

Synthesis of the Antimicrobial Peptide Murepavadin Using Novel Coupling Agents


Abstract
:1. Introduction
2. Materials and Methods
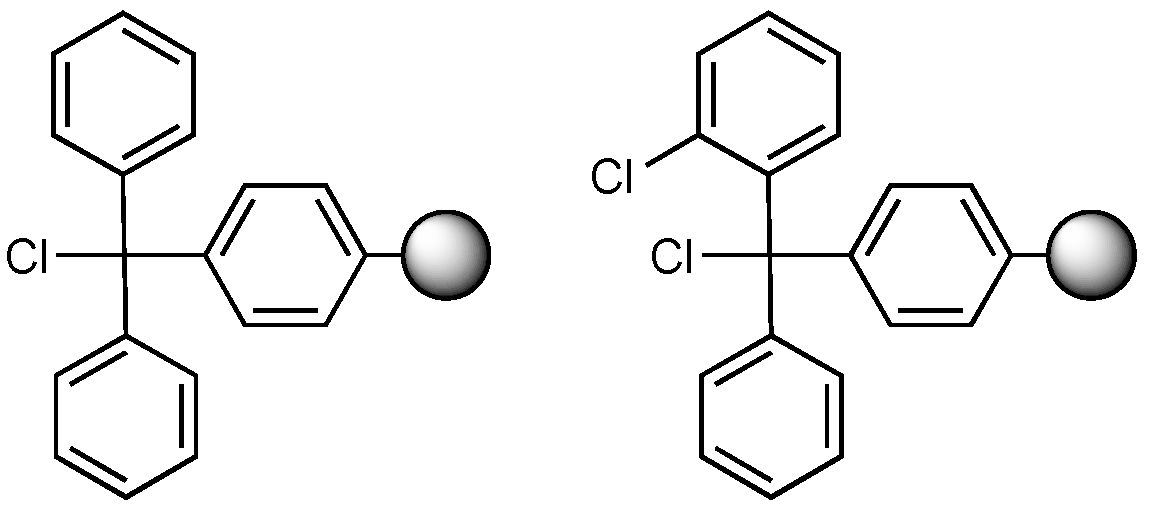
2.1. Chemicals
2.2. Synthesis of K-Oxy-B
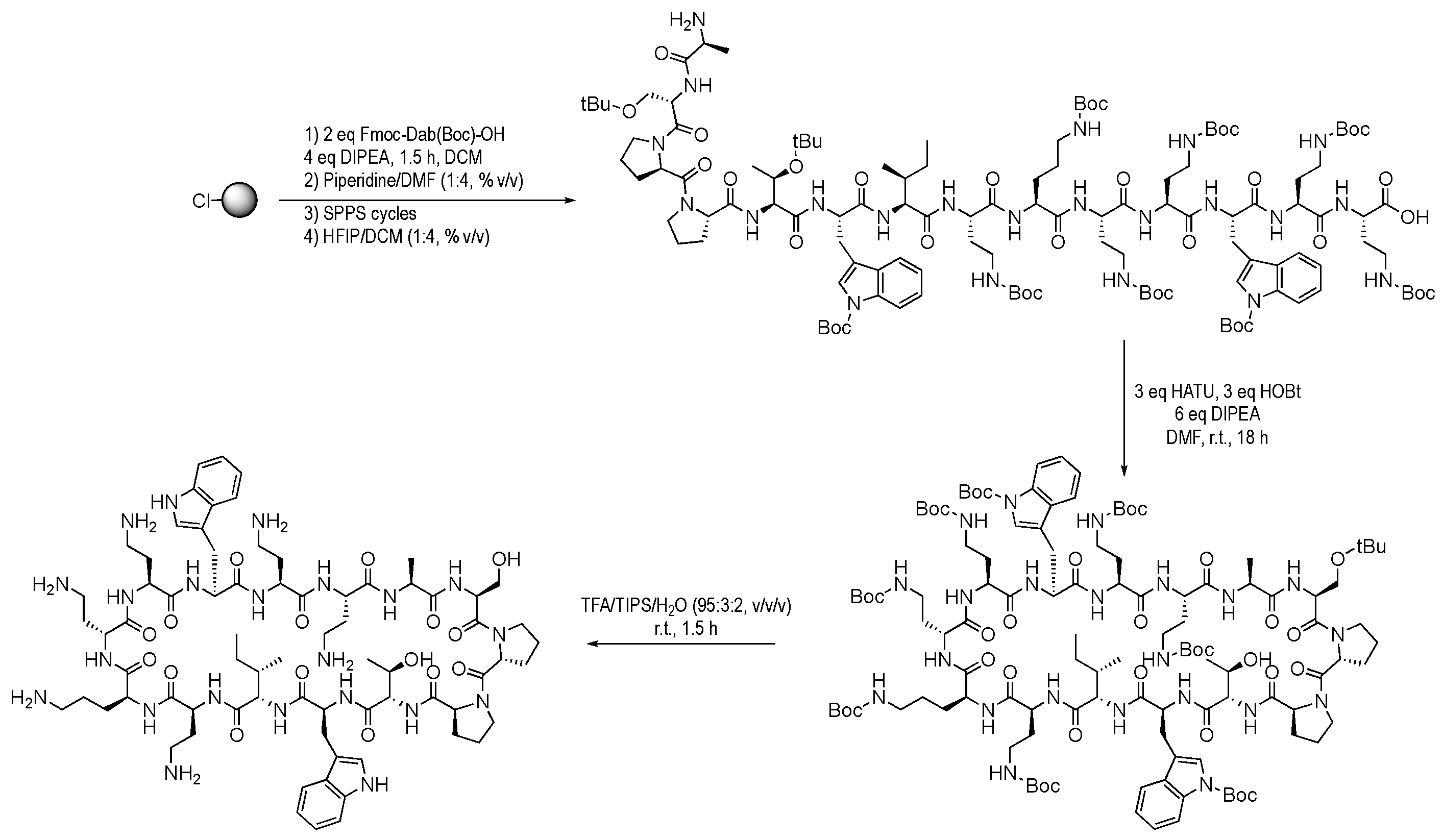
2.3. Peptide Synthesis
2.4. Minimum Inhibitory Concentration Determination
3. Results and Discussion
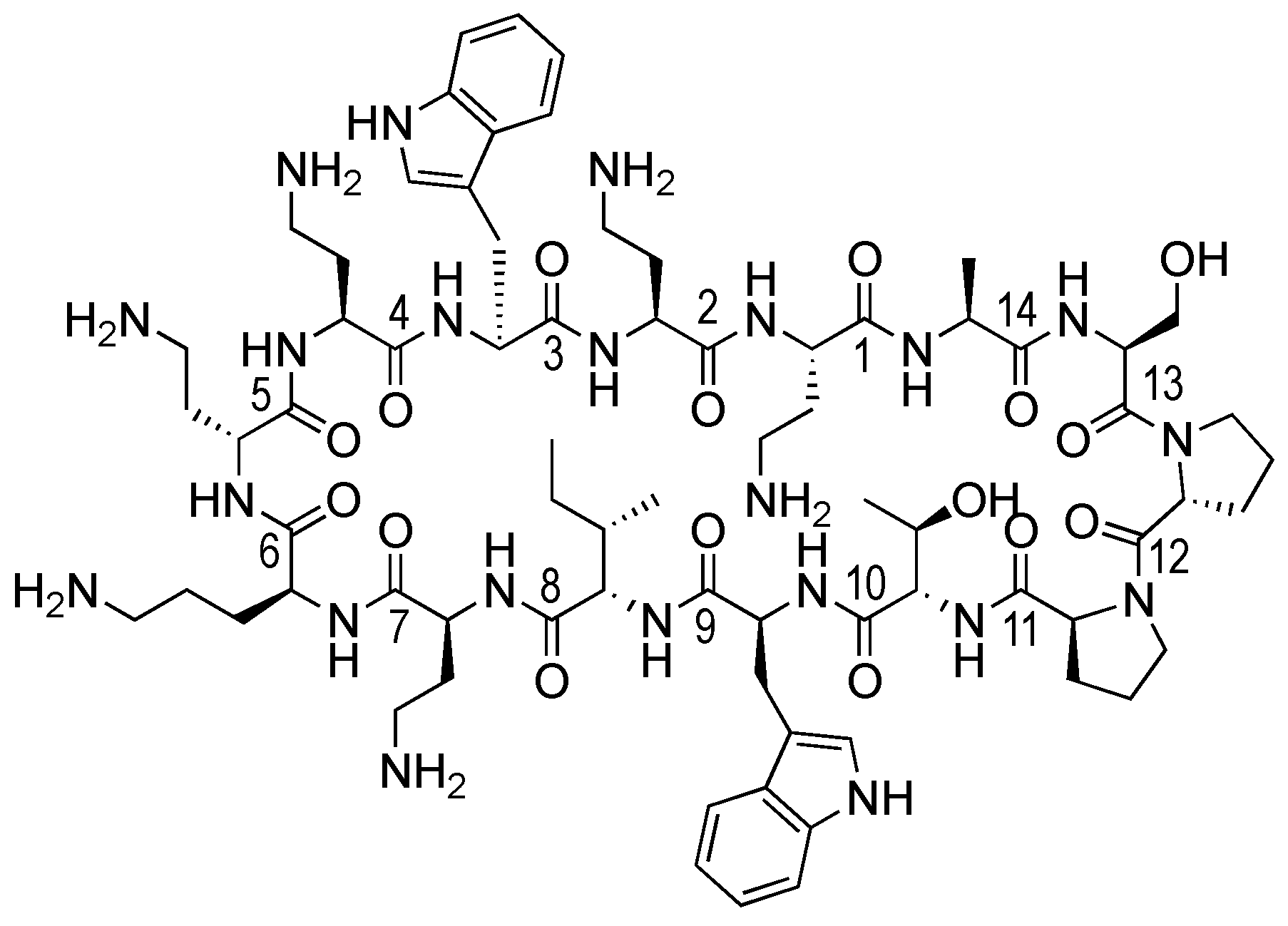
3.1. Murepavadin Synthesis
3.2. Antimicrobial Activity Determination of the Different Murepavadin Batches
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lau, J.L.; Dunn, M.K. Therapeutic Peptides: Historical Perspectives, Current Development Trends, and Future Directions. Bioorg. Med. Chem. 2018, 26, 2700–2707. [Google Scholar] [CrossRef] [PubMed]
- Al Musaimi, O.; Al Shaer, D.; Albericio, F.; de la Torre, B.G. 2020 FDA TIDES (Peptides and Oligonucleotides) Harvest. Pharmaceuticals 2021, 14, 145. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wang, N.; Zhang, W.; Cheng, X.; Yan, Z.; Shao, G.; Wang, X.; Wang, R.; Fu, C. Therapeutic Peptides: Current Applications and Future Directions. Signal. Transduct. Target. Ther. 2022, 7, 48. [Google Scholar] [CrossRef] [PubMed]
- Merrifield, R.B. Solid Phase Peptide Synthesis. I. The Synthesis of a Tetrapeptide. J. Am. Chem. Soc. 1963, 85, 2149–2154. [Google Scholar] [CrossRef]
- Segovia, R.; Díaz-Lobo, M.; Cajal, Y.; Vilaseca, M.; Rabanal, F. Linker-Free Synthesis of Antimicrobial Peptides Using a Novel Cleavage Reagent: Characterisation of the Molecular and Ionic Composition by NanoESI-HR MS. Pharmaceutics 2023, 15, 1310. [Google Scholar] [CrossRef]
- El-Faham, A.; Albericio, F. Peptide Coupling Reagents, More than a Letter Soup. Chem. Rev. 2011, 111, 6557–6602. [Google Scholar] [CrossRef] [PubMed]
- Al-Warhi, T.I.; Al-Hazimi, H.M.A.; El-Faham, A. Recent Development in Peptide Coupling Reagents. J. Saudi Chem. Soc. 2012, 16, 97–116. [Google Scholar] [CrossRef]
- Albericio, F.; El-Faham, A. Choosing the Right Coupling Reagent for Peptides: A Twenty-Five-Year Journey. Org. Process Res. Dev. 2018, 22, 760–772. [Google Scholar] [CrossRef]
- Carpino, L.A.; Imazumi, H.; El-Faham, A.; Ferrer, F.J.; Zhang, C.; Lee, Y.; Foxman, B.M.; Henklein, P.; Hanay, C.; Wenschuh, H.; et al. The Uronium/Guanidinium Peptide Coupling Reagents: Finally the True Uronium Salts. Angew. Chem. Int. Ed. 2002, 41, 441–445. [Google Scholar] [CrossRef]
- Coste, J.; Lenguyen, D.; Castro, B. PyBOP®: A New Peptide Coupling Reagent Devoid of Toxic by-Product. Tetrahedron. Lett. 1990, 31, 205–208. [Google Scholar] [CrossRef]
- Carpino, L.A.; El-Faham, A.; Minorb, C.A.; Albericio, F. Advantageous Applications of Azabenzotriazole (Triazolopyridine)-Based Coupling Reagents to Solid-Phase Peptide Synthesis. J. Chem. Soc. Chem. Commun. 1994, 201–203. [Google Scholar] [CrossRef]
- Carpino, L.A. 1-Hydroxy-7-Azabenzotriazole. An Efficient Peptide Coupling Additive. J. Chem. Soc. Chem. Commun. 1970, 103, 858. [Google Scholar] [CrossRef]
- Spengler, J.; Fernandez-Llamazares, A.I.; Albericio, F. Use of an Internal Reference for the Quantitative HPLC-UV Analysis of Solid-Phase Reactions: A Case Study of 2-Chlorotrityl Chloride Resin. ACS Comb. Sci. 2013, 15, 229–234. [Google Scholar] [CrossRef]
- Wehrstedt, K.D.; Wandrey, P.A.; Heitkamp, D. Explosive Properties of 1-Hydroxybenzotriazoles. J. Hazard. Mater. 2005, 126, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Itoh, M. Racemization Suppression by the Use of Ethyl 2-Hydroximino-2-Cyanoacetate and Its Amide. Bull. Chem. Soc. Jpn. 1973, 46, 2219–2221. [Google Scholar] [CrossRef]
- Subirós-Funosas, R.; Prohens, R.; Barbas, R.; El-Faham, A.; Albericio, F. Oxyma: An Efficient Additive for Peptide Synthesis to Replace the Benzotriazole-Based HOBt and HOAt with a Lower Risk of Explosion. Chem.—A Eur. J. 2009, 15, 9394–9403. [Google Scholar] [CrossRef]
- El-Faham, A.; Albericio, F. COMU: A Third Generation of Uronium-Type Coupling Reagents. J. Pept. Sci. 2010, 16, 6–9. [Google Scholar] [CrossRef] [PubMed]
- Cherkupally, P.; Acosta, G.A.; Nieto-Rodriguez, L.; Spengler, J.; Rodriguez, H.; Khattab, S.N.; El-Faham, A.; Shamis, M.; Luxembourg, Y.; Prohens, R.; et al. K-Oxyma: A Strong Acylation-Promoting, 2-Ctc Resin-Friendly Coupling Additive. Eur. J. Org. Chem. 2013, 28, 6372–6378. [Google Scholar] [CrossRef]
- McFarland, A.D.; Buser, J.Y.; Embry, M.C.; Held, C.B.; Kolis, S.P. Generation of Hydrogen Cyanide from the Reaction of Oxyma (Ethyl Cyano(Hydroxyimino)Acetate) and DIC (Diisopropylcarbodiimide). Org. Process. Res. Dev. 2019, 23, 2099–2105. [Google Scholar] [CrossRef]
- Manne, S.R.; Sharma, A.; Sazonovas, A.; El-Faham, A.; de la Torre, B.G.; Albericio, F. Understanding OxymaPure as a Peptide Coupling Additive: A Guide to New Oxyma Derivatives. ACS Omega 2022, 7, 6007–6023. [Google Scholar] [CrossRef]
- Manne, S.R.; Akintayo, D.C.; Luna, O.; El-Faham, A.; De La Torre, B.G.; Albericio, F. Tert-Butylethylcarbodiimide as an Efficient Substitute for Diisopropylcarbodiimide in Solid-Phase Peptide Synthesis: Understanding the Side Reaction of Carbodiimides with OxymaPure. Org. Process. Res. Dev. 2022, 26, 2894–2899. [Google Scholar] [CrossRef]
- Manne, S.R.; Luna, O.; Acosta, G.A.; Royo, M.; El-Faham, A.; Orosz, G.; De La Torre, B.G.; Albericio, F. Amide Formation: Choosing the Safer Carbodiimide in Combination with OxymaPure to Avoid HCN Release. Org. Lett. 2021, 23, 6900–6904. [Google Scholar] [CrossRef] [PubMed]
- Rabanal, F.; Grau-Campistany, A.; Vila-Farrés, X.; Gonzalez-Linares, J.; Borràs, M.; Vila, J.; Manresa, A.; Cajal, Y. A Bioinspired Peptide Scaffold with High Antibiotic Activity and Low in Vivo Toxicity. Sci. Rep. 2015, 5, 10558. [Google Scholar] [CrossRef] [PubMed]
- Grau-Campistany, A.; Manresa, Á.; Pujol, M.; Rabanal, F.; Cajal, Y. Tryptophan-Containing Lipopeptide Antibiotics Derived from Polymyxin B with Activity against Gram Positive and Gram Negative Bacteria. Biochim. Biophys. Acta Biomembr. 2016, 1858, 333–343. [Google Scholar] [CrossRef] [PubMed]
- Segovia, R.; Solé, J.; Marqués, A.M.; Cajal, Y.; Rabanal, F. Unveiling the Membrane and Cell Wall Action of Antimicrobial Cyclic Lipopeptides: Modulation of the Spectrum of Activity. Pharmaceutics 2021, 13, 2180. [Google Scholar] [CrossRef] [PubMed]
- Rabanal, F.; Cajal, Y. Recent Advances and Perspectives in the Design and Development of Polymyxins. Nat. Prod. Rep. 2017, 34, 886–908. [Google Scholar] [CrossRef] [PubMed]
- Rudilla, H.; Pérez-Guillén, I.; Rabanal, F.; Sierra, J.M.; Vinuesa, T.; Viñas, M. Novel Synthetic Polymyxins Kill Gram-Positive Bacteria. J. Antimicrob. Chemother. 2018, 73, 3385–3390. [Google Scholar] [CrossRef] [PubMed]
- Clausell, A.; Rabanal, F.; Garcia-Subirats, M.; Alsina, M.A.; Cajal, Y. Synthesis and Membrane Action of Polymyxin B Analogues. Luminescence 2005, 20, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Clausell, A.; Rabanal, F.; Garcia-Subirats, M.; Alsina, M.A.; Cajal, Y. Membrane Association and Contact Formation by a Synthetic Analogue of Polymyxin B and Its Fluorescent Derivatives. J. Phys. Chem. B 2006, 110, 4465–4471. [Google Scholar] [CrossRef]
- Priorization of Pathogens to Guide Discovery, Research and Development of New Antibiotics for Drug-Resistant Bacterial Infections, Including Tuberculosis; World Health Organization: Geneva, Switzerland, 2017.
- Martin-Loeches, I.; Dale, G.E.; Torres, A. Murepavadin: A New Antibiotic Class in the Pipeline. Expert Rev. Anti Infect. Ther. 2018, 16, 259–268. [Google Scholar] [CrossRef]
- Shankaramma, S.C.; Athanassiou, Z.; Zerbe, O.; Moehle, K.; Mouton, C.; Bernardini, F.; Vrijbloed, J.W.; Obrecht, D.; Robinson, J.A. Macrocyclic Hairpin Mimetics of the Cationic Antimicrobial Peptide Protegrin I: A New Family of Broad-Spectrum Antibiotics. ChemBioChem 2002, 3, 1126–1133. [Google Scholar] [CrossRef]
- Robinson, J.A.; Shankaramma, S.C.; Jetter, P.; Kienzl, U.; Schwendener, R.A.; Vrijbloed, J.W.; Obrecht, D. Properties and Structure-Activity Studies of Cyclic β-Hairpin Peptidomimetics Based on the Cationic Antimicrobial Peptide Protegrin I. Bioorg. Med. Chem. 2005, 13, 2055–2064. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, J.; Patora-Komisarska, K.; Moehle, K.; Obrecht, D.; Robinson, J.A. Structural Studies of β-Hairpin Peptidomimetic Antibiotics That Target LptD in Pseudomonas sp. Bioorg. Med. Chem. 2013, 21, 5806–5810. [Google Scholar] [CrossRef] [PubMed]
- Andolina, G.; Bencze, L.C.; Zerbe, K.; Müller, M.; Steinmann, J.; Kocherla, H.; Mondal, M.; Sobek, J.; Moehle, K.; Malojčić, G.; et al. A Peptidomimetic Antibiotic Interacts with the Periplasmic Domain of LptD from Pseudomonas aeruginosa. ACS Chem. Biol. 2018, 13, 666–675. [Google Scholar] [CrossRef]
- Sader, H.S.; Dale, G.E.; Rhomberg, P.R.; Flamm, R.K. Antimicrobial Activity of Murepavadin Tested against Clinical Isolates of Pseudomonas aeruginosa from the United States, Europe, and China. Antimicrob. Agents Chemother. 2018, 62, e00311–e00318. [Google Scholar] [CrossRef]
- Werneburg, M.; Zerbe, K.; Juhas, M.; Bigler, L.; Stalder, U.; Kaech, A.; Ziegler, U.; Obrecht, D.; Eberl, L.; Robinson, J.A. Inhibition of Lipopolysaccharide Transport to the Outer Membrane in Pseudomonas aeruginosa by Peptidomimetic Antibiotics. ChemBioChem 2012, 13, 1767–1775. [Google Scholar] [CrossRef]
- Srinivas, N.; Jetter, P.; Ueberbacher, B.J.; Werneburg, M.; Zerbe, K.; Steinmann, J.; Van Der Meijden, B.; Bernardini, F.; Lederer, A.; Dias, R.L.A.; et al. Peptidomimetic Antibiotics Target Outer-Membrane Biogenesis in Pseudomonas aeruginosa. Science 2010, 327, 1010–1013. [Google Scholar] [CrossRef]
- Díez-Aguilar, M.; Hernández-García, M.; Morosini, M.I.; Fluit, A.; Tunney, M.M.; Huertas, N.; Del Campo, R.; Obrecht, D.; Bernardini, F.; Ekkelenkamp, M.; et al. Murepavadin Antimicrobial Activity against and Resistance Development in Cystic Fibrosis Pseudomonas aeruginosa Isolates. J. Antimicrob. Chemother. 2021, 76, 984–992. [Google Scholar] [CrossRef]
- Spexis Reports Solid Safety and Pharmacokinetics Results from First-in-Human Study with Inhaled Murepavadin, a Novel Macrocycle Compound. Available online: https://www.globenewswire.com/news-release/2023/01/09/2584785/0/en/Spexis-reports-solid-safety-and-pharmacokinetics-results-from-first-in-human-study-with-inhaled-murepavadin-a-novel-macrocycle-compound.html (accessed on 16 April 2024).
- Obrecht, D.; Luther, A.; Bernardini, F.; Zbinden, P. Beta–Hairpin Peptidomimetics. U.S. Patent 20180044380A1 2018. [Google Scholar]
- Chaudhuri, D.; Ganesan, R.; Vogelaar, A.; Dughbaj, M.A.; Beringer, P.M.; Camarero, J.A. Chemical Synthesis of a Potent Antimicrobial Peptide Murepavadin Using a Tandem Native Chemical Ligation/Desulfurization Reaction. J. Org. Chem. 2021, 86, 15242–15246. [Google Scholar] [CrossRef] [PubMed]
- Mueller, L.K.; Baumruck, A.C.; Zhdanova, H.; Tietze, A.A. Challenges and Perspectives in Chemical Synthesis of Highly Hydrophobic Peptides. Front. Bioeng. Biotechnol. 2020, 8, 162. [Google Scholar] [CrossRef]
- Isidro-Llobet, A.; Kenworthy, M.N.; Mukherjee, S.; Kopach, M.E.; Wegner, K.; Gallou, F.; Smith, A.G.; Roschangar, F. Sustainability Challenges in Peptide Synthesis and Purification: From R&D to Production. J. Org. Chem. 2019, 84, 4615–4628. [Google Scholar] [CrossRef] [PubMed]
- Jad, Y.E.; Khattab, S.N.; De La Torre, B.G.; Govender, T.; Kruger, H.G.; El-Faham, A.; Albericio, F. Oxyma-B, an Excellent Racemization Suppressor for Peptide Synthesis. Org. Biomol. Chem. 2014, 12, 8379–8385. [Google Scholar] [CrossRef] [PubMed]
- Jad, Y.E.; Khattab, S.N.; de la Torre, B.G.; Govender, T.; Kruger, H.G.; El-Faham, A.; Albericio, F. EDC·HCl and Potassium Salts of Oxyma and Oxyma-B as Superior Coupling Cocktails for Peptide Synthesis. Eur. J. Org. Chem. 2015, 2015, 3116–3120. [Google Scholar] [CrossRef]
- O’Neal, K.D.; Chari, M.V.; Mcdonald, C.H.; Cook, R.G.; Li-Yuan, Y.-L.; Morrisett, J.D.; Shearer, W.T. Multiple Cis-Trans Conformers of the Prolactin Receptor Proline-Rich Motif (PRM) Peptide Detected by Reverse-Phase HPLC, CD and NMR Spectroscopy. Biochem. J. 1996, 315, 833–844. [Google Scholar] [CrossRef]
- Sui, Q.; Rabenstein, D.L. Cis/Trans Isomerization of Proline Peptide Bonds in the Backbone of Cyclic Disulfide-Bridged Peptides. Pept. Sci. 2018, 110, e24088. [Google Scholar] [CrossRef]
- Upert, G.; Luther, A.; Obrecht, D.; Ermert, P. Emerging Peptide Antibiotics with Therapeutic Potential. Med. Drug Discov. 2021, 9, 100078. [Google Scholar] [CrossRef]
- Zerbe, K.; Moehle, K.; Robinson, J.A. Protein Epitope Mimetics: From New Antibiotics to Supramolecular Synthetic Vaccines. Acc. Chem. Res. 2017, 50, 1323–1331. [Google Scholar] [CrossRef]
- BacDive The Bacterial Diversity Metadatabase. Pseudomonas aeruginosa DSM 24600. Available online: https://bacdive.dsmz.de/strain/12803 (accessed on 22 February 2024).
- BacDive The Bacterial Diversity Metadatabase. Pseudomonas aeruginosa DSM 25716. Available online: https://bacdive.dsmz.de/strain/12805 (accessed on 22 February 2024).






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Synthesis | Resin | Loading | Coupling Agents | Resin Detachment Yield | Cyclization Yield | Acidolysis Yield | Overall Crude Yield | Crude Purity a |
|---|---|---|---|---|---|---|---|---|
| A | 2-CTC | 0.86 mmol/g | 3 eq DIC 3 eq HOBt | 46% | 96% | 90% | 40% | 58% |
| B | 2-CTC | 0.86 mmol/g | 3 eq DIC 3 eq K-Oxyma | 91% | 93% | 83% | 70% | 64% |
| C | Cl-Trt | 0.45 mmol/g | 3 eq DIC 3 eq HOBt | 41% | 89% | 83% | 30% | 67% |
| D | 2-CTC | 0.63 mmol/g | 3 eq TBEC 3 eq K-Oxyma | 90% | 95% | 88% | 75% | 60% |
| E | 2-CTC | 0.69 mmol/g | 3 eq TBEC 3 eq Oxy-B | 89% | 96% | 89% | 76% | 67% |
| F | 2-CTC | 0.70 mmol/g | 3 eq TBEC 3 eq K-Oxy-B | 93% | 94% | 91% | 80% | 59% |
| Synthesis | Resin | Coupling Agents | Purification Yield | Global Yield |
|---|---|---|---|---|
| A | 2-CTC | 3 eq DIC 3 eq HOBt | 29% | 12% |
| B | 2-CTC | 3 eq DIC 3 eq K-Oxyma | 40% | 28% |
| C | Cl-Trt | 3 eq DIC 3 eq HOBt | 45% | 14% |
| D | 2-CTC | 3 eq TBEC 3 eq K-Oxyma | 36% | 27% |
| E | 2-CTC | 3 eq TBEC 3 eq Oxy-B | 39% | 30% |
| F | 2-CTC | 3 eq TBEC 3 eq K-Oxy-B | 34% | 27% |
| A | B | C | D | E | F | |
|---|---|---|---|---|---|---|
| P. aeruginosa ATCC 27853 | 0.0625 | 0.0625 | 0.0625 | 0.0625 | 0.0625 | 0.0625 |
| P. aeruginosa DSM 24600 | 0.03125 | 0.03125 | 0.03125 | 0.03125 | 0.03125 | 0.03125 |
| P. aeruginosa DSM 25716 | 0.25 | 0.25 | 0.25 | 0.25 | 0.25 | 0.25 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
García-Gros, J.; Cajal, Y.; Marqués, A.M.; Rabanal, F. Synthesis of the Antimicrobial Peptide Murepavadin Using Novel Coupling Agents. Biomolecules 2024, 14, 526. https://doi.org/10.3390/biom14050526
García-Gros J, Cajal Y, Marqués AM, Rabanal F. Synthesis of the Antimicrobial Peptide Murepavadin Using Novel Coupling Agents. Biomolecules. 2024; 14(5):526. https://doi.org/10.3390/biom14050526
Chicago/Turabian StyleGarcía-Gros, Júlia, Yolanda Cajal, Ana Maria Marqués, and Francesc Rabanal. 2024. "Synthesis of the Antimicrobial Peptide Murepavadin Using Novel Coupling Agents" Biomolecules 14, no. 5: 526. https://doi.org/10.3390/biom14050526