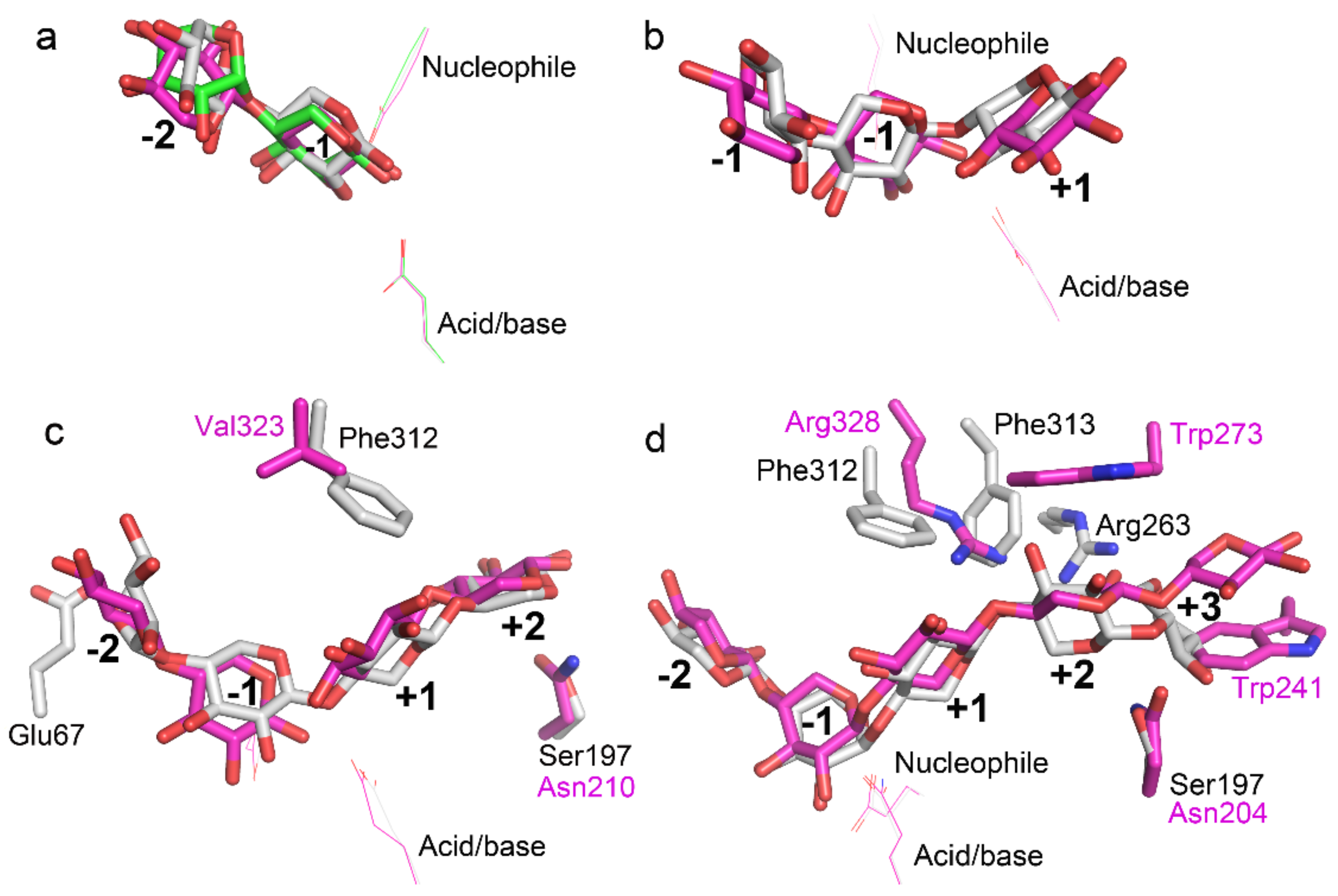
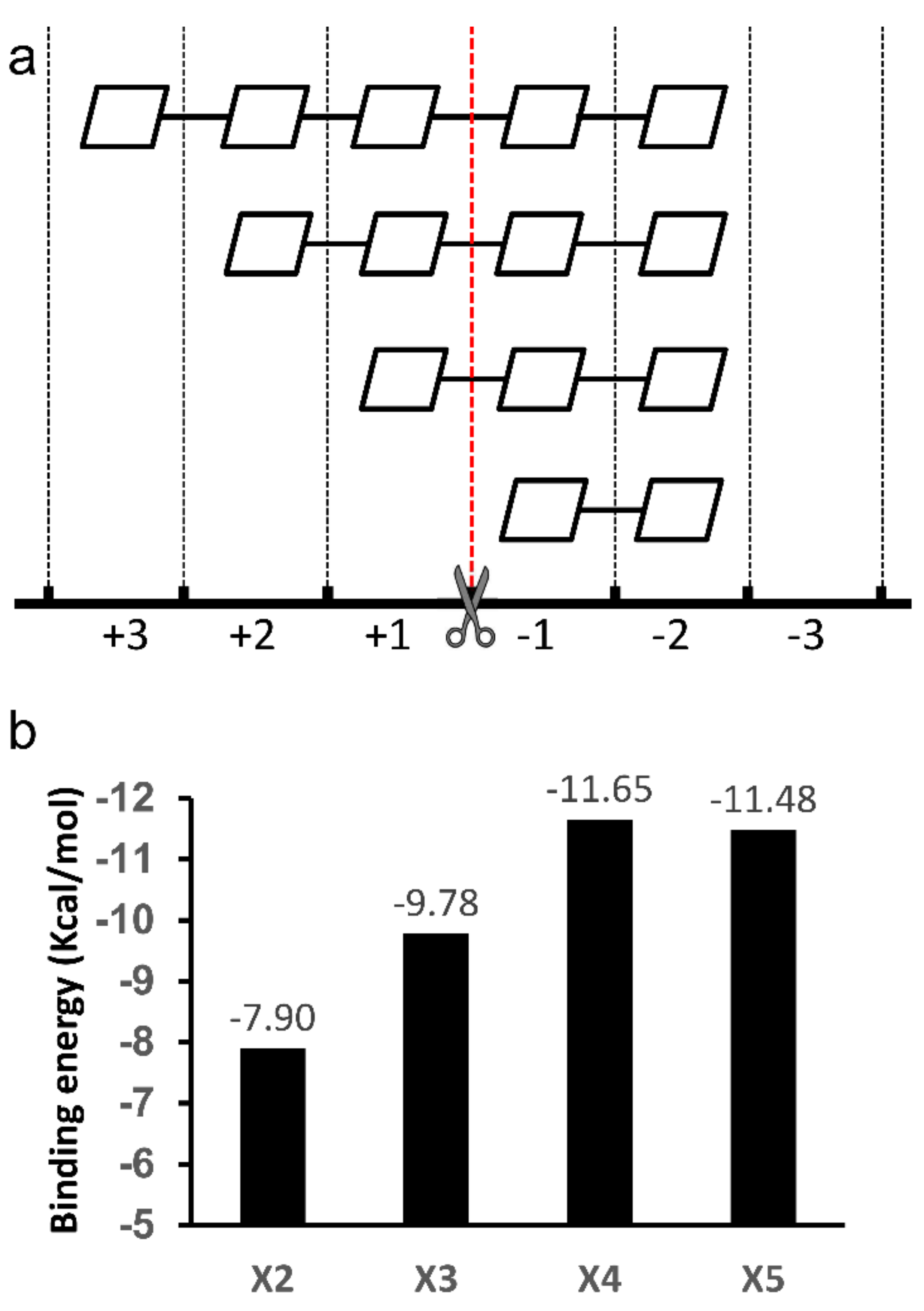
2.2. Docking Structure of X2-Bound TmxB
2.3. Docking Structure of X3-Bound TmxB
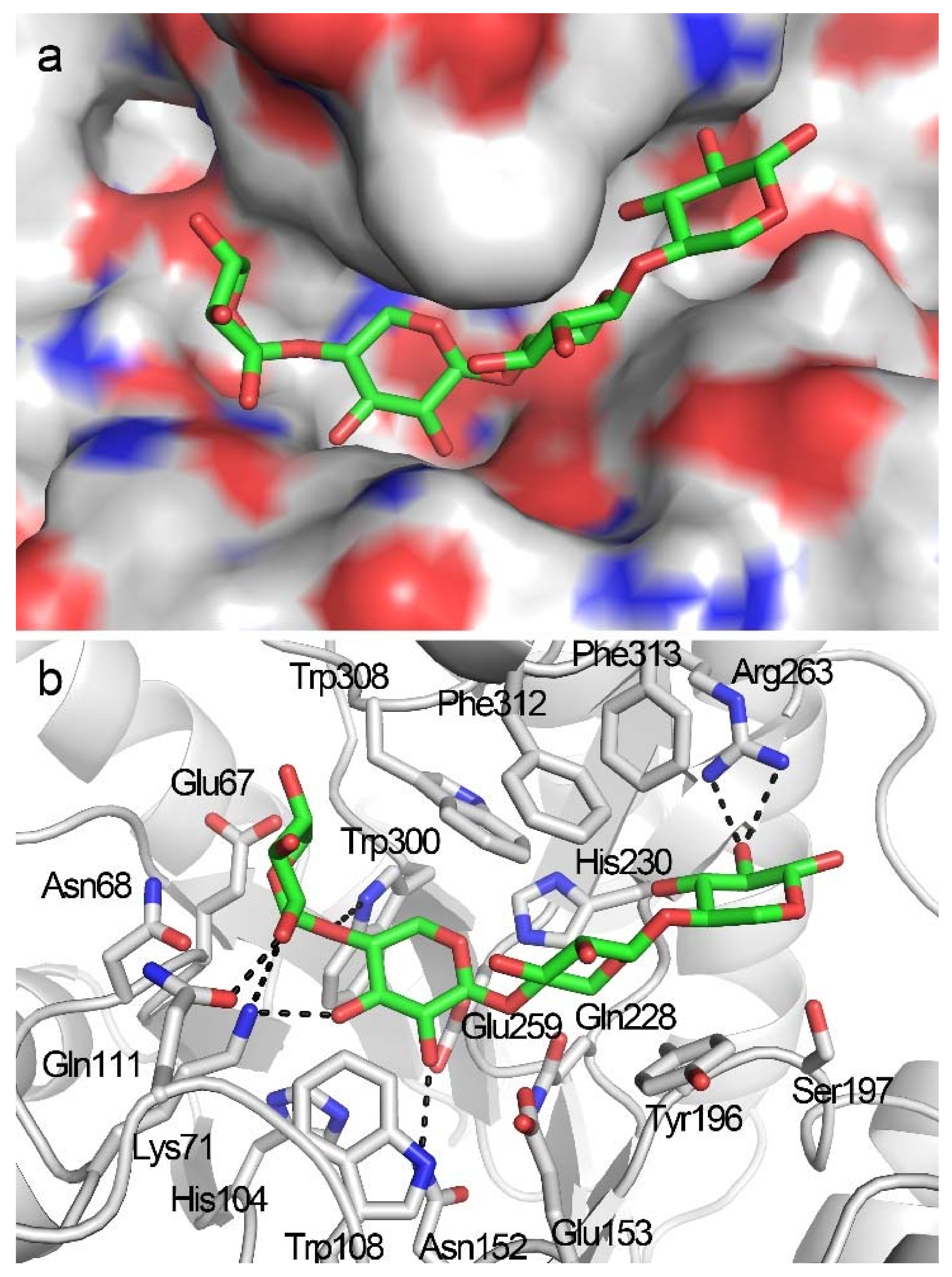
2.4. Docking Structure of X4-Bound TmxB
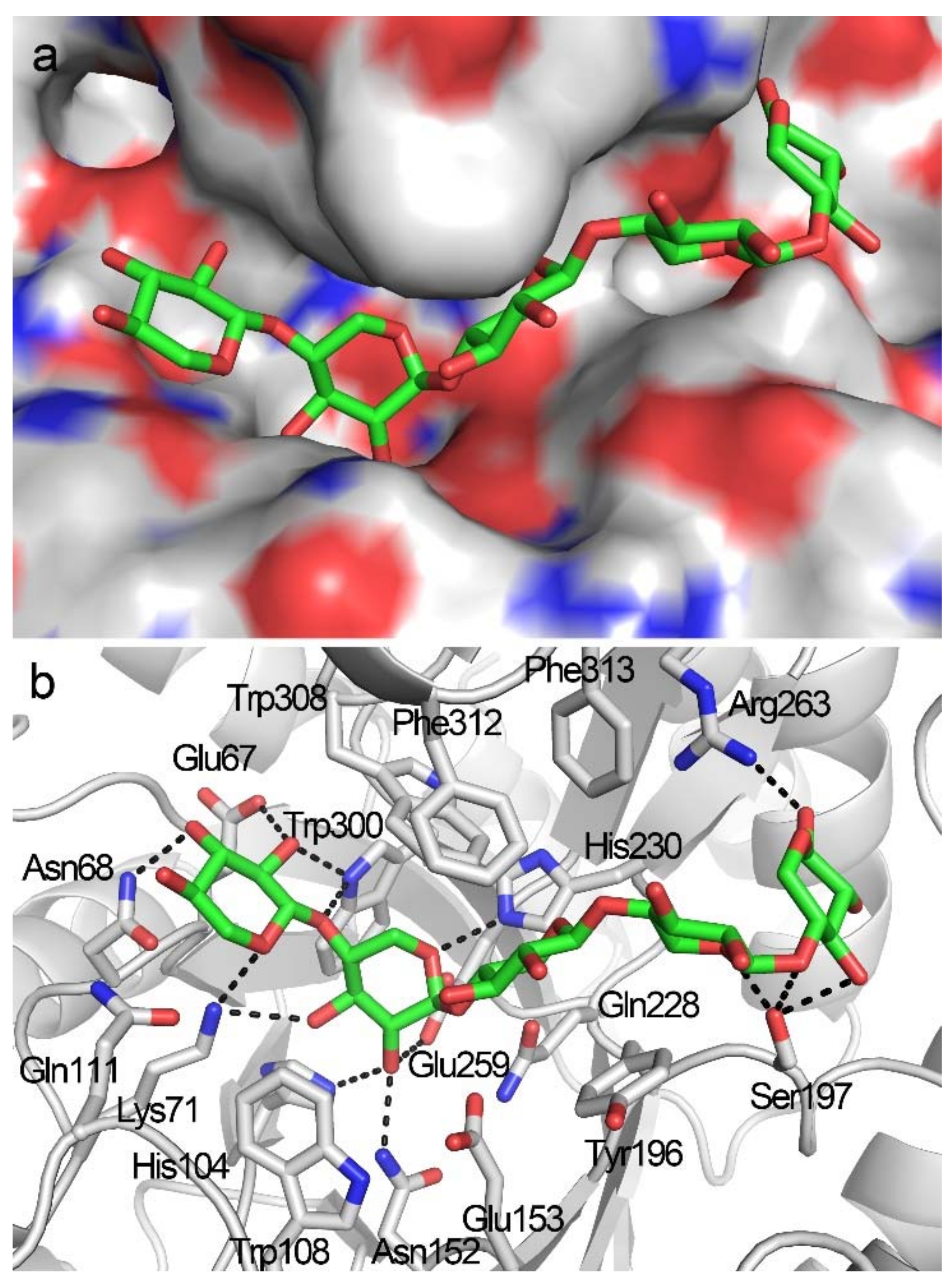
2.5. Docking Structure of TmxB-X5 Complex
3. Discussion
4. Materials and Methods
4.1. Amino Acid Sequence and Structure Comparison of TmxB and Other GH10 Xylanases
4.2. Receptor Molecule Preparation for Docking
4.3. Preparation of the Three-Dimensional Structure of XOS
4.4. Docking Siulations and Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Chakdar, H.; Kumar, M.; Pandiyan, K.; Singh, A.; Nanjappan, K.; Kashyap, P.L.; Srivastava, A.K. Bacterial xylanases: Biology to biotechnology. 3 Biotech 2016, 6, 150. [Google Scholar] [CrossRef] [PubMed]
- Walia, A.; Guleria, S.; Mehta, P.; Chauhan, A.; Parkash, J. Microbial xylanases and their industrial application in pulp and paper biobleaching: A review. 3 Biotech 2017, 7, 11. [Google Scholar] [CrossRef] [PubMed]
- Dhiman, S.S.; Garg, G.; Sharma, J.; Kalia, V.C.; Kang, Y.C.; Lee, J.K. Reduction in acute ecotoxicity of paper mill effluent by sequential application of xylanase and laccase. PLoS ONE 2014, 9, e102581. [Google Scholar] [CrossRef] [PubMed]
- Harris, A.D.; Ramalingam, C. Xylanases and its application in food industry: A review. J. Exp. Sci. 2010, 1, 1–11. [Google Scholar]
- Butt, M.S.; Tahir-Nadeem, M.; Ahmad, Z.; Sultan, M.T. Xylanases and their applications in baking industry. Food Technol. Biotechnol. 2008, 46, 22–31. [Google Scholar]
- Bhalla, A.; Bischoff, K.M.; Sani, R.K. Highly thermostable xylanase production from a thermophilic Geobacillus sp. Strain WsUcF1 utilizing lignocellulosic biomass. Front. Bioeng. Biotechnol. 2015, 16, 84. [Google Scholar] [CrossRef] [PubMed]
- Rhee, M.S.; Wei, L.; Sawhney, N.; Rice, J.D.; John, F.J.S.; Hurlbert, J.C.; Preston, J.F. Engineering the xylan utilization system in Bacillus subtilis for production of acidic xylooligosaccharides. Appl. Environ. Microbiol. 2014, 80, 917–927. [Google Scholar] [CrossRef] [PubMed]
- CAzy. Carbohydrate-Active EnZYmes Homepage. Available online: www.cazy.org (accessed on 15 May 2018).
- Lombard, V.; Golaconda Ramulu, H.; Drula, E.; Coutinho, P.M.; Henrissat, B. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 2014, 42, 490–495. [Google Scholar] [CrossRef] [PubMed]
- Moreira, L.R.; Filho, E.X. Insights into the mechanism of enzymatic hydrolysis of xylan. Appl. Microbiol. Biotechnol. 2016, 100, 5205–5214. [Google Scholar] [CrossRef] [PubMed]
- Dies, G.; Henrissat, B. Structures and mechanisms of glycosyl hydrolases. Structures 1995, 3, 853–859. [Google Scholar]
- Pollet, A.; Delcour, J.A.; Courtin, C.M. Structural determinants of the substrate specificities of xylanases from different glycoside hydrolase families. Crit. Rev. Biotechnol. 2010, 30, 176–191. [Google Scholar] [CrossRef] [PubMed]
- Lo Leggio, L.; Kalogiannis, S.; Bhat, M.K.; Pickersgill, R.W. High resolution structure and sequence of T. aurantiacus xylanase I: Implications for the evolution of thermostability in family 10 xylanases and enzymes with (βα)-barrel architecture. Proteins 1999, 36, 295–306. [Google Scholar] [CrossRef]
- Davies, G.J.; Wilson, K.S.; Henrissat, B. Nomenclature for sugar-binding subsites in glycosyl hydrolases. Biochem. J. 1997, 321, 557–559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Notenboom, V.; Birsan, C.; Nitz, M.; Rose, D.R.; Warren, R.A.; Withers, S.G. Insights into transition state stabilization of the β-1,4-glycosidase Cex by covalent intermediate accumulation in active site mutants. Nat. Struct. Mol. Biol. 1998, 5, 812–818. [Google Scholar] [CrossRef] [PubMed]
- Ducros, V.; Charnock, S.J.; Derewenda, U.; Derewenda, Z.S.; Dauter, Z.; Dupont, C.; Shareck, F.; Morosoli, R.; Kluepfel, D.; Davies, G.J. Substrate specificity in glycoside hydrolase family 10. Structural and kinetic analysis of the Streptomyces lividans xylanase 10A. J. Biol. Chem. 2000, 275, 23020–23026. [Google Scholar] [CrossRef] [PubMed]
- Zolotnitsky, G.; Cogan, U.; Adir, N.; Solomon, V.; Shoham, G.; Shoham, Y. Mapping glycoside hydrolase substrate subsites by isothermal titration calorimetry. Proc. Natl. Acad. Sci. USA 2004, 101, 11275–11280. [Google Scholar] [CrossRef] [PubMed]
- Charnock, S.J.; Spurway, T.D.; Xie, H.; Beylot, M.H.; Virden, R.; Warren, R.A.; Hazlewood, G.P.; Gilbert, H.J. The topology of the substrate binding clefts of glycosyl hydrolase family 10 xylanases are not conserved. J. Biol. Chem. 1998, 273, 32187–32199. [Google Scholar] [CrossRef] [PubMed]
- Winterhalter, C.; Liebl, W. Two extremely thermostable xylanases of the hyperthermophilic bacterium Thermotoga maritima MSB8. Appl. Environ. Microbiol. 1995, 61, 1810–1815. [Google Scholar] [PubMed]
- Zhengqiang, J.; Kobayashi, A.; Ahsan, M.M.; Lite, L.; Kitaoka, M.; Hayashi, K. Characterization of a thermostable family 10 endo-xylanase (XynB) from Thermotoga. maritima that cleaves p-nitrophenyl-β-d-xyloside. J. Biosci. Bioeng. 2001, 92, 423–428. [Google Scholar] [CrossRef]
- Kumasaka, T.; Kaneko, T.; Morokuma, C.; Yatsunami, R.; Sato, T.; Nakamura, S.; Tanaka, N. Structural basis of the substrate subsite and the highly thermal stability of xylanase 10B from Thermotoga. maritima MSB8. Proteins 2005, 61, 999–1009. [Google Scholar]
- Benson, D.A.; Cavanaugh, M.; Clark, K.; Karsch-Mizrachi, I.; Lipman, D.J.; Ostell, J.; Sayers, E.W. GenBank. Nucleic Acids Res. 2013, 41, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, Y.; Tu, T.; Penttinen, L.; Xue, X.; Wang, X.; Yi, Z.; Gong, L.; Rouvinen, J.; Luo, H.; Hakulinen, N.; Yao, B.; Su, X. Insights into the roles of non-catalytic residues in the active site of a GH10 xylanase with activity on cellulose. J. Biol. Chem. 2017, 292, 19315–19327. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- RCSB. PDB Homepage. Available online: www.rcsb.org/pdb/home/home.do (accessed on 20 May 2018).
- Sievers, F.; Wilm, A.; Dineen, D.G.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Pundir, S.; Martin, M.J.; O’Donovan, C. UniProt protein knowledgebase. Methods Mol. Biol. 2017, 1558, 41–55. [Google Scholar] [PubMed]
- Huey, R.; Morris, G.M.; Olson, A.J.; Goodsell, D.S. A semiempirical free energy force field with charge-based desolvation. J. Comput. Chem. 2007, 28, 1145–1152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laskowski, R.A.; Swindells, M.B. LigPlot+: Multiple ligand-protein interaction diagrams for drug discovery. J. Chem. Inf. Model. 2011, 51, 2778–2786. [Google Scholar] [CrossRef] [PubMed]








© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, J.; Han, Z. Understanding the Positional Binding and Substrate Interaction of a Highly Thermostable GH10 Xylanase from Thermotoga maritima by Molecular Docking. Biomolecules 2018, 8, 64. https://doi.org/10.3390/biom8030064
Yang J, Han Z. Understanding the Positional Binding and Substrate Interaction of a Highly Thermostable GH10 Xylanase from Thermotoga maritima by Molecular Docking. Biomolecules. 2018; 8(3):64. https://doi.org/10.3390/biom8030064
Chicago/Turabian StyleYang, Jiangke, and Zhenggang Han. 2018. "Understanding the Positional Binding and Substrate Interaction of a Highly Thermostable GH10 Xylanase from Thermotoga maritima by Molecular Docking" Biomolecules 8, no. 3: 64. https://doi.org/10.3390/biom8030064