
Biological Activities of Reactive Oxygen and Nitrogen Species: Oxidative Stress versus Signal Transduction

{kind=link}
Abstract
:1. Introduction
2. Chemical Basics
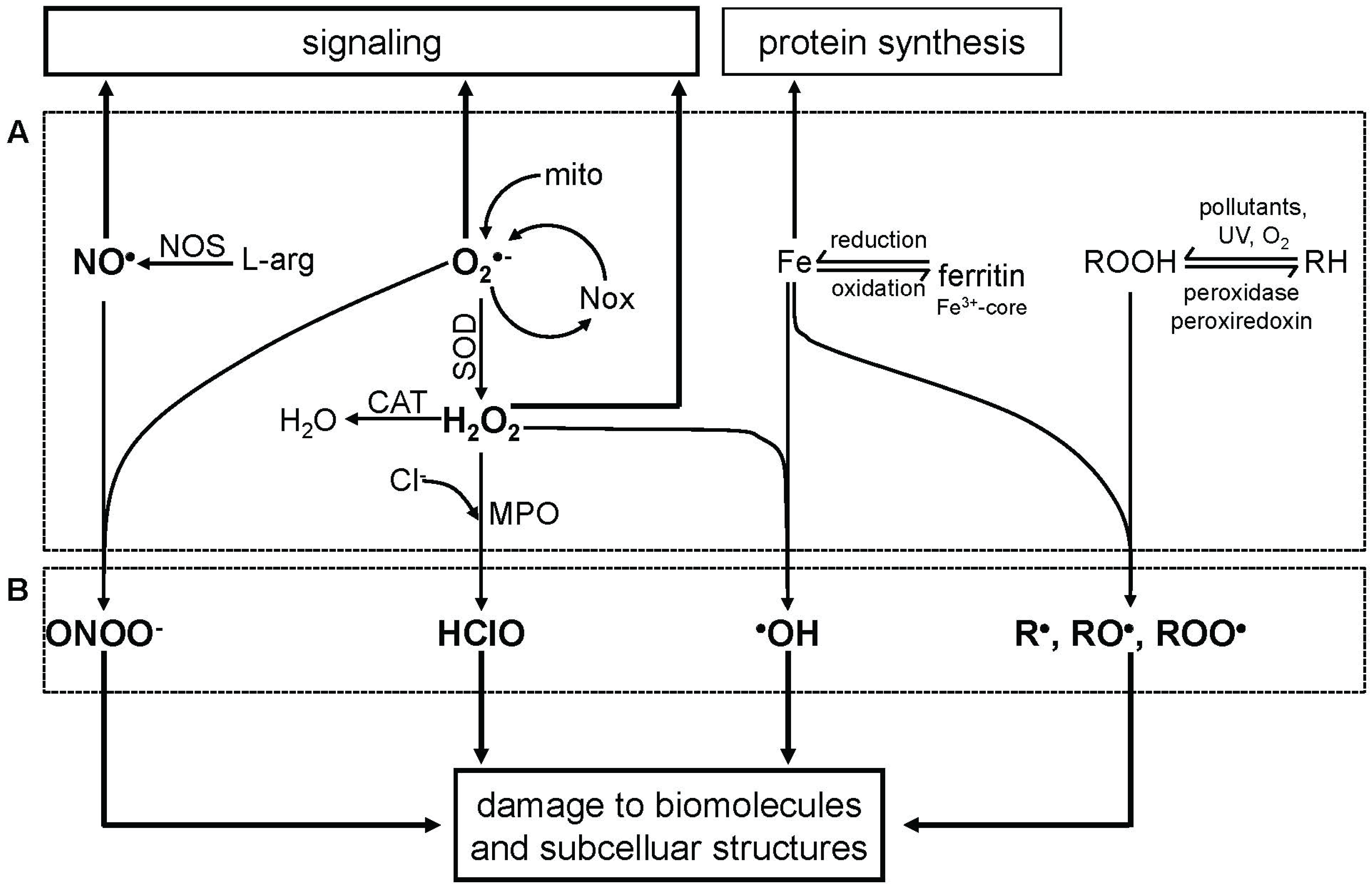
3. Oxidative Stress
4. Signaling
5. Superoxide Radical
- Alteration of mitochondrial function, particularly of complexes I and III, by pharmacological or genetic modulation, which has an effect on signaling pathways.
- The correlation of a certain O2•− level with an effective signaling cascade.
- Application of mitochondria-targeted antioxidants (mtAOX) or radical scavengers has an effect on signaling pathways.
- Genetic manipulation of mitochondrial SOD and cytoplasmic SOD decreases the efficiency of specific signaling cascades.
6. Hydrogen Peroxide
- Exogenous H2O2 has a direct effect on signaling cascades.
- The H2O2 level correlates with the effectiveness of intracellular signal transduction.
- Genetic manipulation of catalase has an effect on signaling.
- Upregulation of MnSOD and/or Cu/ZnSOD activates signaling.
7. Secondary RONS
8. Conclusions

Acknowledgments
Conflicts of Interest
References
- Wertz, J.E.; Bolton, J.R. Electron Spin Resonance: Elementary Theory and Practical Applications; John Wiley & Sons: New York, NY, USA, 1972. [Google Scholar]
- Fenton, H.J.H. Oxidation of tartaric acid in presence of iron. J. Chem. Soc. Trans. 1894, 65, 899–910. [Google Scholar] [CrossRef]
- Koppenol, W.H. The centennial of the Fenton reaction. Free Radic. Biol. Med. 1993, 15, 645–651. [Google Scholar] [CrossRef] [PubMed]
- Haber, F.; Weiss, J. The catalytic decomposition of hydrogen peroxide. Proc. R. Soc. 1934, 147, 332–351. [Google Scholar] [CrossRef]
- Koppenol, W.H. The Haber-Weiss cycle—70 Years later. Redox Rep. 1994, 6, 229–234. [Google Scholar] [CrossRef]
- Kehrer, J.P. The Haber-Weiss reaction and mechanisms of toxicity. Toxicology 2000, 149, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Ullrich, V.; Kissner, R. Redox signaling: Bioinorganic chemistry at its best. J. Inorg. Biochem. 2006, 100, 2079–2086. [Google Scholar] [CrossRef] [PubMed]
- Semenov, N.N. Some Problems in Chemical Kinetics and Reactivity; Princeton University Press: Princeton, NJ, USA, 1959. [Google Scholar]
- Vladimirov, Y.A.; Olenev, V.; Suslova, T.; Cheremisina, Z. Lipid peroxidation in mitochondrial membrane. Adv. Lipid Res. 1980, 17, 173–249. [Google Scholar] [PubMed]
- Gilbert, D.L. Oxygen and Living Processes: An Interdisciplinary Approach; Springer: New York, NY, USA, 1981. [Google Scholar]
- McCord, J.M.; Fridovich, I. The reduction of cytochrome c by hypoxanthine and xanthine oxidase. Biochem. J. 1968, 243, 5753–5760. [Google Scholar]
- Mccord, J.M.; Fridovich, I. Superoxide dismutase: An enzymic function for erythrocuprein (hemocuprein). J. Biol. Chem. 1969, 244, 6049–6055. [Google Scholar] [PubMed]
- Fridovich, I. Superoxide anion radical, superoxide dismutases, and related matters. J. Biol. Chem. 1997, 272, 18515–18517. [Google Scholar] [CrossRef] [PubMed]
- Boveris, B.A.; Oshino, N.; Chance, B. The cellular production of hydrogen peroxide. Biochem. J. 1972, 128, 617–630. [Google Scholar] [PubMed]
- Chance, B.; Sies, H.; Boveris, A. Hydroperoxide metabolism in mammalian organs. Physiol. Rev. 1979, 59, 527–605. [Google Scholar] [PubMed]
- Halliwell, B. Free radicals, reactive oxygen species and human disease: A critical evaluation with special reference to atherosclerosis. Br. J. Exp. Pathol. 1989, 70, 737–757. [Google Scholar] [PubMed]
- Azizova, O.A.; Panasenko, O.; Volnov, T.; Vladimirov, Y.A. Free radical lipid oxidation affects cholesterol transfer between lipoproteins and erythrocytes. Free Radic. Biol. Med. 1989, 7, 251–257. [Google Scholar] [CrossRef] [PubMed]
- Lyras, L.; Perry, R.H.; Perry, E.K.; Ince, P.G.; Jenner, A.; Jenner, P.; Halliwell, B. Oxidative damage to proteins, lipids, and DNA in cortical brain regions from patients with dementia with Lewy bodies. J. Neurochem. 1998, 71, 302–312. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B.; Aruoma, O.I. DNA damage by oxygen-derived species. FEBS Lett. 1991, 281, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Cadet, J.; Douki, T.; Ravanat, J.-L. Oxidatively generated base damage to cellular DNA. Free Radic. Biol. Med. 2010, 49, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Sies, H. Oxidative stress: Introductory remarks. In Oxidative Stress; Academic Press: London, UK, 1985; pp. 1–8. [Google Scholar]
- Sies, H. Oxidative stress: Oxidants and antioxidants. Exp. Physiol. 1997, 82, 291–295. [Google Scholar] [CrossRef] [PubMed]
- Rosen, H.; Klebanoff, S.J.; Wang, Y.; Brot, N.; Heinecke, J.W.; Fu, X. Methionine oxidation contributes to bacterial killing by the myeloperoxidase system of neutrophils. Proc. Natl. Acad. Sci. USA 2009, 106, 18686–18691. [Google Scholar] [CrossRef] [PubMed]
- Schwab, L.; Goroncy, L.; Palaniyandi, S.; Gautam, S.; Triantafyllopoulou, A.; Mocsai, A.; Reichardt, W.; Karlsson, F.J.; Radhakrishnan, S.V.; Hanke, K.; et al. Neutrophil granulocytes recruited upon translocation of intestinal bacteria enhance graft-versus-host disease via tissue damage. Nat. Med. 2014, 20, 648–654. [Google Scholar] [CrossRef] [PubMed]
- Arthur, M.; Kowalski-Saunders, P.; Gurney, S.; Tolcher, R.; Bull, F.; Wright, R. Reduction of ferricytochrome C may underestimate superoxide production by monocytes. J. Immunol. Methods 1987, 98, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Cadet, J.; Wagner, J.R. Oxidatively generated base damage to cellular DNA by hydroxyl radical and one-electron oxidants: Similarities and differences. Arch. Biochem. Biophys. 2014, 557, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Salgo, M.G.; Squadrito, G.L.; Pryor, W.A. Peroxynitrite causes apoptosis in rat thymocytes. Biochem. Biophys. Res. Commun. 1995, 215, 1111–1118. [Google Scholar] [CrossRef] [PubMed]
- Panasenko, O.M.; Evgina, S.A.; Driomina, E.S.; Sharov, V.S.; Sergienko, V.I.; Vladimirov, Y.A. Hypochlorite induces lipoproteins and lipid peroxidation in blood phospholipid liposomes. Free Radic. Biol. Med. 1995, 19, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Chevion, M. A site-specific mechanism for free radical induced biological damage: The essential role of redox-active transition metals. Free Radic. Biol. Med. 1988, 5, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Roy, B.; Hecht, S.M. Hairpin DNA sequences bound strongly by bleomycin exhibit enhanced double-strand cleavage. J. Am. Chem. Soc. 2014, 136, 4382–4393. [Google Scholar] [CrossRef]
- Violi, F.; Milite, M.T.; Medica, I.C.; La, Á. Nitric oxide and its role in lipid peroxidation. Diabetes Metab. Res. Rev. 1999, 15, 283–288. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.; Harrison, D.G. Endothelial dysfunction in cardiovascular disease. The role of oxidant stress. Circ. Res. 2000, 87, 840–844. [Google Scholar] [CrossRef] [PubMed]
- Yung, L.M.; Leung, F.P.; Yao, X.; Chen, Z.-Y.; Huang, Y. Reactive oxygen species in vascular wall. Cardiovasc. Hematol. Disord. Drug Targets 2006, 6, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Schildknecht, S.; Ullrich, V. Peroxynitrite as regulator of vascular prostanoid synthesis. Arch. Biochem. Biophys. 2009, 484, 183–189. [Google Scholar] [CrossRef] [PubMed]
- Samokyszyns, V.M.; Miller, D.M.; Reif, D.W.; Austq, S.D. Inhibition of superoxide and ferritin-dependent lipid peroxidation by ceruloplasmin. J. Biol. Chem. 1989, 264, 21–26. [Google Scholar] [PubMed]
- Marklund, S.L. Extracellular superoxide dismutase and other superoxide dismutase isoenzymes in tissues from nine mammalian species. Biochem. J. 1984, 222, 649–655. [Google Scholar] [PubMed]
- Marklund, S.L. Extracellular superoxide dismutase in human tissues and human cell lines. J. Clin. Invest. 1984, 74, 1398–1403. [Google Scholar] [CrossRef] [PubMed]
- Fattman, C.L.; Schaefer, L.M.; Oury, T.D. Extracellular superoxide dismutase in biology and medicine. Free Radic. Biol. Med. 2003, 35, 236–256. [Google Scholar] [CrossRef] [PubMed]
- Fukai, T.; Ushio-Fukai, M. Superoxide dismutases: Role in redox signaling, vascular function, and diseases. Antioxid. Redox Signal. 2011, 15, 1583–1606. [Google Scholar] [CrossRef] [PubMed]
- Sandstrom, J.; Nilsson, P.; Karlsson, K.; Marklund, S.L. 10-Fold increase in human plasma extracellular superoxide dismutase content caused by a mutation in heparin-binding domain. J. Biol. Chem. 1994, 269, 19163–19166. [Google Scholar] [PubMed]
- Stamler, J.S.; Carolina, N. Redox signaling: Nitrosylation and related target interactions of nitric oxide. Cell 1994, 76, 931–936. [Google Scholar] [CrossRef]
- Foster, M.W.; McMahon, T.J.; Stamler, J.S. S-nitrosylation in health and disease. Trends Mol. Med. 2003, 9, 160–168. [Google Scholar] [CrossRef] [PubMed]
- Guikema, B.; Lu, Q.I.; Heuil, D.J. Chemical considerations and biological selectivity of protein nitrosation: Implications for NO-mediated signal transduction. Antioxid. Redox Signal. 2005, 7, 593–606. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, K.A.; Sawa, T.; Akaike, T. Protein cysteine S-guanylation and electrophilic signal transduction by endogenous nitro-nucleotides. Amino Acids 2011, 41, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Piantadosi, C.A. Regulation of mitochondrial processes by protein S-nitrosylation. Biochim. Biophys. Acta 2012, 1820, 712–721. [Google Scholar] [CrossRef] [PubMed]
- Sha, Y.; Marshall, H.E. S-nitrosylation in the regulation of gene transcription. Biochim. Biophys. Acta 2012, 1820, 701–711. [Google Scholar] [CrossRef] [PubMed]
- Sawa, T.; Zaki, M.H.; Okamoto, T.; Akuta, T.; Tokutomi, Y.; Kim-Mitsuyama, S.; Ihara, H.; Kobayashi, A.; Yamamoto, M.; Fujii, S.; et al. Protein S-guanylation by the biological signal 8-nitroguanosine 3',5'-cyclic monophosphate. Nat. Chem. Biol. 2007, 3, 727–735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daiber, A. Redox signaling (cross-talk) from and to mitochondria involves mitochondrial pores and reactive oxygen species. Biochim. Biophys. Acta 2010, 1797, 897–906. [Google Scholar] [CrossRef] [PubMed]
- Dikalov, S.I.; Nazarewicz, R.R. Angiotensin II-induced production of mitochondrial reactive oxygen species: Potential mechanisms and relevance for cardiovascular disease. Antioxid. Redox Signal. 2013, 19, 1085–1094. [Google Scholar] [CrossRef] [PubMed]
- Schulz, E.; Wenzel, P.; Münzel, T.; Daiber, A. Mitochondrial redox signaling: Interaction of mitochondrial reactive oxygen species with other sources of oxidative stress. Antioxid. Redox Signal. 2014, 20, 308–324. [Google Scholar] [CrossRef] [PubMed]
- Bashan, N.; Kovsan, J.; Kachko, I.; Ovadia, H.; Rudich, A. Positive and negative regulation of insulin signaling by reactive oxygen and nitrogen species. Physiol. Rev. 2009, 89, 27–71. [Google Scholar] [CrossRef] [PubMed]
- Cash, T.P.; Pan, Y.; Simon, M.C. Reactive oxygen species and cellular oxygen sensing. Free Radic. Biol. Med. 2007, 43, 1219–1225. [Google Scholar] [CrossRef] [PubMed]
- Klimova, T.; Chandel, N.S. Mitochondrial complex III regulates hypoxic activation of HIF. Cell Death Differ. 2008, 15, 660–666. [Google Scholar] [CrossRef] [PubMed]
- Thannickal, V.J.; Fanburg, B.L. Reactive oxygen species in cell signaling. Am. J. Physiol. Lung Cell. Mol. Physiol. 2000, 279, L1005–L1028. [Google Scholar] [PubMed]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Bulua, A.C.; Simon, A.; Maddipati, R.; Pelletier, M.; Park, H.; Kim, K.-Y.; Sack, M.N.; Kastner, D.L.; Siegel, R.M. Mitochondrial reactive oxygen species promote production of proinflammatory cytokines and are elevated in TNFR1-associated periodic syndrome (TRAPS). Exp. Med. 2011, 208, 519–533. [Google Scholar] [CrossRef]
- Weidinger, A.; Müllebner, A.; Paier-Pourani, J.; Banerjee, A.; Miller, I.; Lauterböck, L.; Duvigneau, J.C.; Skulachev, V.P.; Redl, H.; Kozlov, A.V.; et al. Vicious inducible nitric oxide synthase-mitochondrial reactive oxygen species cycle accelerates inflammatory response and causes liver injury in rats. Antioxid. Redox Signal. 2015, 22, 572–586. [Google Scholar] [CrossRef] [PubMed]
- Kröller-Schön, S.; Steven, S.; Kossmann, S.; Scholz, A.; Daub, S.; Oelze, M.; Xia, N.; Hausding, M.; Mikhed, Y.; Zinssius, E.; et al. Molecular mechanisms of the crosstalk between mitochondria and NADPH oxidase through reactive oxygen species-studies in white blood cells and in animal models. Antioxid. Redox Signal. 2014, 20, 247–266. [Google Scholar] [CrossRef] [PubMed]
- Dikalova, A.E.; Bikineyeva, A.T.; Budzyn, K.; Nazarewicz, R.R.; McCann, L.; Lewis, W.; Harrison, D.G.; Dikalov, S.I. Therapeutic targeting of mitochondrial superoxide in hypertension. Circ. Res. 2010, 107, 106–116. [Google Scholar] [CrossRef] [PubMed]
- Ullevig, S.; Zhao, Q.; Lee, C.F.; Seok Kim, H.; Zamora, D.; Asmis, R. NADPH oxidase 4 mediates monocyte priming and accelerated chemotaxis induced by metabolic stress. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 415–426. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Stouffs, M.; Serrander, L.; Banfi, B.; Bettiol, E.; Charnay, Y.; Steger, K.; Krause, K.-H.; Jaconi, M.E. The NADPH oxidase NOX4 drives cardiac differentiation: Role in regulating cardiac transcription factors and map kinase activation. Mol. Biol. Cell 2006, 17, 3978–3988. [Google Scholar] [CrossRef] [PubMed]
- Crosas-Molist, E.; Bertran, E.; Sancho, P.; López-Luque, J.; Fernando, J.; Sánchez, A.; Fernández, M.; Navarro, E.; Fabregat, I. The NADPH oxidase NOX4 inhibits hepatocyte proliferation and liver cancer progression. Free Radic. Biol. Med. 2014, 69, 338–347. [Google Scholar] [CrossRef] [PubMed]
- Crestani, B.; Besnard, V.; Boczkowski, J. Signalling pathways from NADPH oxidase-4 to idiopathic pulmonary fibrosis. Int. J. Biochem. Cell Biol. 2011, 43, 1086–1089. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Ghosh, P.; Wan, R.; Ouyang, X.; Cheng, H.; Mattson, M.; Cheng, A. Permeability transition pore-mediated mitochondrial superoxide flashes. Neurobiol. Aging 2014, 35, 975–989. [Google Scholar] [CrossRef] [PubMed]
- Lustgarten, M.S.; Bhattacharya, A.; Muller, F.L.; Jang, Y.C.; Shimizu, T.; Shirasawa, T.; Richardson, A.; van Remmen, H. Complex I generated, mitochondrial matrix-directed superoxide is released from the mitochondria through voltage dependent anion channels. Biochem. Biophys. Res. Commun. 2012, 422, 515–521. [Google Scholar] [CrossRef] [PubMed]
- Piskernik, C.; Haindl, S.; Behling, T.; Gerald, Z.; Kehrer, I.; Redl, H.; Kozlov, A.V. Antimycin A and lipopolysaccharide cause the leakage of superoxide radicals from rat liver mitochondria. Biochim. Biophys. Acta 2008, 1782, 280–285. [Google Scholar] [CrossRef] [PubMed]
- Chen, K. Beyond LDL oxidation: ROS in vascular signal transduction. Free Radic. Biol. Med. 2003, 35, 117–132. [Google Scholar] [CrossRef]
- Chen, K.; Thomas, S.R.; Albano, A.; Murphy, M.P.; Keaney, J.F. Mitochondrial function is required for hydrogen peroxide-induced growth factor receptor transactivation and downstream signaling. J. Biol. Chem. 2004, 279, 35079–35086. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zang, Q.S.; Liu, Z.; Wu, Q.; Maass, D.; Dulan, G.; Shaul, P.W.; Melito, L.; Frantz, D.E.; Kilgore, J.A.; et al. Regulation of VEGF-induced endothelial cell migration by mitochondrial reactive oxygen species. Am. J. Physiol. 2011, 301, C695–C704. [Google Scholar] [CrossRef]
- Schmidt, K.N.; Amstad, P.; Cerutti, P.; Baeuerle, P.A. The roles of hydrogen peroxide and superoxide as messengers in the activation of transcription factor NF-κB. Chem. Biol. 1995, 2, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Brunelle, J.K.; Bell, E.L.; Quesada, N.M.; Vercauteren, K.; Tiranti, V.; Zeviani, M.; Scarpulla, R.C.; Chandel, N.S. Oxygen sensing requires mitochondrial ROS but not oxidative phosphorylation. Cell Metab. 2005, 1, 409–414. [Google Scholar] [CrossRef] [PubMed]
- West, A.P.; Brodsky, I.E.; Rahner, C.; Woo, D.K.; Erdjument-Bromage, H.; Tempst, P.; Walsh, M.C.; Choi, Y.; Shadel, G.S.; Ghosh, S.; et al. TLR signalling augments macrophage bactericidal activity through mitochondrial ROS. Nature 2011, 472, 476–480. [Google Scholar] [CrossRef] [PubMed]
- Hoarau, E.; Chandra, V.; Rustin, P.; Scharfmann, R.; Duvillie, B. Pro-oxidant/antioxidant balance controls pancreatic β-cell differentiation through the ERK1/2 pathway. Cell Death Dis. 2014. [Google Scholar] [CrossRef] [Green Version]
- Henriksen, E.J. Effects of H2O2 on insulin signaling the glucose transport system in mammalian skeletal muscle. Methods Enzymol. 2013, 528, 269–278. [Google Scholar] [PubMed]
- Marinho, H.S.; Real, C.; Cyrne, L.; Soares, H.; Antunes, F. Hydrogen peroxide sensing, signaling and regulation of transcription factors. Redox Biol. 2014, 2, 535–562. [Google Scholar] [CrossRef] [PubMed]
- Bretón-Romero, R.; Lamas, S. Hydrogen peroxide signaling in vascular endothelial cells. Redox Biol. 2014, 2, 529–534. [Google Scholar] [CrossRef] [PubMed]
- Garlid, A.O.; Jaburek, M.; Jacobs, J.P.; Garlid, K.D. Mitochondrial reactive oxygen species: Which ROS signals cardioprotection? Am. J. Physiol. Heart Circ. Physiol. 2013, 305, H960–H968. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.-X.; Liu, M.; Yan, W.; Zhou, Z.-Z.; Xia, Y.-J.; Tu, W.; Li, P.-Y.; Tian, D.-A. β3 Integrin promotes TGF-β1/H2O2/HOCl-mediated induction of metastatic phenotype of hepatocellular carcinoma cells by enhancing TGF-β1 signaling. PLOS ONE 2013, 8, e79857. [Google Scholar] [CrossRef] [PubMed]
- Klotz, L.-O.; Schroeder, P.; Sies, H. Peroxynitrite signaling: Receptor tyrosine kinases and activation of stress-responsive pathways. Free Radic. Biol. Med. 2002, 33, 737–743. [Google Scholar] [CrossRef] [PubMed]
- Szabó, C.; Ischiropoulos, H.; Radi, R. Peroxynitrite: Biochemistry, pathophysiology and development of therapeutics. Nat. Rev. 2007, 6, 662–680. [Google Scholar]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Weidinger, A.; Kozlov, A.V. Biological Activities of Reactive Oxygen and Nitrogen Species: Oxidative Stress versus Signal Transduction. Biomolecules 2015, 5, 472-484. https://doi.org/10.3390/biom5020472
Weidinger A, Kozlov AV. Biological Activities of Reactive Oxygen and Nitrogen Species: Oxidative Stress versus Signal Transduction. Biomolecules. 2015; 5(2):472-484. https://doi.org/10.3390/biom5020472
Chicago/Turabian StyleWeidinger, Adelheid, and Andrey V. Kozlov. 2015. "Biological Activities of Reactive Oxygen and Nitrogen Species: Oxidative Stress versus Signal Transduction" Biomolecules 5, no. 2: 472-484. https://doi.org/10.3390/biom5020472