

The Role of the Complement System in the Pathogenesis of Infectious Forms of Hemolytic Uremic Syndrome

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
- HUS caused by hemorrhagic Shiga toxin-producing E. coli (STEC-HUS);
- Secondary HUS (due to cancer, organ and tissue transplantation, medications, autoimmune disorders, malignant hypertension, and HIV infection);
- HUS associated with infections caused by the H1N1 influenza virus and S. pneumoniae;
- HUS associated with cobalamin C defect;
- HUS associated with mutations in the DGKE gene;
- HUS caused by dysregulation of the alternative complement pathway (mutations in complement genes and antibodies to factor H);
- HUS of unknown etiology.
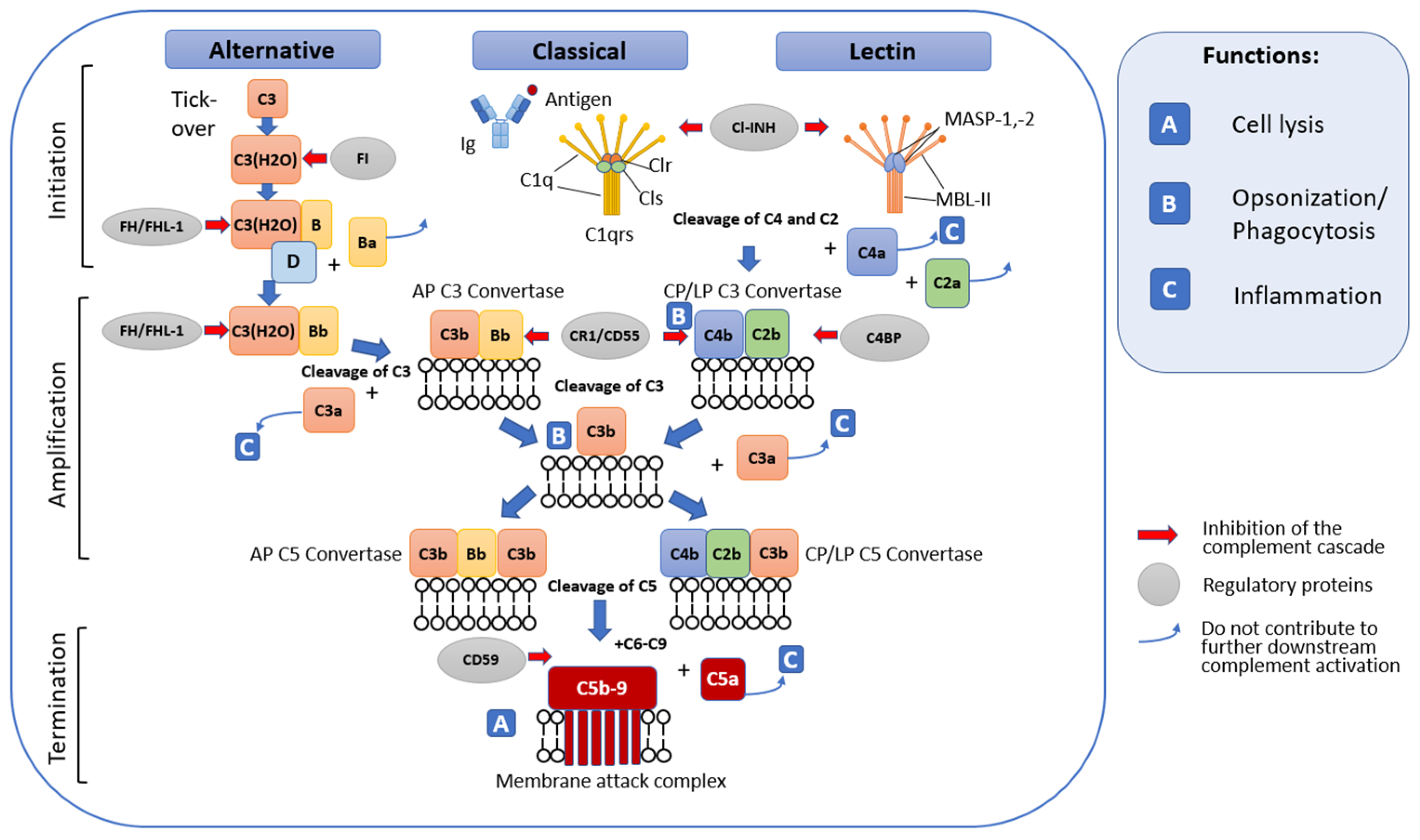
2. Complement System
2.1. General Terminal Stage of Complement Activation
2.2. Complement Regulatory Mechanisms
3. Interactions of the Complement System with the Blood Coagulation System
3.1. Blood Coagulation System
3.1.1. Tissue Factor Pathway
3.1.2. The Internal Contact Activation Pathway
3.2. Synergism in the Functioning of the Complement System and the Blood Coagulation System as a Key Factor in Thrombus Formation in HUS
4. Bacterial Infections That Cause HUS
4.1. Hemorrhagic Shiga Toxin-Producing E. coli
4.1.1. Pathogenetic Mechanisms of STEC-HUS
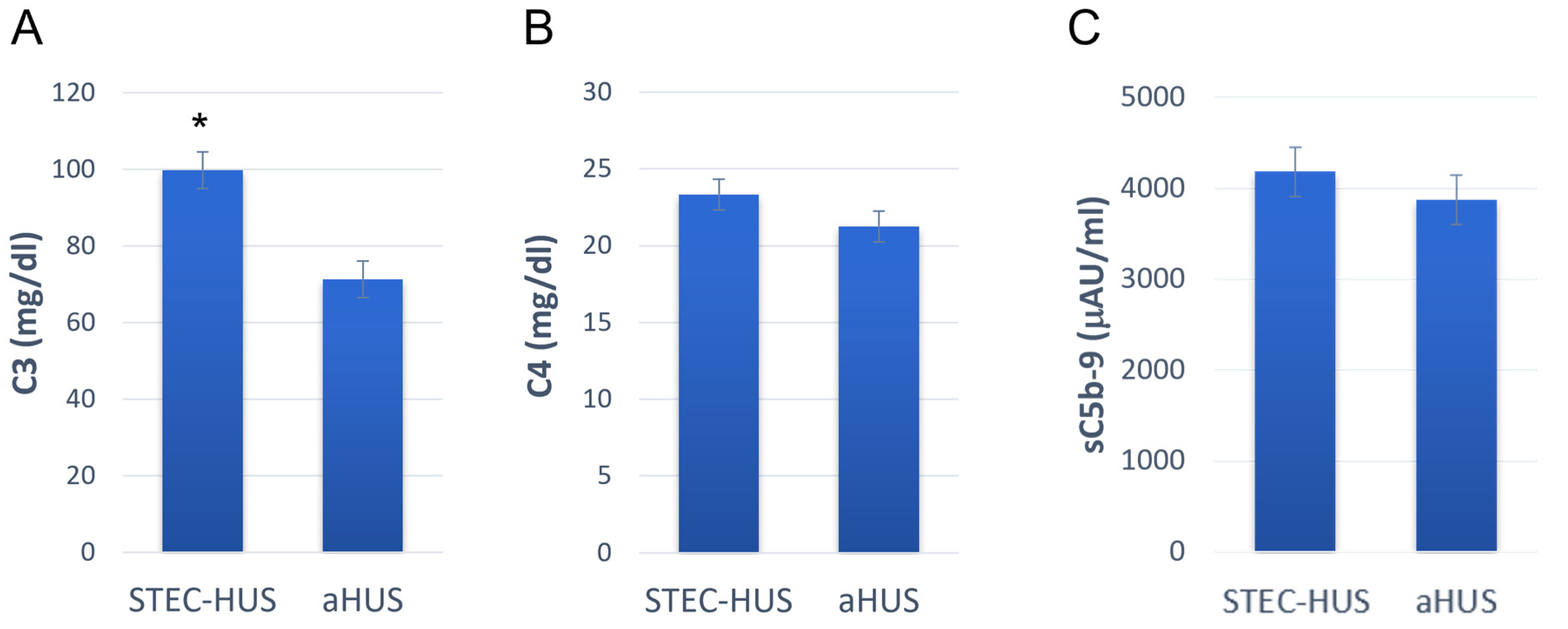
4.1.2. The Role of the Complement System in the Pathogenesis of STEC-HUS
4.1.3. Pathways of Complement Activation in STEC-HUS
4.1.4. Targets of the Complement System in the Pathogenesis of STEC-HUS
4.1.5. Immunometabolic Alterations in STEC-HUS
4.2. Shigella dysenteriae
4.3. Streptococcus pneumoniae and Other Neuraminidase-Producing Bacteria
4.4. Bordetella pertussis
4.5. Salmonella typhi
4.6. Other Bacterial Infections
5. Viral Infections That Cause HUS
5.1. Influenza Virus
5.2. Human Immunodeficiency Viruses (HIV)
5.3. Enteroviral Infections
5.4. SARS-CoV-2
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Aigner, C.; Schmidt, A.; Gaggl, M.; Sunder-Plassmann, G. An updated classification of thrombotic microangiopathies and treatment of complement gene variant-mediated thrombotic microangiopathy. Clin. Kidney J. 2019, 12, 333–337. [Google Scholar] [CrossRef] [PubMed]
- Brocklebank, V.; Wood, K.M.; Kavanagh, D. Thrombotic microangiopathy and the kidney. Clin. J. Am. Soc. Nephrol. CJASN 2018, 13, 300–317. [Google Scholar] [CrossRef] [PubMed]
- Fakhouri, F.; Fila, M.; Hummel, A.; Ribes, D.; Sellier-Leclerc, A.L.; Ville, S.; Pouteil-Noble, C.; Coindre, J.P.; Le Quintrec, M.; Rondeau, E.; et al. Eculizumab discontinuation in children and adults with atypical hemolytic-uremic syndrome: A prospective multicenter study. Blood 2021, 137, 2438–2449. [Google Scholar] [CrossRef] [PubMed]
- Loirat, C.; Fakhouri, F.; Ariceta, G.; Besbas, N.; Bitzan, M.; Bjerre, A.; Coppo, R.; Emma, F.; Johnson, S.; Karpman, D.; et al. An international consensus approach to the management of atypical hemolytic uremic syndrome in children. Pediatr. Nephrol. 2016, 31, 15–39. [Google Scholar] [CrossRef] [PubMed]
- Warwicker, P.; Goodship, T.H.; Donne, R.L.; Pirson, Y.; Nicholls, A.; Ward, R.M.; Turnpenny, P.; Goodship, J.A. Genetic studies into inherited and sporadic hemolytic uremic syndrome. Kidney Int. 1998, 53, 836–844. [Google Scholar] [CrossRef] [PubMed]
- Zipfel, P.F.; Skerka, C. Complement regulators and inhibitory proteins. Nat. Rev. Immunol. 2009, 9, 729–740. [Google Scholar] [CrossRef] [PubMed]
- Cserhalmi, M.; Papp, A.; Brandus, B.; Uzonyi, B.; Jozsi, M. Regulation of regulators: Role of the complement factor h-related proteins. Semin. Immunol. 2019, 45, 101341. [Google Scholar] [CrossRef]
- Wurzner, R.; Joysey, V.C.; Lachmann, P.J. Complement component c7. Assessment of in vivo synthesis after liver transplantation reveals that hepatocytes do not synthesize the majority of human c7. J. Immunol. 1994, 152, 4624–4629. [Google Scholar] [CrossRef]
- Carroll, M.C. The role of complement and complement receptors in induction and regulation of immunity. Annu. Rev. Immunol. 1998, 16, 545–568. [Google Scholar] [CrossRef]
- Arbore, G.; Kemper, C.; Kolev, M. Intracellular complement—The complosome—In immune cell regulation. Mol. Immunol. 2017, 89, 2–9. [Google Scholar] [CrossRef]
- Bhakdi, S.; Tranum-Jensen, J. C5b-9 assembly: Average binding of one c9 molecule to c5b-8 without poly-c9 formation generates a stable transmembrane pore. J. Immunol. 1986, 136, 2999–3005. [Google Scholar] [CrossRef] [PubMed]
- Podack, E.R.; Tschoop, J.; Muller-Eberhard, H.J. Molecular organization of c9 within the membrane attack complex of complement. Induction of circular c9 polymerization by the c5b-8 assembly. J. Exp. Med. 1982, 156, 268–282. [Google Scholar] [CrossRef] [PubMed]
- Ward, P.A.; Newman, L.J. A neutrophil chemotactic factor from human c′5. J. Immunol. 1969, 102, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Ogden, C.A.; Elkon, K.B. Role of complement and other innate immune mechanisms in the removal of apoptotic cells. Curr. Dir. Autoimmun. 2006, 9, 120–142. [Google Scholar] [CrossRef] [PubMed]
- Anliker-Ort, M.; Dingemanse, J.; van den Anker, J.; Kaufmann, P. Treatment of rare inflammatory kidney diseases: Drugs targeting the terminal complement pathway. Front. Immunol. 2020, 11, 599417. [Google Scholar] [CrossRef] [PubMed]
- Dempsey, P.W.; Allison, M.E.; Akkaraju, S.; Goodnow, C.C.; Fearon, D.T. C3d of complement as a molecular adjuvant: Bridging innate and acquired immunity. Science 1996, 271, 348–350. [Google Scholar] [CrossRef]
- Chen, J.Y.; Cortes, C.; Ferreira, V.P. Properdin: A multifaceted molecule involved in inflammation and diseases. Mol. Immunol. 2018, 102, 58–72. [Google Scholar] [CrossRef]
- Mukherjee, P.; Thomas, S.; Pasinetti, G.M. Complement anaphylatoxin c5a neuroprotects through regulation of glutamate receptor subunit 2 in vitro and in vivo. J. Neuroinflamm. 2008, 5, 5. [Google Scholar] [CrossRef]
- Ling, M.; Murali, M. Analysis of the complement system in the clinical immunology laboratory. Clin. Lab. Med. 2019, 39, 579–590. [Google Scholar] [CrossRef]
- Garred, P.; Genster, N.; Pilely, K.; Bayarri-Olmos, R.; Rosbjerg, A.; Ma, Y.J.; Skjoedt, M.O. A journey through the lectin pathway of complement-mbl and beyond. Immunol. Rev. 2016, 274, 74–97. [Google Scholar] [CrossRef]
- Pouw, R.B.; Ricklin, D. Tipping the balance: Intricate roles of the complement system in disease and therapy. Semin. Immunopathol. 2021, 43, 757–771. [Google Scholar] [CrossRef] [PubMed]
- Bossi, F.; Fischetti, F.; Pellis, V.; Bulla, R.; Ferrero, E.; Mollnes, T.E.; Regoli, D.; Tedesco, F. Platelet-activating factor and kinin-dependent vascular leakage as a novel functional activity of the soluble terminal complement complex. J. Immunol. 2004, 173, 6921–6927. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Yang, C.; Jin, N.; Xie, Z.; Tang, Y.; Fei, L.; Jia, Z.; Wu, Y. Terminal complement complex c5b-9-treated human monocyte-derived dendritic cells undergo maturation and induce th1 polarization. Eur. J. Immunol. 2007, 37, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Jozsi, M.; Barlow, P.N.; Meri, S. Editorial: Function and dysfunction of complement factor h. Front. Immunol. 2021, 12, 831044. [Google Scholar] [CrossRef] [PubMed]
- Johnson, E.; Berge, V.; Hogasen, K. Formation of the terminal complement complex on agarose beads: Further evidence that vitronectin (complement s-protein) inhibits c9 polymerization. Scand. J. Immunol. 1994, 39, 281–285. [Google Scholar] [CrossRef] [PubMed]
- Tschopp, J.; Chonn, A.; Hertig, S.; French, L.E. Clusterin, the human apolipoprotein and complement inhibitor, binds to complement c7, c8 beta, and the b domain of c9. J. Immunol. 1993, 151, 2159–2165. [Google Scholar] [CrossRef]
- Pryzdial, E.L.G.; Leatherdale, A.; Conway, E.M. Coagulation and complement: Key innate defense participants in a seamless web. Front. Immunol. 2022, 13, 918775. [Google Scholar] [CrossRef]
- Lenoir, G.; D’Ambrosio, J.M.; Dieudonne, T.; Copic, A. Transport pathways that contribute to the cellular distribution of phosphatidylserine. Front. Cell Dev. Biol. 2021, 9, 737907. [Google Scholar] [CrossRef]
- Protty, M.B.; Jenkins, P.V.; Collins, P.W.; O’Donnell, V.B. The role of procoagulant phospholipids on the surface of circulating blood cells in thrombosis and haemostasis. Open Biol. 2022, 12, 210318. [Google Scholar] [CrossRef]
- Heijnen, H.; van der Sluijs, P. Platelet secretory behaviour: As diverse as the granules … Or not? J. Thromb. Haemost. 2015, 13, 2141–2151. [Google Scholar] [CrossRef]
- Komiyama, Y.; Pedersen, A.H.; Kisiel, W. Proteolytic activation of human factors ix and x by recombinant human factor viia: Effects of calcium, phospholipids, and tissue factor. Biochemistry 1990, 29, 9418–9425. [Google Scholar] [CrossRef] [PubMed]
- Pryzdial, E.L.G. Maestro tissue factor reaches new height. Blood 2017, 130, 1604–1605. [Google Scholar] [CrossRef] [PubMed]
- Lu, G.; Broze, G.J., Jr.; Krishnaswamy, S. Formation of factors ixa and xa by the extrinsic pathway: Differential regulation by tissue factor pathway inhibitor and antithrombin iii. J. Biol. Chem. 2004, 279, 17241–17249. [Google Scholar] [CrossRef] [PubMed]
- Kamikubo, Y.; Mendolicchio, G.L.; Zampolli, A.; Marchese, P.; Rothmeier, A.S.; Orje, J.N.; Gale, A.J.; Krishnaswamy, S.; Gruber, A.; Ostergaard, H.; et al. Selective factor viii activation by the tissue factor-factor viia-factor xa complex. Blood 2017, 130, 1661–1670. [Google Scholar] [CrossRef] [PubMed]
- Mast, A.E.; Ruf, W. Regulation of coagulation by tissue factor pathway inhibitor: Implications for hemophilia therapy. J. Thromb. Haemost. JTH 2022, 20, 1290–1300. [Google Scholar] [CrossRef] [PubMed]
- Olson, S.T.; Richard, B.; Izaguirre, G.; Schedin-Weiss, S.; Gettins, P.G. Molecular mechanisms of antithrombin-heparin regulation of blood clotting proteinases. A paradigm for understanding proteinase regulation by serpin family protein proteinase inhibitors. Biochimie 2010, 92, 1587–1596. [Google Scholar] [CrossRef] [PubMed]
- Schuijt, T.J.; Bakhtiari, K.; Daffre, S.; Deponte, K.; Wielders, S.J.; Marquart, J.A.; Hovius, J.W.; van der Poll, T.; Fikrig, E.; Bunce, M.W.; et al. Factor xa activation of factor v is of paramount importance in initiating the coagulation system: Lessons from a tick salivary protein. Circulation 2013, 128, 254–266. [Google Scholar] [CrossRef] [PubMed]
- Krishnaswamy, S.; Nesheim, M.E.; Pryzdial, E.L.; Mann, K.G. Assembly of prothrombinase complex. Methods Enzymol. 1993, 222, 260–280. [Google Scholar] [CrossRef]
- Shamanaev, A.; Emsley, J.; Gailani, D. Proteolytic activity of contact factor zymogens. J. Thromb. Haemost. JTH 2021, 19, 330–341. [Google Scholar] [CrossRef]
- Mailer, R.K.; Rangaswamy, C.; Konrath, S.; Emsley, J.; Renne, T. An update on factor xii-driven vascular inflammation. Biochim. Biophys. Acta Mol. Cell Res. 2022, 1869, 119166. [Google Scholar] [CrossRef]
- Kannemeier, C.; Shibamiya, A.; Nakazawa, F.; Trusheim, H.; Ruppert, C.; Markart, P.; Song, Y.; Tzima, E.; Kennerknecht, E.; Niepmann, M.; et al. Extracellular rna constitutes a natural procoagulant cofactor in blood coagulation. Proc. Natl. Acad. Sci. USA 2007, 104, 6388–6393. [Google Scholar] [CrossRef] [PubMed]
- von Bruhl, M.L.; Stark, K.; Steinhart, A.; Chandraratne, S.; Konrad, I.; Lorenz, M.; Khandoga, A.; Tirniceriu, A.; Coletti, R.; Kollnberger, M.; et al. Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. J. Exp. Med. 2012, 209, 819–835. [Google Scholar] [CrossRef] [PubMed]
- Muller, F.; Mutch, N.J.; Schenk, W.A.; Smith, S.A.; Esterl, L.; Spronk, H.M.; Schmidbauer, S.; Gahl, W.A.; Morrissey, J.H.; Renne, T. Platelet polyphosphates are proinflammatory and procoagulant mediators in vivo. Cell 2009, 139, 1143–1156. [Google Scholar] [CrossRef] [PubMed]
- Renne, T. The procoagulant and proinflammatory plasma contact system. Semin. Immunopathol. 2012, 34, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Pryzdial, E.L.G.; Lee, F.M.H.; Lin, B.H.; Carter, R.L.R.; Tegegn, T.Z.; Belletrutti, M.J. Blood coagulation dissected. Transfus. Apher. Sci. 2018, 57, 449–457. [Google Scholar] [CrossRef] [PubMed]
- Maas, C.; Renne, T. Coagulation factor xii in thrombosis and inflammation. Blood 2018, 131, 1903–1909. [Google Scholar] [CrossRef]
- Brown, N.J.; Gainer, J.V.; Stein, C.M.; Vaughan, D.E. Bradykinin stimulates tissue plasminogen activator release in human vasculature. Hypertension 1999, 33, 1431–1435. [Google Scholar] [CrossRef]
- Yin, W.; Ghebrehiwet, B.; Weksler, B.; Peerschke, E.I. Classical pathway complement activation on human endothelial cells. Mol. Immunol. 2007, 44, 2228–2234. [Google Scholar] [CrossRef]
- Nayak, A.; Ferluga, J.; Tsolaki, A.G.; Kishore, U. The non-classical functions of the classical complement pathway recognition subcomponent c1q. Immunol. Lett. 2010, 131, 139–150. [Google Scholar] [CrossRef]
- Hamad, O.A.; Nilsson, P.H.; Wouters, D.; Lambris, J.D.; Ekdahl, K.N.; Nilsson, B. Complement component c3 binds to activated normal platelets without preceding proteolytic activation and promotes binding to complement receptor 1. J. Immunol. 2010, 184, 2686–2692. [Google Scholar] [CrossRef]
- Saggu, G.; Cortes, C.; Emch, H.N.; Ramirez, G.; Worth, R.G.; Ferreira, V.P. Identification of a novel mode of complement activation on stimulated platelets mediated by properdin and c3(h2o). J. Immunol. 2013, 190, 6457–6467. [Google Scholar] [CrossRef] [PubMed]
- Monsinjon, T.; Gasque, P.; Chan, P.; Ischenko, A.; Brady, J.J.; Fontaine, M.C. Regulation by complement c3a and c5a anaphylatoxins of cytokine production in human umbilical vein endothelial cells. FASEB J. 2003, 17, 1003–1014. [Google Scholar] [CrossRef] [PubMed]
- Propson, N.E.; Roy, E.R.; Litvinchuk, A.; Kohl, J.; Zheng, H. Endothelial c3a receptor mediates vascular inflammation and blood-brain barrier permeability during aging. J. Clin. Investig. 2021, 131, e140966. [Google Scholar] [CrossRef] [PubMed]
- Shivshankar, P.; Li, Y.D.; Mueller-Ortiz, S.L.; Wetsel, R.A. In response to complement anaphylatoxin peptides c3a and c5a, human vascular endothelial cells migrate and mediate the activation of b-cells and polarization of t-cells. FASEB J. 2020, 34, 7540–7560. [Google Scholar] [CrossRef]
- Foreman, K.E.; Vaporciyan, A.A.; Bonish, B.K.; Jones, M.L.; Johnson, K.J.; Glovsky, M.M.; Eddy, S.M.; Ward, P.A. C5a-induced expression of p-selectin in endothelial cells. J. Clin. Investig. 1994, 94, 1147–1155. [Google Scholar] [CrossRef]
- Fang, W.; Guo, Z.H.; Zhang, B.Q.; Wu, X.F.; Li, P.; Lv, F.L.; Su, L. [Effect of c5a on expression of thrombomodulin in endothelial cells in vitro]. Zhongguo Wei Zhong Bing Ji Jiu Yi Xue Chin. Crit. Care Med. Zhongguo Weizhongbing Jijiuyixue 2009, 21, 168–171. [Google Scholar]
- Bongoni, A.K.; Lu, B.; McRae, J.L.; Salvaris, E.J.; Toonen, E.J.M.; Vikstrom, I.; Baz Morelli, A.; Pearse, M.J.; Cowan, P.J. Complement-mediated damage to the glycocalyx plays a role in renal ischemia-reperfusion injury in mice. Transplant. Direct 2019, 5, e341. [Google Scholar] [CrossRef]
- Wojta, J.; Huber, K.; Valent, P. New aspects in thrombotic research: Complement induced switch in mast cells from a profibrinolytic to a prothrombotic phenotype. Pathophysiol. Haemost. Thromb. 2003, 33, 438–441. [Google Scholar] [CrossRef]
- Gulla, K.C.; Gupta, K.; Krarup, A.; Gal, P.; Schwaeble, W.J.; Sim, R.B.; O’Connor, C.D.; Hajela, K. Activation of mannan-binding lectin-associated serine proteases leads to generation of a fibrin clot. Immunology 2010, 129, 482–495. [Google Scholar] [CrossRef]
- Krarup, A.; Gulla, K.C.; Gal, P.; Hajela, K.; Sim, R.B. The action of mbl-associated serine protease 1 (masp1) on factor xiii and fibrinogen. Biochim. et Biophys. Acta 2008, 1784, 1294–1300. [Google Scholar] [CrossRef]
- Hamilton, K.K.; Hattori, R.; Esmon, C.T.; Sims, P.J. Complement proteins c5b-9 induce vesiculation of the endothelial plasma membrane and expose catalytic surface for assembly of the prothrombinase enzyme complex. J. Biol. Chem. 1990, 265, 3809–3814. [Google Scholar] [CrossRef] [PubMed]
- Wiedmer, T.; Esmon, C.T.; Sims, P.J. Complement proteins c5b-9 stimulate procoagulant activity through platelet prothrombinase. Blood 1986, 68, 875–880. [Google Scholar] [CrossRef] [PubMed]
- Amara, U.; Flierl, M.A.; Rittirsch, D.; Klos, A.; Chen, H.; Acker, B.; Bruckner, U.B.; Nilsson, B.; Gebhard, F.; Lambris, J.D.; et al. Molecular intercommunication between the complement and coagulation systems. J. Immunol. 2010, 185, 5628–5636. [Google Scholar] [CrossRef] [PubMed]
- Huber-Lang, M.; Sarma, J.V.; Zetoune, F.S.; Rittirsch, D.; Neff, T.A.; McGuire, S.R.; Lambris, J.D.; Warner, R.L.; Flierl, M.A.; Hoesel, L.M.; et al. Generation of c5a in the absence of c3: A new complement activation pathway. Nat. Med. 2006, 12, 682–687. [Google Scholar] [CrossRef] [PubMed]
- Krisinger, M.J.; Goebeler, V.; Lu, Z.; Meixner, S.C.; Myles, T.; Pryzdial, E.L.; Conway, E.M. Thrombin generates previously unidentified c5 products that support the terminal complement activation pathway. Blood 2012, 120, 1717–1725. [Google Scholar] [CrossRef] [PubMed]
- Polley, M.J.; Nachman, R. The human complement system in thrombin-mediated platelet function. J. Exp. Med. 1978, 147, 1713–1726. [Google Scholar] [CrossRef]
- Polley, M.J.; Nachman, R.L. Human complement in thrombin-mediated platelet function: Uptake of the c5b-9 complex. J. Exp. Med. 1979, 150, 633–645. [Google Scholar] [CrossRef]
- Dobo, J.; Szakacs, D.; Oroszlan, G.; Kortvely, E.; Kiss, B.; Boros, E.; Szasz, R.; Zavodszky, P.; Gal, P.; Pal, G. Masp-3 is the exclusive pro-factor d activator in resting blood: The lectin and the alternative complement pathways are fundamentally linked. Sci. Rep. 2016, 6, 31877. [Google Scholar] [CrossRef]
- Oroszlan, G.; Kortvely, E.; Szakacs, D.; Kocsis, A.; Dammeier, S.; Zeck, A.; Ueffing, M.; Zavodszky, P.; Pal, G.; Gal, P.; et al. Masp-1 and masp-2 do not activate pro-factor d in resting human blood, whereas masp-3 is a potential activator: Kinetic analysis involving specific masp-1 and masp-2 inhibitors. J. Immunol. 2016, 196, 857–865. [Google Scholar] [CrossRef]
- Lidington, E.A.; Haskard, D.O.; Mason, J.C. Induction of decay-accelerating factor by thrombin through a protease-activated receptor 1 and protein kinase c-dependent pathway protects vascular endothelial cells from complement-mediated injury. Blood 2000, 96, 2784–2792. [Google Scholar] [CrossRef]
- Foley, J.H.; Walton, B.L.; Aleman, M.M.; O’Byrne, A.M.; Lei, V.; Harrasser, M.; Foley, K.A.; Wolberg, A.S.; Conway, E.M. Complement activation in arterial and venous thrombosis is mediated by plasmin. eBioMedicine 2016, 5, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Ward, P.A. A plasmin-split fragment of c′3 as a new chemotactic factor. J. Exp. Med. 1967, 126, 189–206. [Google Scholar] [CrossRef] [PubMed]
- Mannes, M.; Dopler, A.; Zolk, O.; Lang, S.J.; Halbgebauer, R.; Hochsmann, B.; Skerra, A.; Braun, C.K.; Huber-Lang, M.; Schrezenmeier, H.; et al. Complement inhibition at the level of c3 or c5: Mechanistic reasons for ongoing terminal pathway activity. Blood 2021, 137, 443–455. [Google Scholar] [CrossRef] [PubMed]
- Wetsel, R.A.; Kolb, W.P. Expression of c5a-like biological activities by the fifth component of human complement (c5) upon limited digestion with noncomplement enzymes without release of polypeptide fragments. J. Exp. Med. 1983, 157, 2029–2048. [Google Scholar] [CrossRef] [PubMed]
- DiScipio, R.G. The activation of the alternative pathway c3 convertase by human plasma kallikrein. Immunology 1982, 45, 587–595. [Google Scholar] [PubMed]
- Saito, A. Plasma kallikrein is activated on dermatan sulfate and cleaves factor h. Biochem. Biophys. Res. Commun. 2008, 370, 646–650. [Google Scholar] [CrossRef]
- Ellis, V.; Scully, M.; MacGregor, I.; Kakkar, V. Inhibition of human factor xa by various plasma protease inhibitors. Biochim. Biophys. Acta 1982, 701, 24–31. [Google Scholar] [CrossRef]
- Osterud, B.; Miller-Andersson, M.; Abildgaard, U.; Prydz, H. The effect of antithrombin iii on the activity of the coagulation factors vii, ix and x. Thromb. Haemost. 1976, 35, 295–304. [Google Scholar] [CrossRef]
- Parej, K.; Dobo, J.; Zavodszky, P.; Gal, P. The control of the complement lectin pathway activation revisited: Both c1-inhibitor and antithrombin are likely physiological inhibitors, while alpha2-macroglobulin is not. Mol. Immunol. 2013, 54, 415–422. [Google Scholar] [CrossRef]
- Ziccardi, R.J. Activation of the early components of the classical complement pathway under physiologic conditions. J. Immunol. 1981, 126, 1769–1773. [Google Scholar] [CrossRef]
- Rossi, V.; Cseh, S.; Bally, I.; Thielens, N.M.; Jensenius, J.C.; Arlaud, G.J. Substrate specificities of recombinant mannan-binding lectin-associated serine proteases-1 and -2. J. Biol. Chem. 2001, 276, 40880–40887. [Google Scholar] [CrossRef] [PubMed]
- Kerr, F.K.; Thomas, A.R.; Wijeyewickrema, L.C.; Whisstock, J.C.; Boyd, S.E.; Kaiserman, D.; Matthews, A.Y.; Bird, P.I.; Thielens, N.M.; Rossi, V.; et al. Elucidation of the substrate specificity of the masp-2 protease of the lectin complement pathway and identification of the enzyme as a major physiological target of the serpin, c1-inhibitor. Mol. Immunol. 2008, 45, 670–677. [Google Scholar] [CrossRef] [PubMed]
- Ratnoff, O.D. Some relationships among hemostasis, fibrinolytic phenomena, immunity, and the inflammatory response. Adv. Immunol. 1969, 10, 145–227. [Google Scholar] [CrossRef] [PubMed]
- Maroney, S.A.; Ellery, P.E.; Mast, A.E. Alternatively spliced isoforms of tissue factor pathway inhibitor. Thromb. Res. 2010, 125 (Suppl. 1), S52–S56. [Google Scholar] [CrossRef]
- Mast, A.E. Tissue factor pathway inhibitor: Multiple anticoagulant activities for a single protein. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Keizer, M.P.; Pouw, R.B.; Kamp, A.M.; Patiwael, S.; Marsman, G.; Hart, M.H.; Zeerleder, S.; Kuijpers, T.W.; Wouters, D. Tfpi inhibits lectin pathway of complement activation by direct interaction with masp-2. Eur. J. Immunol. 2015, 45, 544–550. [Google Scholar] [CrossRef] [PubMed]
- Puy, C.; Pang, J.; Reitsma, S.E.; Lorentz, C.U.; Tucker, E.I.; Gailani, D.; Gruber, A.; Lupu, F.; McCarty, O.J.T. Cross-talk between the complement pathway and the contact activation system of coagulation: Activated factor xi neutralizes complement factor h. J. Immunol. 2021, 206, 1784–1792. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.J.; Liu, D.T.; Tam, P.O.; Chan, W.M.; Liu, K.; Chong, K.K.; Lam, D.S.; Pang, C.P. Association of complement factor h polymorphisms with exudative age-related macular degeneration. Mol. Vis. 2006, 12, 1536–1542. [Google Scholar] [CrossRef]
- Thangaraj, S.S.; Christiansen, S.H.; Graversen, J.H.; Sidelmann, J.J.; Hansen, S.W.K.; Bygum, A.; Gram, J.B.; Palarasah, Y. Contact activation-induced complex formation between complement factor h and coagulation factor xiia. J. Thromb. Haemost. JTH 2020, 18, 876–884. [Google Scholar] [CrossRef]
- Feng, S.; Liang, X.; Cruz, M.A.; Vu, H.; Zhou, Z.; Pemmaraju, N.; Dong, J.F.; Kroll, M.H.; Afshar-Kharghan, V. The interaction between factor h and von willebrand factor. PLoS ONE 2013, 8, e73715. [Google Scholar] [CrossRef]
- Nolasco, L.; Nolasco, J.; Feng, S.; Afshar-Kharghan, V.; Moake, J. Human complement factor h is a reductase for large soluble von willebrand factor multimers—Brief report. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 2524–2528. [Google Scholar] [CrossRef] [PubMed]
- Rayes, J.; Roumenina, L.T.; Dimitrov, J.D.; Repesse, Y.; Ing, M.; Christophe, O.; Jokiranta, T.S.; Halbwachs-Mecarelli, L.; Borel-Derlon, A.; Kaveri, S.V.; et al. The interaction between factor h and vwf increases factor h cofactor activity and regulates vwf prothrombotic status. Blood 2014, 123, 121–125. [Google Scholar] [CrossRef] [PubMed]
- Turner, N.; Nolasco, L.; Nolasco, J.; Sartain, S.; Moake, J. Thrombotic microangiopathies and the linkage between von willebrand factor and the alternative complement pathway. Semin. Thromb. Hemost. 2014, 40, 544–550. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.; Liang, X.; Kroll, M.H.; Chung, D.W.; Afshar-Kharghan, V. Von willebrand factor is a cofactor in complement regulation. Blood 2015, 125, 1034–1037. [Google Scholar] [CrossRef] [PubMed]
- Bajzar, L.; Manuel, R.; Nesheim, M.E. Purification and characterization of tafi, a thrombin-activable fibrinolysis inhibitor. J. Biol. Chem. 1995, 270, 14477–14484. [Google Scholar] [CrossRef] [PubMed]
- Campbell, W.D.; Lazoura, E.; Okada, N.; Okada, H. Inactivation of c3a and c5a octapeptides by carboxypeptidase r and carboxypeptidase n. Microbiol. Immunol. 2002, 46, 131–134. [Google Scholar] [CrossRef] [PubMed]
- Delvaeye, M.; Noris, M.; De Vriese, A.; Esmon, C.T.; Esmon, N.L.; Ferrell, G.; Del-Favero, J.; Plaisance, S.; Claes, B.; Lambrechts, D.; et al. Thrombomodulin mutations in atypical hemolytic-uremic syndrome. N. Engl. J. Med. 2009, 361, 345–357. [Google Scholar] [CrossRef]
- Heurich, M.; Preston, R.J.; O’Donnell, V.B.; Morgan, B.P.; Collins, P.W. Thrombomodulin enhances complement regulation through strong affinity interactions with factor h and c3b-factor h complex. Thromb. Res. 2016, 145, 84–92. [Google Scholar] [CrossRef]
- Tateishi, K.; Imaoka, M.; Matsushita, M. Dual modulating functions of thrombomodulin in the alternative complement pathway. Biosci. Trends 2016, 10, 231–234. [Google Scholar] [CrossRef]
- Bu, F.; Maga, T.; Meyer, N.C.; Wang, K.; Thomas, C.P.; Nester, C.M.; Smith, R.J. Comprehensive genetic analysis of complement and coagulation genes in atypical hemolytic uremic syndrome. J. Am. Soc. Nephrol. JASN 2014, 25, 55–64. [Google Scholar] [CrossRef]
- Majowicz, S.E.; Scallan, E.; Jones-Bitton, A.; Sargeant, J.M.; Stapleton, J.; Angulo, F.J.; Yeung, D.H.; Kirk, M.D. Global incidence of human shiga toxin-producing Escherichia coli infections and deaths: A systematic review and knowledge synthesis. Foodborne Pathog. Dis. 2014, 11, 447–455. [Google Scholar] [CrossRef] [PubMed]
- Rivas, M.; Chinen, I.; Miliwebsky, E.; Masana, M. Risk factors for shiga toxin-producing escherichia coli-associated human diseases. Microbiol. Spectr. 2014, 2. [Google Scholar] [CrossRef] [PubMed]
- Karpman, D.; Loos, S.; Tati, R.; Arvidsson, I. Haemolytic uraemic syndrome. J. Intern. Med. 2017, 281, 123–148. [Google Scholar] [CrossRef] [PubMed]
- Bowen, E.E.; Coward, R.J. Advances in our understanding of the pathogenesis of hemolytic uremic syndromes. Am. J. Physiol. Ren. Physiol. 2018, 314, F454–F461. [Google Scholar] [CrossRef] [PubMed]
- Garg, A.X.; Suri, R.S.; Barrowman, N.; Rehman, F.; Matsell, D.; Rosas-Arellano, M.P.; Salvadori, M.; Haynes, R.B.; Clark, W.F. Long-term renal prognosis of diarrhea-associated hemolytic uremic syndrome: A systematic review, meta-analysis, and meta-regression. JAMA 2003, 290, 1360–1370. [Google Scholar] [CrossRef] [PubMed]
- Rosales, A.; Hofer, J.; Zimmerhackl, L.B.; Jungraithmayr, T.C.; Riedl, M.; Giner, T.; Strasak, A.; Orth-Holler, D.; Wurzner, R.; Karch, H.; et al. Need for long-term follow-up in enterohemorrhagic escherichia coli-associated hemolytic uremic syndrome due to late-emerging sequelae. Clin. Infect. Dis. 2012, 54, 1413–1421. [Google Scholar] [CrossRef] [PubMed]
- Frankel, G.; Phillips, A.D. Attaching effacing escherichia coli and paradigms of tir-triggered actin polymerization: Getting off the pedestal. Cell Microbiol. 2008, 10, 549–556. [Google Scholar] [CrossRef]
- Abreu, A.G.; Fraga, T.R.; Granados Martinez, A.P.; Kondo, M.Y.; Juliano, M.A.; Juliano, L.; Navarro-Garcia, F.; Isaac, L.; Barbosa, A.S.; Elias, W.P. The serine protease pic from enteroaggregative escherichia coli mediates immune evasion by the direct cleavage of complement proteins. J. Infect. Dis. 2015, 212, 106–115. [Google Scholar] [CrossRef]
- Ayala-Lujan, J.L.; Vijayakumar, V.; Gong, M.; Smith, R.; Santiago, A.E.; Ruiz-Perez, F. Broad spectrum activity of a lectin-like bacterial serine protease family on human leukocytes. PLoS ONE 2014, 9, e107920. [Google Scholar] [CrossRef]
- Orth, D.; Ehrlenbach, S.; Brockmeyer, J.; Khan, A.B.; Huber, G.; Karch, H.; Sarg, B.; Lindner, H.; Wurzner, R. Espp, a serine protease of enterohemorrhagic escherichia coli, impairs complement activation by cleaving complement factors c3/c3b and c5. Infect. Immun. 2010, 78, 4294–4301. [Google Scholar] [CrossRef]
- Weiss, A.; Joerss, H.; Brockmeyer, J. Structural and functional characterization of cleavage and inactivation of human serine protease inhibitors by the bacterial spate protease esppalpha from enterohemorrhagic E. coli. PLoS ONE 2014, 9, e111363. [Google Scholar] [CrossRef] [PubMed]
- Djafari, S.; Ebel, F.; Deibel, C.; Kramer, S.; Hudel, M.; Chakraborty, T. Characterization of an exported protease from shiga toxin-producing escherichia coli. Mol. Microbiol. 1997, 25, 771–784. [Google Scholar] [CrossRef] [PubMed]
- Lathem, W.W.; Bergsbaken, T.; Welch, R.A. Potentiation of c1 esterase inhibitor by stce, a metalloprotease secreted by escherichia coli o157:H7. J. Exp. Med. 2004, 199, 1077–1087. [Google Scholar] [CrossRef] [PubMed]
- Yu, A.C.; Worrall, L.J.; Strynadka, N.C. Structural insight into the bacterial mucinase stce essential to adhesion and immune evasion during enterohemorrhagic E. coli infection. Structure 2012, 20, 707–717. [Google Scholar] [CrossRef] [PubMed]
- Castuma, C.E.; Huang, R.; Kornberg, A.; Reusch, R.N. Inorganic polyphosphates in the acquisition of competence in Escherichia coli. J. Biol. Chem. 1995, 270, 12980–12983. [Google Scholar] [CrossRef] [PubMed]
- Wat, J.M.; Foley, J.H.; Krisinger, M.J.; Ocariza, L.M.; Lei, V.; Wasney, G.A.; Lameignere, E.; Strynadka, N.C.; Smith, S.A.; Morrissey, J.H.; et al. Polyphosphate suppresses complement via the terminal pathway. Blood 2014, 123, 768–776. [Google Scholar] [CrossRef]
- Stavrou, E.; Schmaier, A.H. Factor xii: What does it contribute to our understanding of the physiology and pathophysiology of hemostasis & thrombosis. Thromb. Res. 2010, 125, 210–215. [Google Scholar] [CrossRef]
- Kleinschnitz, C.; Stoll, G.; Bendszus, M.; Schuh, K.; Pauer, H.U.; Burfeind, P.; Renne, C.; Gailani, D.; Nieswandt, B.; Renne, T. Targeting coagulation factor xii provides protection from pathological thrombosis in cerebral ischemia without interfering with hemostasis. J. Exp. Med. 2006, 203, 513–518. [Google Scholar] [CrossRef]
- Renne, T.; Pozgajova, M.; Gruner, S.; Schuh, K.; Pauer, H.U.; Burfeind, P.; Gailani, D.; Nieswandt, B. Defective thrombus formation in mice lacking coagulation factor xii. J. Exp. Med. 2005, 202, 271–281. [Google Scholar] [CrossRef]
- Tarr, P.I.; Gordon, C.A.; Chandler, W.L. Shiga-toxin-producing escherichia coli and haemolytic uraemic syndrome. Lancet 2005, 365, 1073–1086. [Google Scholar] [CrossRef]
- Rutjes, N.W.; Binnington, B.A.; Smith, C.R.; Maloney, M.D.; Lingwood, C.A. Differential tissue targeting and pathogenesis of verotoxins 1 and 2 in the mouse animal model. Kidney Int. 2002, 62, 832–845. [Google Scholar] [CrossRef] [PubMed]
- Scheutz, F.; Teel, L.D.; Beutin, L.; Pierard, D.; Buvens, G.; Karch, H.; Mellmann, A.; Caprioli, A.; Tozzoli, R.; Morabito, S.; et al. Multicenter evaluation of a sequence-based protocol for subtyping shiga toxins and standardizing stx nomenclature. J. Clin. Microbiol. 2012, 50, 2951–2963. [Google Scholar] [CrossRef] [PubMed]
- Gallegos, K.M.; Conrady, D.G.; Karve, S.S.; Gunasekera, T.S.; Herr, A.B.; Weiss, A.A. Shiga toxin binding to glycolipids and glycans. PLoS ONE 2012, 7, e30368. [Google Scholar] [CrossRef] [PubMed]
- Clayton, F.; Pysher, T.J.; Lou, R.; Kohan, D.E.; Denkers, N.D.; Tesh, V.L.; Taylor, F.B., Jr.; Siegler, R.L. Lipopolysaccharide upregulates renal shiga toxin receptors in a primate model of hemolytic uremic syndrome. Am. J. Nephrol. 2005, 25, 536–540. [Google Scholar] [CrossRef] [PubMed]
- Louise, C.B.; Obrig, T.G. Shiga toxin-associated hemolytic uremic syndrome: Combined cytotoxic effects of shiga toxin and lipopolysaccharide (endotoxin) on human vascular endothelial cells in vitro. Infect. Immun. 1992, 60, 1536–1543. [Google Scholar] [CrossRef]
- Ikeda, M.; Ito, S.; Honda, M. Hemolytic uremic syndrome induced by lipopolysaccharide and shiga-like toxin. Pediatr. Nephrol. 2004, 19, 485–489. [Google Scholar] [CrossRef] [PubMed]
- Keepers, T.R.; Psotka, M.A.; Gross, L.K.; Obrig, T.G. A murine model of hus: Shiga toxin with lipopolysaccharide mimics the renal damage and physiologic response of human disease. J. Am. Soc. Nephrol. 2006, 17, 3404–3414. [Google Scholar] [CrossRef]
- Zanchi, C.; Zoja, C.; Morigi, M.; Valsecchi, F.; Liu, X.Y.; Rottoli, D.; Locatelli, M.; Buelli, S.; Pezzotta, A.; Mapelli, P.; et al. Fractalkine and cx3cr1 mediate leukocyte capture by endothelium in response to shiga toxin. J. Immunol. 2008, 181, 1460–1469. [Google Scholar] [CrossRef]
- O’Brien, A.D.; Tesh, V.L.; Donohue-Rolfe, A.; Jackson, M.P.; Olsnes, S.; Sandvig, K.; Lindberg, A.A.; Keusch, G.T. Shiga toxin: Biochemistry, genetics, mode of action, and role in pathogenesis. Curr. Top. Microbiol. Immunol. 1992, 180, 65–94. [Google Scholar] [CrossRef]
- Lingwood, C.A. Role of verotoxin receptors in pathogenesis. Trends Microbiol. 1996, 4, 147–153. [Google Scholar] [CrossRef]
- Sandvig, K.; Garred, O.; Prydz, K.; Kozlov, J.V.; Hansen, S.H.; van Deurs, B. Retrograde transport of endocytosed shiga toxin to the endoplasmic reticulum. Nature 1992, 358, 510–512. [Google Scholar] [CrossRef] [PubMed]
- Endo, Y.; Tsurugi, K.; Yutsudo, T.; Takeda, Y.; Ogasawara, T.; Igarashi, K. Site of action of a vero toxin (vt2) from escherichia coli o157:H7 and of shiga toxin on eukaryotic ribosomes. Rna n-glycosidase activity of the toxins. Eur. J. Biochem. 1988, 171, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.S.; Koo, S.; Jeong, D.G.; Tesh, V.L. Shiga toxins as multi-functional proteins: Induction of host cellular stress responses, role in pathogenesis and therapeutic applications. Toxins 2016, 8, 77. [Google Scholar] [CrossRef]
- Petruzziello-Pellegrini, T.N.; Moslemi-Naeini, M.; Marsden, P.A. New insights into shiga toxin-mediated endothelial dysfunction in hemolytic uremic syndrome. Virulence 2013, 4, 556–563. [Google Scholar] [CrossRef] [PubMed]
- Tesh, V.L. Activation of cell stress response pathways by shiga toxins. Cell Microbiol. 2012, 14, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Zoja, C.; Buelli, S.; Morigi, M. Shiga toxin-associated hemolytic uremic syndrome: Pathophysiology of endothelial dysfunction. Pediatr. Nephrol. 2010, 25, 2231–2240. [Google Scholar] [CrossRef] [PubMed]
- Brigotti, M.; Tazzari, P.L.; Ravanelli, E.; Carnicelli, D.; Rocchi, L.; Arfilli, V.; Scavia, G.; Minelli, F.; Ricci, F.; Pagliaro, P.; et al. Clinical relevance of shiga toxin concentrations in the blood of patients with hemolytic uremic syndrome. Pediatr. Infect. Dis. J. 2011, 30, 486–490. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Ardissino, G.; Patfield, S.; Cheng, L.W.; Silva, C.J.; Brigotti, M. An improved method for the sensitive detection of shiga toxin 2 in human serum. Toxins 2018, 10, 59. [Google Scholar] [CrossRef]
- Obrig, T.G.; Karpman, D. Shiga toxin pathogenesis: Kidney complications and renal failure. Curr. Top. Microbiol. Immunol. 2012, 357, 105–136. [Google Scholar] [CrossRef]
- Brigotti, M.; Carnicelli, D.; Arfilli, V.; Tamassia, N.; Borsetti, F.; Fabbri, E.; Tazzari, P.L.; Ricci, F.; Pagliaro, P.; Spisni, E.; et al. Identification of tlr4 as the receptor that recognizes shiga toxins in human neutrophils. J. Immunol. 2013, 191, 4748–4758. [Google Scholar] [CrossRef]
- Stahl, A.L.; Arvidsson, I.; Johansson, K.E.; Chromek, M.; Rebetz, J.; Loos, S.; Kristoffersson, A.C.; Bekassy, Z.D.; Morgelin, M.; Karpman, D. A novel mechanism of bacterial toxin transfer within host blood cell-derived microvesicles. PLoS Pathog. 2015, 11, e1004619. [Google Scholar] [CrossRef] [PubMed]
- Villysson, A.; Tontanahal, A.; Karpman, D. Microvesicle involvement in shiga toxin-associated infection. Toxins 2017, 9, 376. [Google Scholar] [CrossRef] [PubMed]
- Matussek, A.; Lauber, J.; Bergau, A.; Hansen, W.; Rohde, M.; Dittmar, K.E.; Gunzer, M.; Mengel, M.; Gatzlaff, P.; Hartmann, M.; et al. Molecular and functional analysis of shiga toxin-induced response patterns in human vascular endothelial cells. Blood 2003, 102, 1323–1332. [Google Scholar] [CrossRef]
- Morigi, M.; Micheletti, G.; Figliuzzi, M.; Imberti, B.; Karmali, M.A.; Remuzzi, A.; Remuzzi, G.; Zoja, C. Verotoxin-1 promotes leukocyte adhesion to cultured endothelial cells under physiologic flow conditions. Blood 1995, 86, 4553–4558. [Google Scholar] [CrossRef] [PubMed]
- Zoja, C.; Angioletti, S.; Donadelli, R.; Zanchi, C.; Tomasoni, S.; Binda, E.; Imberti, B.; te Loo, M.; Monnens, L.; Remuzzi, G.; et al. Shiga toxin-2 triggers endothelial leukocyte adhesion and transmigration via nf-kappab dependent up-regulation of il-8 and mcp-1. Kidney Int. 2002, 62, 846–856. [Google Scholar] [CrossRef] [PubMed]
- Morigi, M.; Galbusera, M.; Binda, E.; Imberti, B.; Gastoldi, S.; Remuzzi, A.; Zoja, C.; Remuzzi, G. Verotoxin-1-induced up-regulation of adhesive molecules renders microvascular endothelial cells thrombogenic at high shear stress. Blood 2001, 98, 1828–1835. [Google Scholar] [CrossRef] [PubMed]
- Lo, N.C.; Turner, N.A.; Cruz, M.A.; Moake, J. Interaction of shiga toxin with the a-domains and multimers of von willebrand factor. J. Biol. Chem. 2013, 288, 33118–33123. [Google Scholar] [CrossRef] [PubMed]
- Dettmar, A.K.; Binder, E.; Greiner, F.R.; Liebau, M.C.; Kurschat, C.E.; Jungraithmayr, T.C.; Saleem, M.A.; Schmitt, C.P.; Feifel, E.; Orth-Holler, D.; et al. Protection of human podocytes from shiga toxin 2-induced phosphorylation of mitogen-activated protein kinases and apoptosis by human serum amyloid p component. Infect. Immun. 2014, 82, 1872–1879. [Google Scholar] [CrossRef]
- Ergonul, Z.; Clayton, F.; Fogo, A.B.; Kohan, D.E. Shigatoxin-1 binding and receptor expression in human kidneys do not change with age. Pediatr. Nephrol. 2003, 18, 246–253. [Google Scholar] [CrossRef]
- Hughes, A.K.; Stricklett, P.K.; Schmid, D.; Kohan, D.E. Cytotoxic effect of shiga toxin-1 on human glomerular epithelial cells. Kidney Int. 2000, 57, 2350–2359. [Google Scholar] [CrossRef]
- Morigi, M.; Buelli, S.; Zanchi, C.; Longaretti, L.; Macconi, D.; Benigni, A.; Moioli, D.; Remuzzi, G.; Zoja, C. Shigatoxin-induced endothelin-1 expression in cultured podocytes autocrinally mediates actin remodeling. Am. J. Pathol. 2006, 169, 1965–1975. [Google Scholar] [CrossRef] [PubMed]
- Hughes, A.K.; Stricklett, P.K.; Kohan, D.E. Shiga toxin-1 regulation of cytokine production by human glomerular epithelial cells. Nephron 2001, 88, 14–23. [Google Scholar] [CrossRef]
- Keir, L.S.; Saleem, M.A. Current evidence for the role of complement in the pathogenesis of shiga toxin haemolytic uraemic syndrome. Pediatr. Nephrol. 2014, 29, 1895–1902. [Google Scholar] [CrossRef] [PubMed]
- Mele, C.; Remuzzi, G.; Noris, M. Hemolytic uremic syndrome. Semin. Immunopathol. 2014, 36, 399–420. [Google Scholar] [CrossRef] [PubMed]
- Orth-Holler, D.; Wurzner, R. Role of complement in enterohemorrhagic escherichia coli-induced hemolytic uremic syndrome. Semin. Thromb. Hemost. 2014, 40, 503–507. [Google Scholar] [CrossRef]
- Zoja, C.; Buelli, S.; Morigi, M. Shiga toxin triggers endothelial and podocyte injury: The role of complement activation. Pediatr. Nephrol. 2019, 34, 379–388. [Google Scholar] [CrossRef] [PubMed]
- Koster, F.T.; Boonpucknavig, V.; Sujaho, S.; Gilman, R.H.; Rahaman, M.M. Renal histopathology in the hemolytic-uremic syndrome following shigellosis. Clin. Nephrol. 1984, 21, 126–133. [Google Scholar]
- Monnens, L.; Molenaar, J.; Lambert, P.H.; Proesmans, W.; van Munster, P. The complement system in hemolytic-uremic syndrome in childhood. Clin. Nephrol. 1980, 13, 168–171. [Google Scholar]
- Robson, W.L.; Leung, A.K.; Fick, G.H.; McKenna, A.I. Hypocomplementemia and leukocytosis in diarrhea-associated hemolytic uremic syndrome. Nephron 1992, 62, 296–299. [Google Scholar] [CrossRef]
- Stahl, A.L.; Sartz, L.; Karpman, D. Complement activation on platelet-leukocyte complexes and microparticles in enterohemorrhagic escherichia coli-induced hemolytic uremic syndrome. Blood 2011, 117, 5503–5513. [Google Scholar] [CrossRef]
- Thurman, J.M.; Marians, R.; Emlen, W.; Wood, S.; Smith, C.; Akana, H.; Holers, V.M.; Lesser, M.; Kline, M.; Hoffman, C.; et al. Alternative pathway of complement in children with diarrhea-associated hemolytic uremic syndrome. Clin. J. Am. Soc. Nephrol. CJASN 2009, 4, 1920–1924. [Google Scholar] [CrossRef] [PubMed]
- Ferraris, J.R.; Ferraris, V.; Acquier, A.B.; Sorroche, P.B.; Saez, M.S.; Ginaca, A.; Mendez, C.F. Activation of the alternative pathway of complement during the acute phase of typical haemolytic uraemic syndrome. Clin. Exp. Immunol. 2015, 181, 118–125. [Google Scholar] [CrossRef] [PubMed]
- Arvidsson, I.; Rebetz, J.; Loos, S.; Herthelius, M.; Kristoffersson, A.C.; Englund, E.; Chromek, M.; Karpman, D. Early terminal complement blockade and c6 deficiency are protective in enterohemorrhagic escherichia coli-infected mice. J. Immunol. 2016, 197, 1276–1286. [Google Scholar] [CrossRef] [PubMed]
- Orth, D.; Khan, A.B.; Naim, A.; Grif, K.; Brockmeyer, J.; Karch, H.; Joannidis, M.; Clark, S.J.; Day, A.J.; Fidanzi, S.; et al. Shiga toxin activates complement and binds factor h: Evidence for an active role of complement in hemolytic uremic syndrome. J. Immunol. 2009, 182, 6394–6400. [Google Scholar] [CrossRef] [PubMed]
- Poolpol, K.; Orth-Holler, D.; Speth, C.; Zipfel, P.F.; Skerka, C.; de Cordoba, S.R.; Brockmeyer, J.; Bielaszewska, M.; Wurzner, R. Interaction of shiga toxin 2 with complement regulators of the factor h protein family. Mol. Immunol. 2014, 58, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Ehrlenbach, S.; Rosales, A.; Posch, W.; Wilflingseder, D.; Hermann, M.; Brockmeyer, J.; Karch, H.; Satchell, S.C.; Wurzner, R.; Orth-Holler, D. Shiga toxin 2 reduces complement inhibitor cd59 expression on human renal tubular epithelial and glomerular endothelial cells. Infect. Immun. 2013, 81, 2678–2685. [Google Scholar] [CrossRef]
- Iwaki, D.; Kanno, K.; Takahashi, M.; Endo, Y.; Matsushita, M.; Fujita, T. The role of mannose-binding lectin-associated serine protease-3 in activation of the alternative complement pathway. J. Immunol. 2011, 187, 3751–3758. [Google Scholar] [CrossRef]
- Ozaki, M.; Kang, Y.; Tan, Y.S.; Pavlov, V.I.; Liu, B.; Boyle, D.C.; Kushak, R.I.; Skjoedt, M.O.; Grabowski, E.F.; Taira, Y.; et al. Human mannose-binding lectin inhibitor prevents shiga toxin-induced renal injury. Kidney Int. 2016, 90, 774–782. [Google Scholar] [CrossRef]
- Geelen, J.; van den Biggelaar, M.; Linssen, P.; van der Velden, T.; Mertens, K.; Monnens, L. The effect of shiga toxin on weibel-palade bodies in primary human endothelial cells. Nephron Extra 2014, 4, 101–107. [Google Scholar] [CrossRef]
- Morigi, M.; Galbusera, M.; Gastoldi, S.; Locatelli, M.; Buelli, S.; Pezzotta, A.; Pagani, C.; Noris, M.; Gobbi, M.; Stravalaci, M.; et al. Alternative pathway activation of complement by shiga toxin promotes exuberant c3a formation that triggers microvascular thrombosis. J. Immunol. 2011, 187, 172–180. [Google Scholar] [CrossRef]
- Del Conde, I.; Cruz, M.A.; Zhang, H.; Lopez, J.A.; Afshar-Kharghan, V. Platelet activation leads to activation and propagation of the complement system. J. Exp. Med. 2005, 201, 871–879. [Google Scholar] [CrossRef]
- Nolasco, L.H.; Turner, N.A.; Bernardo, A.; Tao, Z.; Cleary, T.G.; Dong, J.F.; Moake, J.L. Hemolytic uremic syndrome-associated shiga toxins promote endothelial-cell secretion and impair adamts13 cleavage of unusually large von willebrand factor multimers. Blood 2005, 106, 4199–4209. [Google Scholar] [CrossRef]
- Grabowski, E.F.; Kushak, R.I.; Liu, B.; Ingelfinger, J.R. Shiga toxin downregulates tissue factor pathway inhibitor, modulating an increase in the expression of functional tissue factor on endothelium. Thromb. Res. 2013, 131, 521–528. [Google Scholar] [CrossRef] [PubMed]
- Arvidsson, I.; Stahl, A.L.; Hedstrom, M.M.; Kristoffersson, A.C.; Rylander, C.; Westman, J.S.; Storry, J.R.; Olsson, M.L.; Karpman, D. Shiga toxin-induced complement-mediated hemolysis and release of complement-coated red blood cell-derived microvesicles in hemolytic uremic syndrome. J. Immunol. 2015, 194, 2309–2318. [Google Scholar] [CrossRef] [PubMed]
- Sims, P.J.; Faioni, E.M.; Wiedmer, T.; Shattil, S.J. Complement proteins c5b-9 cause release of membrane vesicles from the platelet surface that are enriched in the membrane receptor for coagulation factor va and express prothrombinase activity. J. Biol. Chem. 1988, 263, 18205–18212. [Google Scholar] [CrossRef] [PubMed]
- Stahl, A.L.; Sartz, L.; Nelsson, A.; Bekassy, Z.D.; Karpman, D. Shiga toxin and lipopolysaccharide induce platelet-leukocyte aggregates and tissue factor release, a thrombotic mechanism in hemolytic uremic syndrome. PLoS ONE 2009, 4, e6990. [Google Scholar] [CrossRef] [PubMed]
- Locatelli, M.; Buelli, S.; Pezzotta, A.; Corna, D.; Perico, L.; Tomasoni, S.; Rottoli, D.; Rizzo, P.; Conti, D.; Thurman, J.M.; et al. Shiga toxin promotes podocyte injury in experimental hemolytic uremic syndrome via activation of the alternative pathway of complement. J. Am. Soc. Nephrol. JASN 2014, 25, 1786–1798. [Google Scholar] [CrossRef] [PubMed]
- Qu, L.; Jiao, B. The interplay between immune and metabolic pathways in kidney disease. Cells 2023, 12, 1584. [Google Scholar] [CrossRef]
- Chi, H. Immunometabolism at the intersection of metabolic signaling, cell fate, and systems immunology. Cell Mol. Immunol. 2022, 19, 299–302. [Google Scholar] [CrossRef]
- Ding, S.; Xu, S.; Ma, Y.; Liu, G.; Jang, H.; Fang, J. Modulatory mechanisms of the nlrp3 inflammasomes in diabetes. Biomolecules 2019, 9, 850. [Google Scholar] [CrossRef]
- Shimizu, M. Pathogenic functions and diagnostic utility of cytokines/chemokines in ehec-hus. Pediatr. Int. 2020, 62, 308–315. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.E.; Kim, J.S.; Choi, I.H.; Tagawa, M.; Kohsaka, T.; Jin, D.K. Cytokine expression in the renal tubular epithelial cells stimulated by shiga toxin 2 of escherichia coli o157:H7. Ren. Fail. 2002, 24, 567–575. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Xiao, Y.; Li, X.; Huang, Y.; Meng, G.; Ren, Z. Activation of the nlrp3 inflammasome contributes to shiga toxin-induced hemolytic uremic syndrome in a mouse model. Front. Immunol. 2020, 11, 619096. [Google Scholar] [CrossRef] [PubMed]
- Asgari, E.; Le Friec, G.; Yamamoto, H.; Perucha, E.; Sacks, S.S.; Kohl, J.; Cook, H.T.; Kemper, C. C3a modulates il-1beta secretion in human monocytes by regulating atp efflux and subsequent nlrp3 inflammasome activation. Blood 2013, 122, 3473–3481. [Google Scholar] [CrossRef] [PubMed]
- Laudisi, F.; Spreafico, R.; Evrard, M.; Hughes, T.R.; Mandriani, B.; Kandasamy, M.; Morgan, B.P.; Sivasankar, B.; Mortellaro, A. Cutting edge: The nlrp3 inflammasome links complement-mediated inflammation and il-1beta release. J. Immunol. 2013, 191, 1006–1010. [Google Scholar] [CrossRef] [PubMed]
- Triantafilou, K.; Hughes, T.R.; Triantafilou, M.; Morgan, B.P. The complement membrane attack complex triggers intracellular Ca2+ fluxes leading to nlrp3 inflammasome activation. J. Cell Sci. 2013, 126, 2903–2913. [Google Scholar] [CrossRef]
- Butler, T. Haemolytic uraemic syndrome during shigellosis. Trans. R. Soc. Trop. Med. Hyg. 2012, 106, 395–399. [Google Scholar] [CrossRef]
- Al-Qarawi, S.; Fontaine, R.E.; Al-Qahtani, M.S. An outbreak of hemolytic uremic syndrome associated with antibiotic treatment of hospital inpatients for dysentery. Emerg. Infect. Dis. 1995, 1, 138–140. [Google Scholar] [CrossRef]
- Bin Saeed, A.A.; El Bushra, H.E.; Al-Hamdan, N.A. Does treatment of bloody diarrhea due to Shigella dysenteriae type 1 with ampicillin precipitate hemolytic uremic syndrome? Emerg. Infect. Dis. 1995, 1, 134–137. [Google Scholar] [CrossRef]
- Bloom, P.D.; MacPhail, A.P.; Klugman, K.; Louw, M.; Raubenheimer, C.; Fischer, C. Haemolytic-uraemic syndrome in adults with resistant Shigella dysenteriae type i. Lancet 1994, 344, 206. [Google Scholar] [CrossRef]
- Houdouin, V.; Doit, C.; Mariani, P.; Brahimi, N.; Loirat, C.; Bourrillon, A.; Bingen, E. A pediatric cluster of Shigella dysenteriae serotype 1 diarrhea with hemolytic uremic syndrome in 2 families from france. Clin. Infect. Dis. 2004, 38, e96–e99. [Google Scholar] [CrossRef] [PubMed]
- Oneko, M.; Nyathi, M.N.; Doehring, E. Post-dysenteric hemolytic uremic syndrome in bulawayo, zimbabwe. Pediatr. Nephrol. 2001, 16, 1142–1145. [Google Scholar] [CrossRef] [PubMed]
- Parsonnet, J.; Greene, K.D.; Gerber, A.R.; Tauxe, R.V.; Vallejo Aguilar, O.J.; Blake, P.A. Shigella dysenteriae type 1 infections in us travellers to mexico, 1988. Lancet 1989, 2, 543–545. [Google Scholar] [CrossRef] [PubMed]
- Rollins, N.C.; Wittenberg, D.F.; Coovadia, H.M.; Pillay, D.G.; Karas, A.J.; Sturm, A.W. Epidemic Shigella dysenteriae type 1 in natal. J. Trop. Pediatr. 1995, 41, 281–284. [Google Scholar] [CrossRef] [PubMed]
- Bennish, M.L.; Khan, W.A.; Begum, M.; Bridges, E.A.; Ahmed, S.; Saha, D.; Salam, M.A.; Acheson, D.; Ryan, E.T. Low risk of hemolytic uremic syndrome after early effective antimicrobial therapy for Shigella dysenteriae type 1 infection in bangladesh. Clin. Infect. Dis. 2006, 42, 356–362. [Google Scholar] [CrossRef] [PubMed]
- Tzipori, S.; Sheoran, A.; Akiyoshi, D.; Donohue-Rolfe, A.; Trachtman, H. Antibody therapy in the management of shiga toxin-induced hemolytic uremic syndrome. Clin. Microbiol. Rev. 2004, 17, 926–941. [Google Scholar] [CrossRef] [PubMed]
- Koster, F.; Levin, J.; Walker, L.; Tung, K.S.; Gilman, R.H.; Rahaman, M.M.; Majid, M.A.; Islam, S.; Williams, R.C., Jr. Hemolytic-uremic syndrome after shigellosis. Relation to endotoxemia and circulating immune complexes. N. Engl. J. Med. 1978, 298, 927–933. [Google Scholar] [CrossRef]
- Butler, T.; Rahman, H.; Al-Mahmud, K.A.; Islam, M.; Bardhan, P.; Kabir, I.; Rahman, M.M. An animal model of haemolytic--uraemic syndrome in shigellosis: Lipopolysaccharides of Shigella dysenteriae i and S. flexneri produce leucocyte-mediated renal cortical necrosis in rabbits. Br. J. Exp. Pathol. 1985, 66, 7–15. [Google Scholar]
- Coats, M.T.; Murphy, T.; Paton, J.C.; Gray, B.; Briles, D.E. Exposure of thomsen-friedenreich antigen in Streptococcus pneumoniae infection is dependent on pneumococcal neuraminidase a. Microb. Pathog. 2011, 50, 343–349. [Google Scholar] [CrossRef]
- Vaith, P.; Uhlenbruck, G. The thomsen agglutination phenomenon: A discovery revisited 50 years later. Z. Immunitatsforschung. Immunobiol. 1978, 154, 1–15. [Google Scholar] [CrossRef]
- Copelovitch, L.; Kaplan, B.S. Streptococcus pneumoniae-associated hemolytic uremic syndrome. Pediatr. Nephrol. 2008, 23, 1951–1956. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.T.; Chi, H.; Lee, H.C.; Chiu, N.C.; Huang, F.Y. T-antigen activation for prediction of pneumococcus-induced hemolytic uremic syndrome and hemolytic anemia. Pediatr. Infect. Dis. J. 2006, 25, 608–610. [Google Scholar] [CrossRef] [PubMed]
- Gomez Delgado, I.; Corvillo, F.; Nozal, P.; Arjona, E.; Madrid, A.; Melgosa, M.; Bravo, J.; Szilagyi, A.; Csuka, D.; Veszeli, N.; et al. Complement genetic variants and fh desialylation in s. Pneumoniae-haemolytic uraemic syndrome. Front. Immunol. 2021, 12, 641656. [Google Scholar] [CrossRef] [PubMed]
- Poschmann, A.; Fischer, K.; Grundmann, A.; Vongjirad, A. Neuraminidase induced hemolytic anemia. Experimental and clinical observations (author’s transl). Monatsschrift Kinderheilkd. 1976, 124, 15–24. [Google Scholar]
- Rose, P.E.; Armour, J.A.; Williams, C.E.; Hill, F.G. Verotoxin and neuraminidase induced platelet aggregating activity in plasma: Their possible role in the pathogenesis of the haemolytic uraemic syndrome. J. Clin. Pathol. 1985, 38, 438–441. [Google Scholar] [CrossRef]
- Gilbert, R.D.; Nagra, A.; Haq, M.R. Does dysregulated complement activation contribute to haemolytic uraemic syndrome secondary to streptococcus pneumoniae? Med. Hypotheses 2013, 81, 400–403. [Google Scholar] [CrossRef]
- Johnson, S.; Waters, A. Is complement a culprit in infection-induced forms of haemolytic uraemic syndrome? Immunobiology 2012, 217, 235–243. [Google Scholar] [CrossRef]
- Martinez, J.; MacDonald, K.A.; Palascak, J.E. The role of sialic acid in the dysfibrinogenemia associated with liver disease: Distribution of sialic acid on the constituent chains. Blood 1983, 61, 1196–1202. [Google Scholar] [CrossRef]
- Martinez, J.; Palascak, J.; Peters, C. Functional and metabolic properties of human asialofibrinogen. J. Lab. Clin. Med. 1977, 89, 367–377. [Google Scholar]
- Voos, K.M.; Cao, W.; Arce, N.A.; Legan, E.R.; Wang, Y.; Shajahan, A.; Azadi, P.; Lollar, P.; Zhang, X.F.; Li, R. Desialylation of o-glycans activates von willebrand factor by destabilizing its autoinhibitory module. J. Thromb. Haemost. JTH 2022, 20, 196–207. [Google Scholar] [CrossRef]
- Sim, D.S.; Mallari, C.R.; Teare, J.M.; Feldman, R.I.; Bauzon, M.; Hermiston, T.W. In vitro characterization of ct-001-a short-acting factor viia with enhanced prohemostatic activity. Res. Pract. Thromb. Haemost. 2021, 5, e12530. [Google Scholar] [CrossRef] [PubMed]
- Chavin, S.I.; Weidner, S.M. Blood clotting factor ix. Loss of activity after cleavage of sialic acid residues. J. Biol. Chem. 1984, 259, 3387–3390. [Google Scholar] [CrossRef] [PubMed]
- Henriksen, A.; Christensen, T.B.; Helgeland, L. On the significance of the carbohydrate moieties of bovine prothrombin for clotting activity. Biochim. et Biophys. Acta 1976, 421, 348–352. [Google Scholar] [CrossRef] [PubMed]
- Nelsestuen, G.L.; Suttie, J.W. Properties of asialo and aglycoprothrombin. Biochem. Biophys. Res. Commun. 1971, 45, 198–203. [Google Scholar] [CrossRef] [PubMed]
- Schwick, G.; Schultze, H.E. Immunochemical experiments with prothrombin and thrombin. Clin. Chim. Acta Int. J. Clin. Chem. 1959, 4, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Eber, S.W.; Polster, H.; Quentin, S.H.; Rumpf, K.W.; Lynen, R. Hemolytic-uremic syndrome in pneumococcal meningitis and infection. Importance of t-transformation. Monatsschrift Kinderheilkd. Organ. Dtsch. Ges. Kinderheilkd. 1993, 141, 219–222. [Google Scholar]
- Loupiac, A.; Elayan, A.; Cailliez, M.; Adra, A.L.; Decramer, S.; Thouret, M.C.; Harambat, J.; Guigonis, V. Diagnosis of streptococcus pneumoniae-associated hemolytic uremic syndrome. Pediatr. Infect. Dis. J. 2013, 32, 1045–1049. [Google Scholar] [CrossRef] [PubMed]
- Herbert, A.P.; Makou, E.; Chen, Z.A.; Kerr, H.; Richards, A.; Rappsilber, J.; Barlow, P.N. Complement evasion mediated by enhancement of captured factor h: Implications for protection of self-surfaces from complement. J. Immunol. 2015, 195, 4986–4998. [Google Scholar] [CrossRef]
- Lu, L.; Ma, Z.; Jokiranta, T.S.; Whitney, A.R.; DeLeo, F.R.; Zhang, J.R. Species-specific interaction of Streptococcus pneumoniae with human complement factor h. J. Immunol. 2008, 181, 7138–7146. [Google Scholar] [CrossRef]
- Bollaert, P.E.; Bauer, P.; Judlin, P.; Laprevote-Heully, M.C.; Lambert, H.; Larcan, A. Hemorrhagic colitis with streptococcus pyogenes preceding hemolytic uremic syndrome during early pregnancy. Nephron 1989, 52, 103–104. [Google Scholar] [CrossRef]
- Izumi, T.; Hyodo, T.; Kikuchi, Y.; Imakiire, T.; Ikenoue, T.; Suzuki, S.; Yoshizawa, N.; Miura, S. An adult with acute poststreptococcal glomerulonephritis complicated by hemolytic uremic syndrome and nephrotic syndrome. Am. J. Kidney Dis. 2005, 46, e59–e63. [Google Scholar] [CrossRef] [PubMed]
- Yildiz, B.; Kural, N.; Yarar, C. Atypical hemolytic uremic syndrome associated with group a beta hemolytic streptococcus. Pediatr. Nephrol. 2004, 19, 943–944; author reply 945. [Google Scholar] [CrossRef]
- Shepherd, A.B.; Palmer, A.L.; Bigler, S.A.; Baliga, R. Hemolytic uremic syndrome associated with group a beta-hemolytic streptococcus. Pediatr. Nephrol. 2003, 18, 949–951. [Google Scholar] [CrossRef] [PubMed]
- Pandiripally, V.; Gregory, E.; Cue, D. Acquisition of regulators of complement activation by streptococcus pyogenes serotype m1. Infect. Immun. 2002, 70, 6206–6214. [Google Scholar] [CrossRef] [PubMed]
- Pandiripally, V.; Wei, L.; Skerka, C.; Zipfel, P.F.; Cue, D. Recruitment of complement factor h-like protein 1 promotes intracellular invasion by group a streptococci. Infect. Immun. 2003, 71, 7119–7128. [Google Scholar] [CrossRef] [PubMed]
- Thern, A.; Stenberg, L.; Dahlback, B.; Lindahl, G. Ig-binding surface proteins of streptococcus pyogenes also bind human c4b-binding protein (c4bp), a regulatory component of the complement system. J. Immunol. 1995, 154, 375–386. [Google Scholar] [CrossRef]
- Herwald, H.; Morgelin, M.; Dahlback, B.; Bjorck, L. Interactions between surface proteins of streptococcus pyogenes and coagulation factors modulate clotting of human plasma. J. Thromb. Haemost. JTH 2003, 1, 284–291. [Google Scholar] [CrossRef]
- Inoue, D.; Oda, T.; Iwama, S.; Hoshino, T.; Mukae, M.; Sakai, T.; Kojima, A.; Uchida, T.; Kojima, T.; Sugisaki, K.; et al. Thrombotic microangiopathy with transiently positive direct coombs test in an adult with poststreptococcal acute glomerulonephritis: A case report. BMC Nephrol. 2022, 23, 56. [Google Scholar] [CrossRef]
- Oda, T.; Yoshizawa, N.; Yamakami, K.; Sakurai, Y.; Takechi, H.; Yamamoto, K.; Oshima, N.; Kumagai, H. The role of nephritis-associated plasmin receptor (naplr) in glomerulonephritis associated with streptococcal infection. J. Biomed. Biotechnol. 2012, 2012, 417675. [Google Scholar] [CrossRef]
- Poschmann, A.; Fischer, K. Exchange transfusion with heparinised fresh blood in necrotising enterocolitis. Lancet 1979, 1, 824–825. [Google Scholar] [CrossRef]
- Seger, R.; Joller, P.; Bird, G.W.; Wingham, J.; Wuest, J.; Kenny, A.; Rapp, A.; Garzoni, D.; Hitzig, W.H.; Duc, G. Necrotising enterocolitis and neuraminidase-producing bacteria. Helv. Paediatr. Acta 1980, 35, 121–128. [Google Scholar] [PubMed]
- Seges, R.A.; Kenny, A.; Bird, G.W.; Wingham, J.; Baals, H.; Stauffer, U.G. Pediatric surgical patients with severe anaerobic infection: Report of 16 t-antigen positive cases and possible hazards of blood transfusion. J. Pediatr. Surg. 1981, 16, 905–910. [Google Scholar] [CrossRef] [PubMed]
- Seitz, R.C.; Poschmann, A.; Hellwege, H.H. Monoclonal antibodies for the detection of desialylation of erythrocyte membranes during haemolytic disease and haemolytic uraemic syndrome caused by the in vivo action of microbial neuraminidase. Glycoconj. J. 1997, 14, 699–706. [Google Scholar] [CrossRef] [PubMed]
- Paddock, C.D.; Sanden, G.N.; Cherry, J.D.; Gal, A.A.; Langston, C.; Tatti, K.M.; Wu, K.H.; Goldsmith, C.S.; Greer, P.W.; Montague, J.L.; et al. Pathology and pathogenesis of fatal Bordetella pertussis infection in infants. Clin. Infect. Dis. 2008, 47, 328–338. [Google Scholar] [CrossRef] [PubMed]
- Berner, R.; Krause, M.F.; Gordjani, N.; Zipfel, P.F.; Boehm, N.; Krueger, M.; Brandis, M.; Zimmerhackl, L.B. Hemolytic uremic syndrome due to an altered factor h triggered by neonatal pertussis. Pediatr. Nephrol. 2002, 17, 190–192. [Google Scholar] [CrossRef] [PubMed]
- Chaturvedi, S.; Licht, C.; Langlois, V. Hemolytic uremic syndrome caused by Bordetella pertussis infection. Pediatr. Nephrol. 2010, 25, 1361–1364. [Google Scholar] [CrossRef]
- Pela, I.; Seracini, D.; Caprioli, A.; Castelletti, F.; Giammanco, A. Hemolytic uremic syndrome in an infant following Bordetella pertussis infection. Eur. J. Clin. Microbiol. Infect. Dis. 2006, 25, 515–517. [Google Scholar] [CrossRef]
- Saida, K.; Ogura, M.; Kano, Y.; Ishimori, S.; Yoshikawa, T.; Nagata, H.; Sato, M.; Kamei, K.; Ishikura, K. Treatment of hemolytic uremic syndrome related to Bordetella pertussis infection—Is plasma exchange or eculizumab use necessary? BMC Nephrol. 2018, 19, 365. [Google Scholar] [CrossRef]
- Jongerius, I.; Schuijt, T.J.; Mooi, F.R.; Pinelli, E. Complement evasion by Bordetella pertussis: Implications for improving current vaccines. J. Mol. Med. 2015, 93, 395–402. [Google Scholar] [CrossRef]
- Barnes, M.G.; Weiss, A.A. Brka protein of Bordetella pertussis inhibits the classical pathway of complement after c1 deposition. Infect. Immun. 2001, 69, 3067–3072. [Google Scholar] [CrossRef]
- Marr, N.; Shah, N.R.; Lee, R.; Kim, E.J.; Fernandez, R.C. Bordetella pertussis autotransporter vag8 binds human c1 esterase inhibitor and confers serum resistance. PLoS ONE 2011, 6, e20585. [Google Scholar] [CrossRef] [PubMed]
- Mooi, F.R.; van Loo, I.H.; van Gent, M.; He, Q.; Bart, M.J.; Heuvelman, K.J.; de Greeff, S.C.; Diavatopoulos, D.; Teunis, P.; Nagelkerke, N.; et al. Bordetella pertussis strains with increased toxin production associated with pertussis resurgence. Emerg. Infect. Dis. 2009, 15, 1206–1213. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, A.; Deme, J.C.; Furlong, E.; Roem, D.; Jongerius, I.; Johnson, S.; Lea, S.M. Molecular basis for Bordetella pertussis interference with complement, coagulation, fibrinolytic, and contact activation systems: The cryo-em structure of the vag8-c1 inhibitor complex. mBio 2021, 12, e02823-20. [Google Scholar] [CrossRef] [PubMed]
- Berggard, K.; Johnsson, E.; Mooi, F.R.; Lindahl, G. Bordetella pertussis binds the human complement regulator c4bp: Role of filamentous hemagglutinin. Infect. Immun. 1997, 65, 3638–3643. [Google Scholar] [CrossRef] [PubMed]
- Berggard, K.; Lindahl, G.; Dahlback, B.; Blom, A.M. Bordetella pertussis binds to human c4b-binding protein (c4bp) at a site similar to that used by the natural ligand c4b. Eur. J. Immunol. 2001, 31, 2771–2780. [Google Scholar] [CrossRef]
- Fernandez, R.C.; Weiss, A.A. Serum resistance in bvg-regulated mutants of Bordetella pertussis. FEMS Microbiol. Lett. 1998, 163, 57–63. [Google Scholar] [CrossRef]
- Zipfel, P.F.; Hallstrom, T.; Riesbeck, K. Human complement control and complement evasion by pathogenic microbes--tipping the balance. Mol. Immunol. 2013, 56, 152–160. [Google Scholar] [CrossRef]
- Amdahl, H.; Jarva, H.; Haanpera, M.; Mertsola, J.; He, Q.; Jokiranta, T.S.; Meri, S. Interactions between Bordetella pertussis and the complement inhibitor factor h. Mol. Immunol. 2011, 48, 697–705. [Google Scholar] [CrossRef]
- Meri, T.; Amdahl, H.; Lehtinen, M.J.; Hyvarinen, S.; McDowell, J.V.; Bhattacharjee, A.; Meri, S.; Marconi, R.; Goldman, A.; Jokiranta, T.S. Microbes bind complement inhibitor factor h via a common site. PLoS Pathog. 2013, 9, e1003308. [Google Scholar] [CrossRef]
- Albaqali, A.; Ghuloom, A.; Al Arrayed, A.; Al Ajami, A.; Shome, D.K.; Jamsheer, A.; Al Mahroos, H.; Jelacic, S.; Tarr, P.I.; Kaplan, B.S.; et al. Hemolytic uremic syndrome in association with typhoid fever. Am. J. Kidney Dis. 2003, 41, 709–713. [Google Scholar] [CrossRef]
- Grossman, N.; Leive, L. Complement activation via the alternative pathway by purified salmonella lipopolysaccharide is affected by its structure but not its o-antigen length. J. Immunol. 1984, 132, 376–385. [Google Scholar] [CrossRef] [PubMed]
- Grossman, N.; Svenson, S.B.; Leive, L.; Lindberg, A.A. Salmonella o antigen-specific oligosaccharide-octyl conjugates activate complement via the alternative pathway at different rates depending on the structure of the o antigen. Mol. Immunol. 1990, 27, 859–865. [Google Scholar] [CrossRef] [PubMed]
- Hart, P.J.; O’Shaughnessy, C.M.; Siggins, M.K.; Bobat, S.; Kingsley, R.A.; Goulding, D.A.; Crump, J.A.; Reyburn, H.; Micoli, F.; Dougan, G.; et al. Differential killing of Salmonella enterica serovar typhi by antibodies targeting vi and lipopolysaccharide o:9 antigen. PLoS ONE 2016, 11, e0145945. [Google Scholar] [CrossRef] [PubMed]
- Robbins, J.D.; Robbins, J.B. Reexamination of the protective role of the capsular polysaccharide (vi antigen) of Salmonella typhi. J. Infect. Dis. 1984, 150, 436–449. [Google Scholar] [CrossRef] [PubMed]
- Lahteenmaki, K.; Kyllonen, P.; Partanen, L.; Korhonen, T.K. Antiprotease inactivation by Salmonella enterica released from infected macrophages. Cell. Microbiol. 2005, 7, 529–538. [Google Scholar] [CrossRef] [PubMed]
- Ramu, P.; Tanskanen, R.; Holmberg, M.; Lahteenmaki, K.; Korhonen, T.K.; Meri, S. The surface protease pgte of Salmonella enterica affects complement activity by proteolytically cleaving c3b, c4b and c5. FEBS Lett. 2007, 581, 1716–1720. [Google Scholar] [CrossRef] [PubMed]
- Riva, R.; Korhonen, T.K.; Meri, S. The outer membrane protease pgte of Salmonella enterica interferes with the alternative complement pathway by cleaving factors b and h. Front. Microbiol. 2015, 6, 63. [Google Scholar] [CrossRef]
- Haiko, J.; Laakkonen, L.; Juuti, K.; Kalkkinen, N.; Korhonen, T.K. The omptins of yersinia pestis and Salmonella enterica cleave the reactive center loop of plasminogen activator inhibitor 1. J. Bacteriol. 2010, 192, 4553–4561. [Google Scholar] [CrossRef]
- Valls Seron, M.; Haiko, J.; DE Groot, P.G.; Korhonen, T.K.; Meijers, J.C. Thrombin-activatable fibrinolysis inhibitor is degraded by Salmonella enterica and yersinia pestis. J. Thromb. Haemost. JTH 2010, 8, 2232–2240. [Google Scholar] [CrossRef]
- de Jong, H.K.; Parry, C.M.; van der Vaart, T.W.; Kager, L.M.; van den Ende, S.J.; Maude, R.R.; Wijedoru, L.; Ghose, A.; Hassan, M.U.; Hossain, M.A.; et al. Activation of coagulation and endothelium with concurrent impairment of anticoagulant mechanisms in patients with typhoid fever. J. Infect. 2018, 77, 60–67. [Google Scholar] [CrossRef]
- Beutin, L.; Strauch, E.; Fischer, I. Isolation of shigella sonnei lysogenic for a bacteriophage encoding gene for production of shiga toxin. Lancet 1999, 353, 1498. [Google Scholar] [CrossRef] [PubMed]
- Lamba, K.; Nelson, J.A.; Kimura, A.C.; Poe, A.; Collins, J.; Kao, A.S.; Cruz, L.; Inami, G.; Vaishampayan, J.; Garza, A.; et al. Shiga toxin 1-producing shigella sonnei infections, california, united states, 2014–2015. Emerg. Infect. Dis. 2016, 22, 679–686. [Google Scholar] [CrossRef] [PubMed]
- Nyholm, O.; Lienemann, T.; Halkilahti, J.; Mero, S.; Rimhanen-Finne, R.; Lehtinen, V.; Salmenlinna, S.; Siitonen, A. Characterization of shigella sonnei isolate carrying shiga toxin 2-producing gene. Emerg. Infect. Dis. 2015, 21, 891–892. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, H. Shiga-toxin-converting bacteriophages. Res. Microbiol. 2001, 152, 687–695. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Castillo, A.; Quiros, P.; Navarro, F.; Miro, E.; Muniesa, M. Shiga toxin 2-encoding bacteriophages in human fecal samples from healthy individuals. Appl. Environ. Microbiol. 2013, 79, 4862–4868. [Google Scholar] [CrossRef] [PubMed]
- Muniesa, M.; Jofre, J. Abundance in sewage of bacteriophages that infect escherichia coli o157:H7 and that carry the shiga toxin 2 gene. Appl. Environ. Microbiol. 1998, 64, 2443–2448. [Google Scholar] [CrossRef] [PubMed]
- Muniesa, M.; Lucena, F.; Jofre, J. Comparative survival of free shiga toxin 2-encoding phages and escherichia coli strains outside the gut. Appl. Environ. Microbiol. 1999, 65, 5615–5618. [Google Scholar] [CrossRef]
- Chan, Y.S.; Ng, T.B. Shiga toxins: From structure and mechanism to applications. Appl. Microbiol. Biotechnol. 2016, 100, 1597–1610. [Google Scholar] [CrossRef]
- Adams, C.; Vose, A.; Edmond, M.B.; Lyckholm, L. Shigella sonnei and hemolytic uremic syndrome: A case report and literature review. IDCases 2017, 8, 6–8. [Google Scholar] [CrossRef]
- Armstrong, S.M.; Wang, C.; Tigdi, J.; Si, X.; Dumpit, C.; Charles, S.; Gamage, A.; Moraes, T.J.; Lee, W.L. Influenza infects lung microvascular endothelium leading to microvascular leak: Role of apoptosis and claudin-5. PLoS ONE 2012, 7, e47323. [Google Scholar] [CrossRef]
- Hutchinson, E.C. Influenza virus. Trends Microbiol. 2018, 26, 809–810. [Google Scholar] [CrossRef] [PubMed]
- Allen, U.; Licht, C. Pandemic h1n1 influenza a infection and (atypical) hus--more than just another trigger? Pediatr. Nephrol. 2011, 26, 3–5. [Google Scholar] [CrossRef] [PubMed]
- Bento, D.; Mapril, J.; Rocha, C.; Marchbank, K.J.; Kavanagh, D.; Barge, D.; Strain, L.; Goodship, T.H.; Meneses-Oliveira, C. Triggering of atypical hemolytic uremic syndrome by influenza a (h1n1). Ren. Fail. 2010, 32, 753–756. [Google Scholar] [CrossRef] [PubMed]
- Caltik, A.; Akyuz, S.G.; Erdogan, O.; Demircin, G. Hemolytic uremic syndrome triggered with a new pandemic virus: Influenza a (h1n1). Pediatr. Nephrol. 2011, 26, 147–148. [Google Scholar] [CrossRef] [PubMed]
- Trachtman, H.; Sethna, C.; Epstein, R.; D’Souza, M.; Rubin, L.G.; Ginocchio, C.C. Atypical hemolytic uremic syndrome associated with h1n1 influenza a virus infection. Pediatr. Nephrol. 2011, 26, 145–146. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, T. Hemolytic uremic syndrome associated with influenza a virus infection. Nephron 2001, 89, 359–360. [Google Scholar] [CrossRef] [PubMed]
- Kobbe, R.; Schild, R.; Christner, M.; Oh, J.; Loos, S.; Kemper, M.J. Case report—Atypical hemolytic uremic syndrome triggered by influenza b. BMC Nephrol. 2017, 18, 96. [Google Scholar] [CrossRef]
- Mittal, N.; Hartemayer, R.; Jandeska, S.; Giordano, L. Steroid responsive atypical hemolytic uremic syndrome triggered by influenza b infection. J. Pediatr. Hematol. Oncol. 2019, 41, e63–e67. [Google Scholar] [CrossRef]
- van Hoeve, K.; Vandermeulen, C.; Van Ranst, M.; Levtchenko, E.; van den Heuvel, L.; Mekahli, D. Occurrence of atypical hus associated with influenza b. Eur. J. Pediatr. 2017, 176, 449–454. [Google Scholar] [CrossRef]
- Boilard, E.; Pare, G.; Rousseau, M.; Cloutier, N.; Dubuc, I.; Levesque, T.; Borgeat, P.; Flamand, L. Influenza virus h1n1 activates platelets through fcgammariia signaling and thrombin generation. Blood 2014, 123, 2854–2863. [Google Scholar] [CrossRef]
- Rondina, M.T.; Brewster, B.; Grissom, C.K.; Zimmerman, G.A.; Kastendieck, D.H.; Harris, E.S.; Weyrich, A.S. In vivo platelet activation in critically ill patients with primary 2009 influenza a(h1n1). Chest 2012, 141, 1490–1495. [Google Scholar] [CrossRef] [PubMed]
- Lambre, C.R.; Kazatchkine, M.D.; Maillet, F.; Thibon, M. Guinea pig erythrocytes, after their contact with influenza virus, acquire the ability to activate the human alternative complement pathway through virus-induced desialation of the cells. J. Immunol. 1982, 128, 629–634. [Google Scholar] [CrossRef] [PubMed]
- Berdal, J.E.; Mollnes, T.E.; Waehre, T.; Olstad, O.K.; Halvorsen, B.; Ueland, T.; Laake, J.H.; Furuseth, M.T.; Maagaard, A.; Kjekshus, H.; et al. Excessive innate immune response and mutant d222g/n in severe a (h1n1) pandemic influenza. J. Infect. 2011, 63, 308–316. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Zhao, G.; Liu, C.; Wu, X.; Guo, Y.; Yu, H.; Song, H.; Du, L.; Jiang, S.; Guo, R.; et al. Inhibition of complement activation alleviates acute lung injury induced by highly pathogenic avian influenza h5n1 virus infection. Am. J. Respir. Cell Mol. Biol. 2013, 49, 221–230. [Google Scholar] [CrossRef] [PubMed]
- Noris, M.; Remuzzi, G. Glomerular diseases dependent on complement activation, including atypical hemolytic uremic syndrome, membranoproliferative glomerulonephritis, and c3 glomerulopathy: Core curriculum 2015. Am. J. Kidney Dis. 2015, 66, 359–375. [Google Scholar] [CrossRef] [PubMed]
- Salvadori, M.; Bertoni, E. Update on hemolytic uremic syndrome: Diagnostic and therapeutic recommendations. World J. Nephrol. 2013, 2, 56–76. [Google Scholar] [CrossRef] [PubMed]
- Thurman, J.M. Complement in kidney disease: Core curriculum 2015. Am. J. Kidney Dis. 2015, 65, 156–168. [Google Scholar] [CrossRef] [PubMed]
- Bitzan, M.; Zieg, J. Influenza-associated thrombotic microangiopathies. Pediatr. Nephrol. 2018, 33, 2009–2025. [Google Scholar] [CrossRef]
- Silecchia, V.; D’Onofrio, G.; Valerio, E.; Rubin, G.; Vidal, E.; Murer, L. Influenza-associated hemolytic uremic syndrome: The pathogenic role of the virus. Clin. Nephrol. Case Stud. 2021, 9, 45–48. [Google Scholar] [CrossRef]
- Boccia, R.V.; Gelmann, E.P.; Baker, C.C.; Marti, G.; Longo, D.L. A hemolytic-uremic syndrome with the acquired immunodeficiency syndrome. Ann. Intern. Med. 1984, 101, 716–717. [Google Scholar] [CrossRef]
- Freist, M.; Garrouste, C.; Szlavik, N.; Coppo, P.; Lautrette, A.; Heng, A.E. Efficacy of eculizumab in an adult patient with hiv-associated hemolytic uremic syndrome: A case report. Medicine 2017, 96, e9358. [Google Scholar] [CrossRef] [PubMed]
- Jin, A.; Boroujerdi-Rad, L.; Shah, G.; Chen, J.L. Thrombotic microangiopathy and human immunodeficiency virus in the era of eculizumab. Clin. Kidney J. 2016, 9, 576–579. [Google Scholar] [CrossRef] [PubMed]
- Huson, M.A.; Wouters, D.; van Mierlo, G.; Grobusch, M.P.; Zeerleder, S.S.; van der Poll, T. Hiv coinfection enhances complement activation during sepsis. J. Infect. Dis. 2015, 212, 474–483. [Google Scholar] [CrossRef] [PubMed]
- Huber, M.; Fischer, M.; Misselwitz, B.; Manrique, A.; Kuster, H.; Niederost, B.; Weber, R.; von Wyl, V.; Gunthard, H.F.; Trkola, A. Complement lysis activity in autologous plasma is associated with lower viral loads during the acute phase of hiv-1 infection. PLoS Med. 2006, 3, e441. [Google Scholar] [CrossRef] [PubMed]
- Senaldi, G.; Peakman, M.; McManus, T.; Davies, E.T.; Tee, D.E.; Vergani, D. Activation of the complement system in human immunodeficiency virus infection: Relevance of the classical pathway to pathogenesis and disease severity. J. Infect. Dis. 1990, 162, 1227–1232. [Google Scholar] [CrossRef] [PubMed]
- Spear, G.T.; Takefman, D.M.; Sullivan, B.L.; Landay, A.L.; Zolla-Pazner, S. Complement activation by human monoclonal antibodies to human immunodeficiency virus. J. Virol. 1993, 67, 53–59. [Google Scholar] [CrossRef]
- Stoiber, H.; Kacani, L.; Speth, C.; Wurzner, R.; Dierich, M.P. The supportive role of complement in hiv pathogenesis. Immunol. Rev. 2001, 180, 168–176. [Google Scholar] [CrossRef]
- Humbert, M.; Dietrich, U. The role of neutralizing antibodies in hiv infection. AIDS Rev. 2006, 8, 51–59. [Google Scholar]
- Ji, X.; Gewurz, H.; Spear, G.T. Mannose binding lectin (mbl) and hiv. Mol. Immunol. 2005, 42, 145–152. [Google Scholar] [CrossRef]
- Ezekowitz, R.A.; Kuhlman, M.; Groopman, J.E.; Byrn, R.A. A human serum mannose-binding protein inhibits in vitro infection by the human immunodeficiency virus. J. Exp. Med. 1989, 169, 185–196. [Google Scholar] [CrossRef]
- Haurum, J.S.; Thiel, S.; Jones, I.M.; Fischer, P.B.; Laursen, S.B.; Jensenius, J.C. Complement activation upon binding of mannan-binding protein to hiv envelope glycoproteins. Aids 1993, 7, 1307–1313. [Google Scholar] [CrossRef] [PubMed]
- Saifuddin, M.; Hart, M.L.; Gewurz, H.; Zhang, Y.; Spear, G.T. Interaction of mannose-binding lectin with primary isolates of human immunodeficiency virus type 1. J. Gen. Virol. 2000, 81, 949–955. [Google Scholar] [CrossRef] [PubMed]
- Hart, M.L.; Saifuddin, M.; Spear, G.T. Glycosylation inhibitors and neuraminidase enhance human immunodeficiency virus type 1 binding and neutralization by mannose-binding lectin. J. Gen. Virol. 2003, 84, 353–360. [Google Scholar] [CrossRef] [PubMed]
- Ying, H.; Ji, X.; Hart, M.L.; Gupta, K.; Saifuddin, M.; Zariffard, M.R.; Spear, G.T. Interaction of mannose-binding lectin with hiv type 1 is sufficient for virus opsonization but not neutralization. AIDS Res. Hum. Retroviruses 2004, 20, 327–335. [Google Scholar] [CrossRef] [PubMed]
- Bajtay, Z.; Speth, C.; Erdei, A.; Dierich, M.P. Cutting edge: Productive hiv-1 infection of dendritic cells via complement receptor type 3 (cr3, cd11b/cd18). J. Immunol. 2004, 173, 4775–4778. [Google Scholar] [CrossRef] [PubMed]
- Pruenster, M.; Wilflingseder, D.; Banki, Z.; Ammann, C.G.; Muellauer, B.; Meyer, M.; Speth, C.; Dierich, M.P.; Stoiber, H. C-type lectin-independent interaction of complement opsonized hiv with monocyte-derived dendritic cells. Eur. J. Immunol. 2005, 35, 2691–2698. [Google Scholar] [CrossRef]
- Prohaszka, Z.; Nemes, J.; Hidvegi, T.; Toth, F.D.; Kerekes, K.; Erdei, A.; Szabo, J.; Ujhelyi, E.; Thielens, N.; Dierich, M.P.; et al. Two parallel routes of the complement-mediated antibody-dependent enhancement of hiv-1 infection. Aids 1997, 11, 949–958. [Google Scholar] [CrossRef]
- Delibrias, C.C.; Kazatchkine, M.D.; Fischer, E. Evidence for the role of cr1 (cd35), in addition to cr2 (cd21), in facilitating infection of human t cells with opsonized hiv. Scand. J. Immunol. 1993, 38, 183–189. [Google Scholar] [CrossRef]
- Kacani, L.; Banki, Z.; Zwirner, J.; Schennach, H.; Bajtay, Z.; Erdei, A.; Stoiber, H.; Dierich, M.P. C5a and c5a(desarg) enhance the susceptibility of monocyte-derived macrophages to hiv infection. J. Immunol. 2001, 166, 3410–3415. [Google Scholar] [CrossRef]
- Speth, C.; Schabetsberger, T.; Mohsenipour, I.; Stockl, G.; Wurzner, R.; Stoiber, H.; Lass-Florl, C.; Dierich, M.P. Mechanism of human immunodeficiency virus-induced complement expression in astrocytes and neurons. J. Virol. 2002, 76, 3179–3188. [Google Scholar] [CrossRef]
- Horakova, E.; Gasser, O.; Sadallah, S.; Inal, J.M.; Bourgeois, G.; Ziekau, I.; Klimkait, T.; Schifferli, J.A. Complement mediates the binding of hiv to erythrocytes. J. Immunol. 2004, 173, 4236–4241. [Google Scholar] [CrossRef] [PubMed]
- Dierich, M.P.; Stoiber, H.; Clivio, A. A “complement-ary” aids vaccine. Nat. Med. 1996, 2, 153–155. [Google Scholar] [CrossRef] [PubMed]
- Saifuddin, M.; Parker, C.J.; Peeples, M.E.; Gorny, M.K.; Zolla-Pazner, S.; Ghassemi, M.; Rooney, I.A.; Atkinson, J.P.; Spear, G.T. Role of virion-associated glycosylphosphatidylinositol-linked proteins cd55 and cd59 in complement resistance of cell line-derived and primary isolates of hiv-1. J. Exp. Med. 1995, 182, 501–509. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, J.; Zimmer, J.P.; Kluxen, B.; Aries, S.; Bogel, M.; Gigli, I.; Schmitz, H. Antibody-dependent complement-mediated cytotoxicity in sera from patients with hiv-1 infection is controlled by cd55 and cd59. J. Clin. Investig. 1995, 96, 1520–1526. [Google Scholar] [CrossRef] [PubMed]
- Stoiber, H.; Pinter, C.; Siccardi, A.G.; Clivio, A.; Dierich, M.P. Efficient destruction of human immunodeficiency virus in human serum by inhibiting the protective action of complement factor h and decay accelerating factor (daf, cd55). J. Exp. Med. 1996, 183, 307–310. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Lupu, F.; Esmon, C.T. Inflammation, innate immunity and blood coagulation. Hamostaseologie 2010, 30, 5–6, 8–9. [Google Scholar] [CrossRef] [PubMed]
- Armah, K.A.; McGinnis, K.; Baker, J.; Gibert, C.; Butt, A.A.; Bryant, K.J.; Goetz, M.; Tracy, R.; Oursler, K.K.; Rimland, D.; et al. Hiv status, burden of comorbid disease, and biomarkers of inflammation, altered coagulation, and monocyte activation. Clin. Infect. Dis. 2012, 55, 126–136. [Google Scholar] [CrossRef] [PubMed]
- Duprez, D.A.; Neuhaus, J.; Kuller, L.H.; Tracy, R.; Belloso, W.; De Wit, S.; Drummond, F.; Lane, H.C.; Ledergerber, B.; Lundgren, J.; et al. Inflammation, coagulation and cardiovascular disease in hiv-infected individuals. PLoS ONE 2012, 7, e44454. [Google Scholar] [CrossRef]
- Kuller, L.H.; Tracy, R.; Belloso, W.; De Wit, S.; Drummond, F.; Lane, H.C.; Ledergerber, B.; Lundgren, J.; Neuhaus, J.; Nixon, D.; et al. Inflammatory and coagulation biomarkers and mortality in patients with hiv infection. PLoS Med. 2008, 5, e203. [Google Scholar] [CrossRef]
- Neuhaus, J.; Jacobs, D.R., Jr.; Baker, J.V.; Calmy, A.; Duprez, D.; La Rosa, A.; Kuller, L.H.; Pett, S.L.; Ristola, M.; Ross, M.J.; et al. Markers of inflammation, coagulation, and renal function are elevated in adults with hiv infection. J. Infect. Dis. 2010, 201, 1788–1795. [Google Scholar] [CrossRef]
- Sandler, N.G.; Wand, H.; Roque, A.; Law, M.; Nason, M.C.; Nixon, D.E.; Pedersen, C.; Ruxrungtham, K.; Lewin, S.R.; Emery, S.; et al. Plasma levels of soluble cd14 independently predict mortality in hiv infection. J. Infect. Dis. 2011, 203, 780–790. [Google Scholar] [CrossRef] [PubMed]
- Brenchley, J.M.; Price, D.A.; Schacker, T.W.; Asher, T.E.; Silvestri, G.; Rao, S.; Kazzaz, Z.; Bornstein, E.; Lambotte, O.; Altmann, D.; et al. Microbial translocation is a cause of systemic immune activation in chronic hiv infection. Nat. Med. 2006, 12, 1365–1371. [Google Scholar] [CrossRef] [PubMed]
- Brenchley, J.M.; Schacker, T.W.; Ruff, L.E.; Price, D.A.; Taylor, J.H.; Beilman, G.J.; Nguyen, P.L.; Khoruts, A.; Larson, M.; Haase, A.T.; et al. Cd4+ t cell depletion during all stages of hiv disease occurs predominantly in the gastrointestinal tract. J. Exp. Med. 2004, 200, 749–759. [Google Scholar] [CrossRef] [PubMed]
- Pandrea, I.; Cornell, E.; Wilson, C.; Ribeiro, R.M.; Ma, D.; Kristoff, J.; Xu, C.; Haret-Richter, G.S.; Trichel, A.; Apetrei, C.; et al. Coagulation biomarkers predict disease progression in siv-infected nonhuman primates. Blood 2012, 120, 1357–1366. [Google Scholar] [CrossRef] [PubMed]
- Funderburg, N.T.; Mayne, E.; Sieg, S.F.; Asaad, R.; Jiang, W.; Kalinowska, M.; Luciano, A.A.; Stevens, W.; Rodriguez, B.; Brenchley, J.M.; et al. Increased tissue factor expression on circulating monocytes in chronic hiv infection: Relationship to in vivo coagulation and immune activation. Blood 2010, 115, 161–167. [Google Scholar] [CrossRef] [PubMed]
- Baker, J.V.; Brummel-Ziedins, K.; Neuhaus, J.; Duprez, D.; Cummins, N.; Dalmau, D.; DeHovitz, J.; Lehmann, C.; Sullivan, A.; Woolley, I.; et al. Hiv replication alters the composition of extrinsic pathway coagulation factors and increases thrombin generation. J. Am. Heart Assoc. 2013, 2, e000264. [Google Scholar] [CrossRef] [PubMed]
- Glasgow, L.A.; Balduzzi, P. Isolation of coxsackie virus group a, type 4, from a patient with hemolytic-uremic syndrome. N. Engl. J. Med. 1965, 273, 754–756. [Google Scholar] [CrossRef]
- Austin, T.W.; Ray, C.G. Coxsackie virus group b infections and the hemolytic-uremic syndrome. J. Infect. Dis. 1973, 127, 698–701. [Google Scholar] [CrossRef]
- De Petris, L.; Gianviti, A.; Caione, D.; Innocenzi, D.; Edefonti, A.; Montini, G.; De Palo, T.; Tozzi, A.E.; Caprioli, A.; Rizzoni, G. Role of non-polio enterovirus infection in pediatric hemolytic uremic syndrome. Pediatr. Nephrol. 2002, 17, 852–855. [Google Scholar] [CrossRef]
- O’Regan, S.; Robitaille, P.; Mongeau, J.G.; McLaughlin, B. The hemolytic uremic syndrome associated with echo 22 infection. Clin. Pediatr. 1980, 19, 125–127. [Google Scholar] [CrossRef]
- Ray, C.G.; Portman, J.N.; Stamm, S.J.; Hickman, R.O. Hemolytic-uremic syndrome and myocarditis. Association with coxsackievirus b infection. Am. J. Dis. Child. 1971, 122, 418–420. [Google Scholar] [CrossRef] [PubMed]
- Ray, C.G.; Tucker, V.L.; Harris, D.J.; Cuppage, F.E.; Chin, T.D. Enteroviruses associated with the hemolytic-uremic syndrome. Pediatrics 1970, 46, 378–388. [Google Scholar] [CrossRef] [PubMed]
- Vecilla, M.C.; Ruiz Moreno, M.; Bernacer, M.; Casado, S.; Rocandio, L. Familial hemolytic-uremic syndrome associated with coxsackie b infection. An. Esp. de Pediatr. 1984, 20, 369–374. [Google Scholar]
- Lee, M.D.; Tzen, C.Y.; Lin, C.C.; Huang, F.Y.; Liu, H.C.; Tsai, J.D. Hemolytic uremic syndrome caused by enteroviral infection. Pediatr. Neonatol. 2013, 54, 207–210. [Google Scholar] [CrossRef] [PubMed]
- Spiller, O.B.; Goodfellow, I.G.; Evans, D.J.; Almond, J.W.; Morgan, B.P. Echoviruses and coxsackie b viruses that use human decay-accelerating factor (daf) as a receptor do not bind the rodent analogues of daf. J. Infect. Dis. 2000, 181, 340–343. [Google Scholar] [CrossRef] [PubMed]
- Fujita, T.; Inoue, T.; Ogawa, K.; Iida, K.; Tamura, N. The mechanism of action of decay-accelerating factor (daf). Daf inhibits the assembly of c3 convertases by dissociating c2a and bb. J. Exp. Med. 1987, 166, 1221–1228. [Google Scholar] [CrossRef]
- Medof, M.E.; Kinoshita, T.; Nussenzweig, V. Inhibition of complement activation on the surface of cells after incorporation of decay-accelerating factor (daf) into their membranes. J. Exp. Med. 1984, 160, 1558–1578. [Google Scholar] [CrossRef]
- Nicholson-Weller, A.; Burge, J.; Fearon, D.T.; Weller, P.F.; Austen, K.F. Isolation of a human erythrocyte membrane glycoprotein with decay-accelerating activity for c3 convertases of the complement system. J. Immunol. 1982, 129, 184–189. [Google Scholar] [CrossRef]
- Nicholson-Weller, A.; Wang, C.E. Structure and function of decay accelerating factor cd55. J. Lab. Clin. Med. 1994, 123, 485–491. [Google Scholar]
- Anderson, D.R.; Carthy, C.M.; Wilson, J.E.; Yang, D.; Devine, D.V.; McManus, B.M. Complement component 3 interactions with coxsackievirus b3 capsid proteins: Innate immunity and the rapid formation of splenic antiviral germinal centers. J. Virol. 1997, 71, 8841–8845. [Google Scholar] [CrossRef]
- Zanone, M.M.; Favaro, E.; Conaldi, P.G.; Greening, J.; Bottelli, A.; Perin, P.C.; Klein, N.J.; Peakman, M.; Camussi, G. Persistent infection of human microvascular endothelial cells by coxsackie b viruses induces increased expression of adhesion molecules. J. Immunol. 2003, 171, 438–446. [Google Scholar] [CrossRef] [PubMed]
- Angus, D.C.; van der Poll, T. Severe sepsis and septic shock. N. Engl. J. Med. 2013, 369, 840–851. [Google Scholar] [CrossRef] [PubMed]
- Richardson, G.M.; Su, S.W.; Iragorri, S. Case report: Diarrhea-associated hemolytic uremic syndrome in the era of COVID-19. Front. Pediatr. 2022, 10, 979850. [Google Scholar] [CrossRef]
- Smarz-Widelska, I.; Syroka-Glowka, M.; Janowska-Jaremek, J.; Koziol, M.M.; Zaluska, W. Atypical hemolytic uremic syndrome after SARS-CoV-2 infection: Report of two cases. Int. J. Environ. Res. Public Health 2022, 19, 11437. [Google Scholar] [CrossRef] [PubMed]
- Dalkiran, T.; Kandur, Y.; Kara, E.M.; Dagoglu, B.; Taner, S.; Oncu, D. Thrombotic microangiopathy in a severe pediatric case of COVID-19. Clin. Med. Insights. Pediatr. 2021, 15, 11795565211049897. [Google Scholar] [CrossRef]
- Helms, J.; Tacquard, C.; Severac, F.; Leonard-Lorant, I.; Ohana, M.; Delabranche, X.; Merdji, H.; Clere-Jehl, R.; Schenck, M.; Fagot Gandet, F.; et al. High risk of thrombosis in patients with severe SARS-CoV-2 infection: A multicenter prospective cohort study. Intensive Care Med. 2020, 46, 1089–1098. [Google Scholar] [CrossRef]
- Magro, C.; Mulvey, J.J.; Berlin, D.; Nuovo, G.; Salvatore, S.; Harp, J.; Baxter-Stoltzfus, A.; Laurence, J. Complement associated microvascular injury and thrombosis in the pathogenesis of severe COVID-19 infection: A report of five cases. Transl. Res. J. Lab. Clin. Med. 2020, 220, 1–13. [Google Scholar] [CrossRef]
- Beltrame, M.H.; Catarino, S.J.; Goeldner, I.; Boldt, A.B.; de Messias-Reason, I.J. The lectin pathway of complement and rheumatic heart disease. Front. Pediatr. 2014, 2, 148. [Google Scholar] [CrossRef]
- Krarup, A.; Wallis, R.; Presanis, J.S.; Gal, P.; Sim, R.B. Simultaneous activation of complement and coagulation by mbl-associated serine protease 2. PLoS ONE 2007, 2, e623. [Google Scholar] [CrossRef]
- Subramaniam, S.; Scharrer, I. Procoagulant activity during viral infections. Front. Biosci. 2018, 23, 1060–1081. [Google Scholar] [CrossRef]
- Teimury, A.; Khameneh, M.T.; Khaledi, E.M. Major coagulation disorders and parameters in COVID-19 patients. Eur. J. Med. Res. 2022, 27, 25. [Google Scholar] [CrossRef] [PubMed]
- Godier, A.; Clausse, D.; Meslin, S.; Bazine, M.; Lang, E.; Huche, F.; Cholley, B.; Hamada, S.R. Major bleeding complications in critically ill patients with COVID-19 pneumonia. J. Thromb. Thrombolysis 2021, 52, 18–21. [Google Scholar] [CrossRef] [PubMed]
- Manne, B.K.; Denorme, F.; Middleton, E.A.; Portier, I.; Rowley, J.W.; Stubben, C.; Petrey, A.C.; Tolley, N.D.; Guo, L.; Cody, M.; et al. Platelet gene expression and function in patients with COVID-19. Blood 2020, 136, 1317–1329. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Avdonin, P.P.; Blinova, M.S.; Generalova, G.A.; Emirova, K.M.; Avdonin, P.V. The Role of the Complement System in the Pathogenesis of Infectious Forms of Hemolytic Uremic Syndrome. Biomolecules 2024, 14, 39. https://doi.org/10.3390/biom14010039
Avdonin PP, Blinova MS, Generalova GA, Emirova KM, Avdonin PV. The Role of the Complement System in the Pathogenesis of Infectious Forms of Hemolytic Uremic Syndrome. Biomolecules. 2024; 14(1):39. https://doi.org/10.3390/biom14010039
Chicago/Turabian StyleAvdonin, Piotr P., Maria S. Blinova, Galina A. Generalova, Khadizha M. Emirova, and Pavel V. Avdonin. 2024. "The Role of the Complement System in the Pathogenesis of Infectious Forms of Hemolytic Uremic Syndrome" Biomolecules 14, no. 1: 39. https://doi.org/10.3390/biom14010039