
Regulation of Endosomal Trafficking by Rab7 and Its Effectors in Neurons: Clues from Charcot–Marie–Tooth 2B Disease

Abstract
:1. Introduction
2. Molecular Basis of Rab7 Function and Disruption in Disease
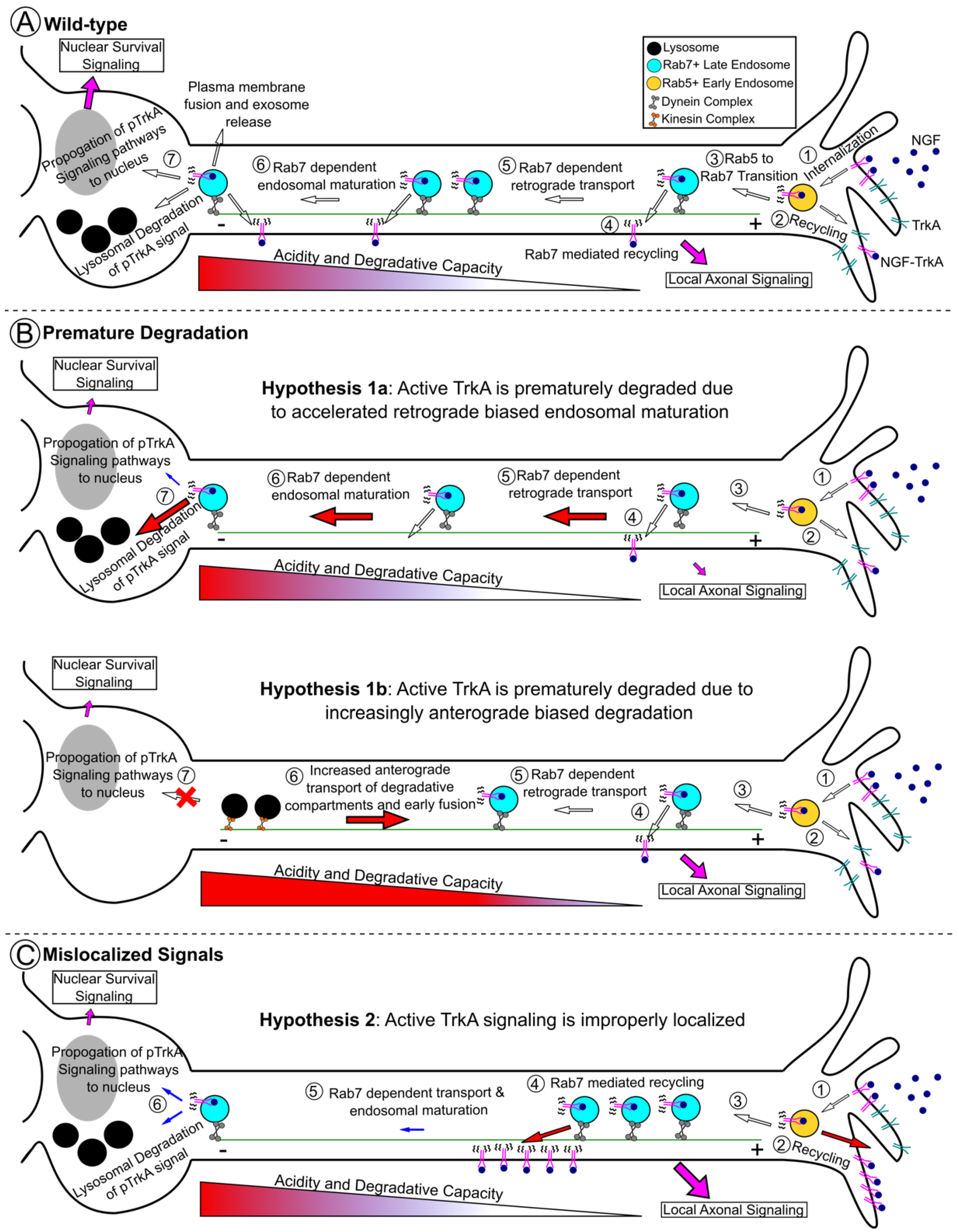
3. Physiologic Control of Rab7 in Neuronal Trafficking and Trophic Signaling
4. Receptor Tyrosine Kinase Signaling in Charcot–Marie–Tooth 2B Models
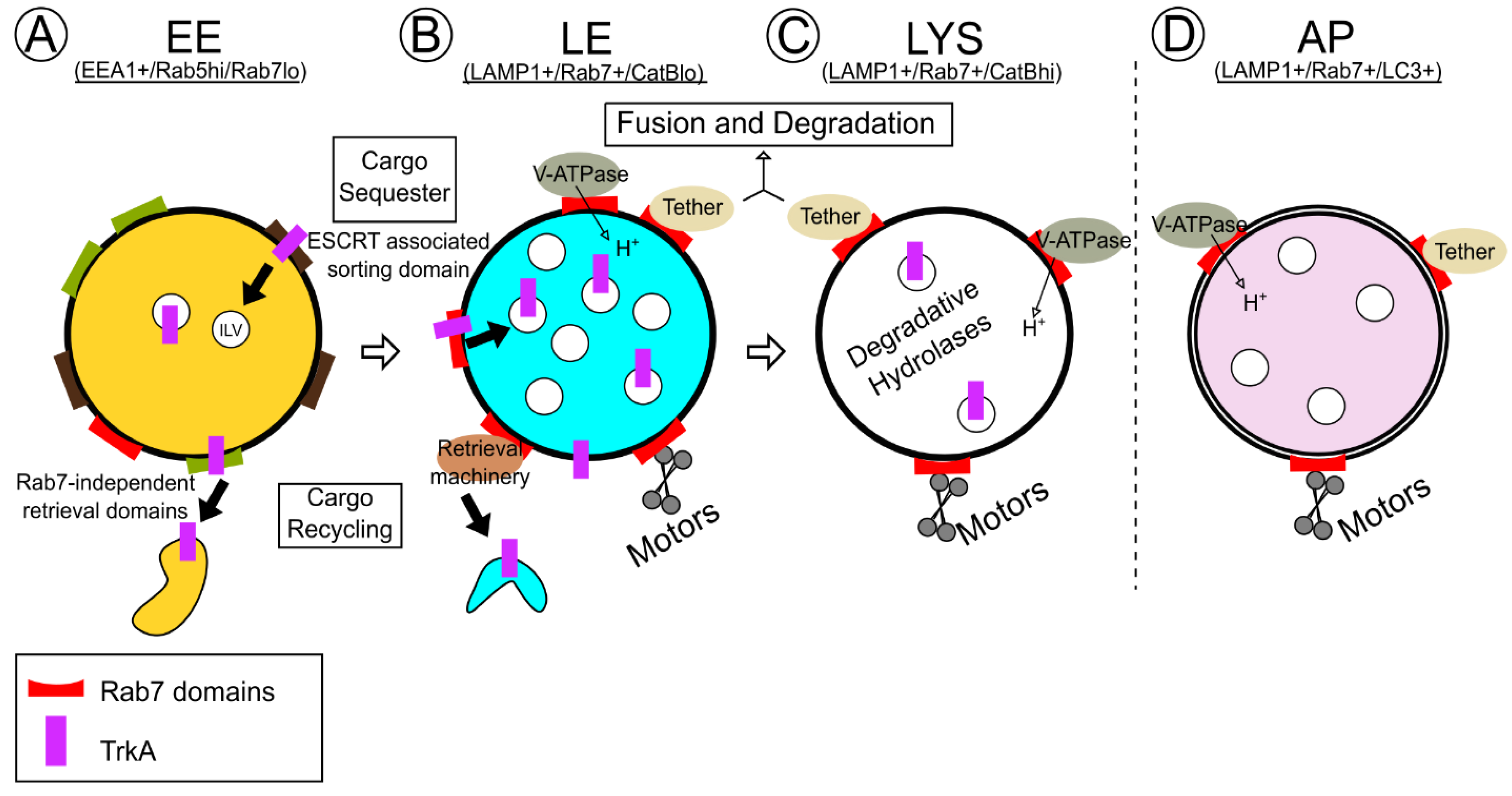
5. Rab7 Effectors and Physiologic TrkA Trafficking and Degradation
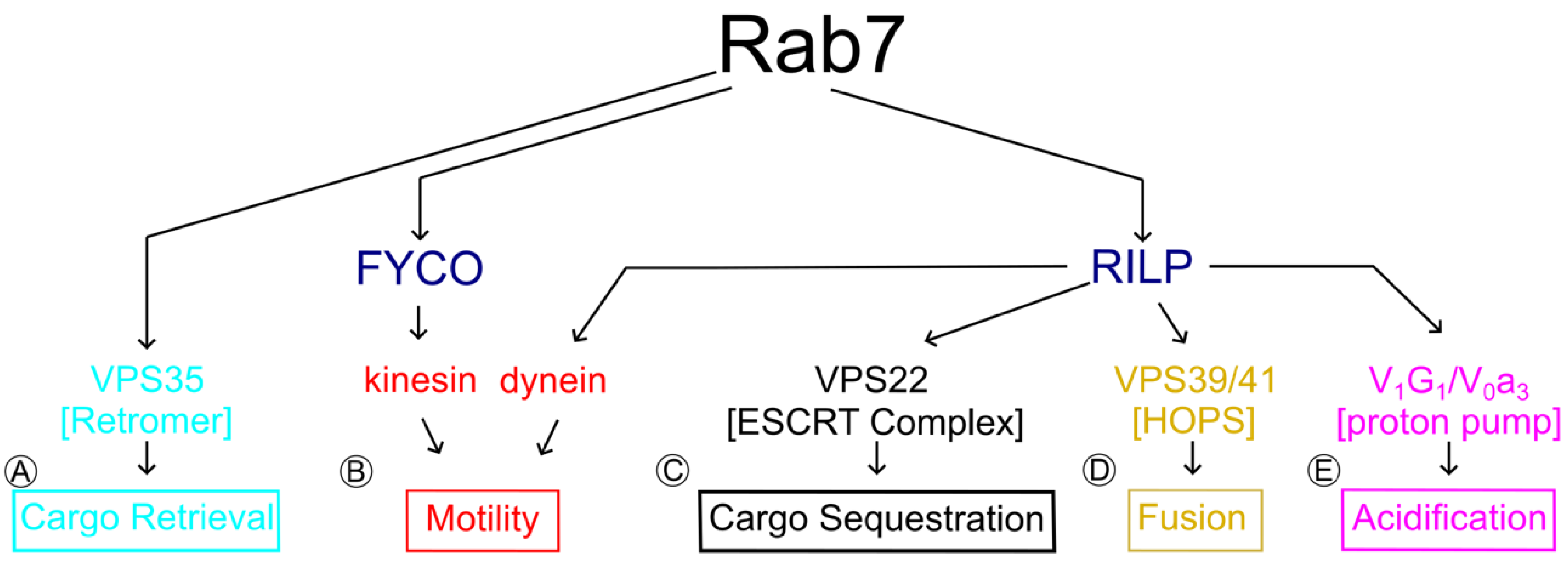
5.1. Rab-Interacting Lysosomal Protein (RILP) and Oxysterol-Related Binding Protein 1 L (ORP1L): Control over Rab7 Motility and Positioning
5.2. V-ATPase
5.3. Vps35: Connections to Both TrkA Sorting and Degradation
5.4. Homotypic Fusion and Protein Sorting (HOPS) Complex for Tethering and Fusion
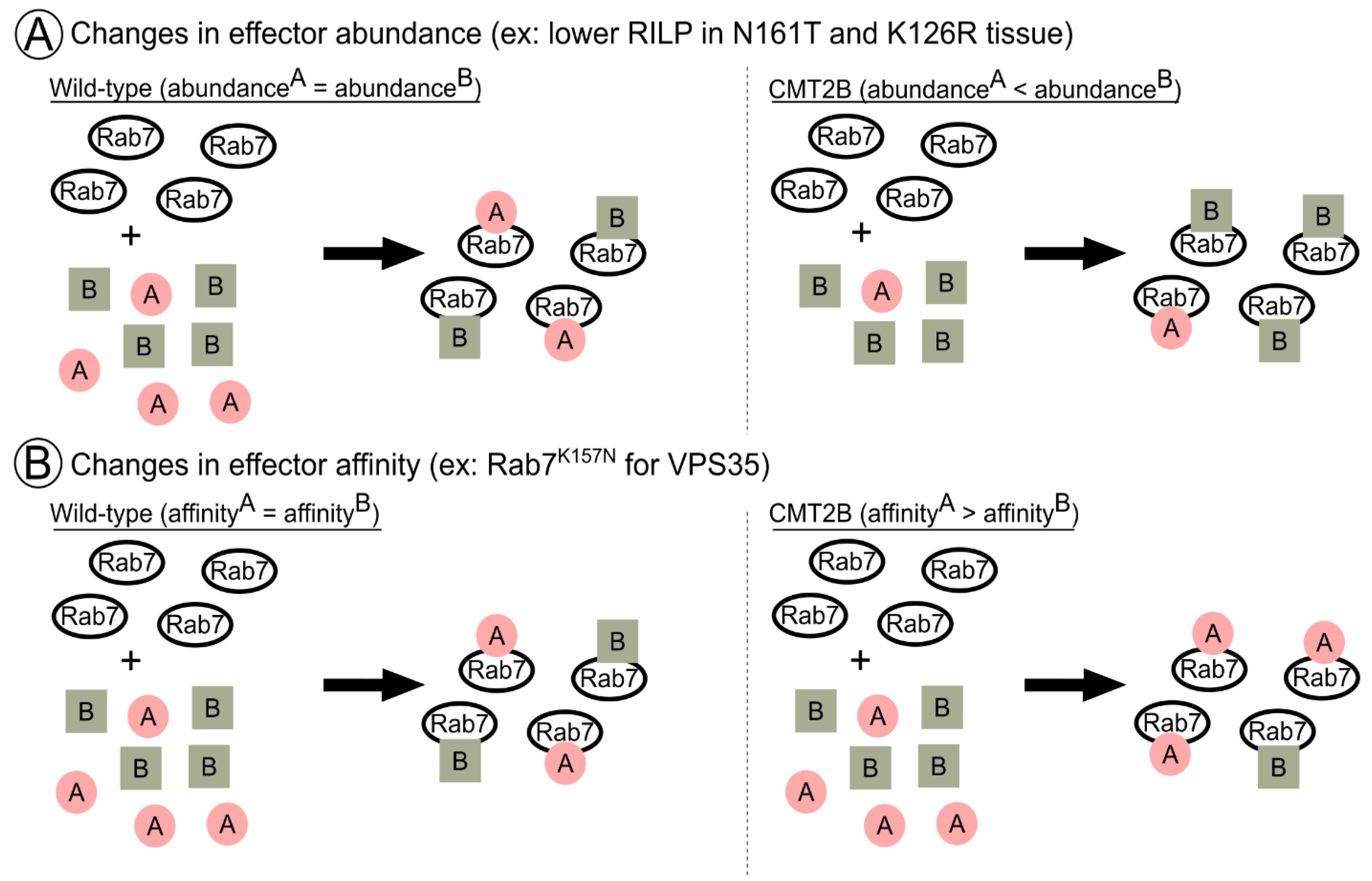
6. A Novel Notion of Effector Balance and Ordering: Is this Disrupted in Disease?
6.1. RILP in CMT2B: A Case for Effector Abundance?
6.2. VPS35 in CMT2B: A Case for Effector Affinity?
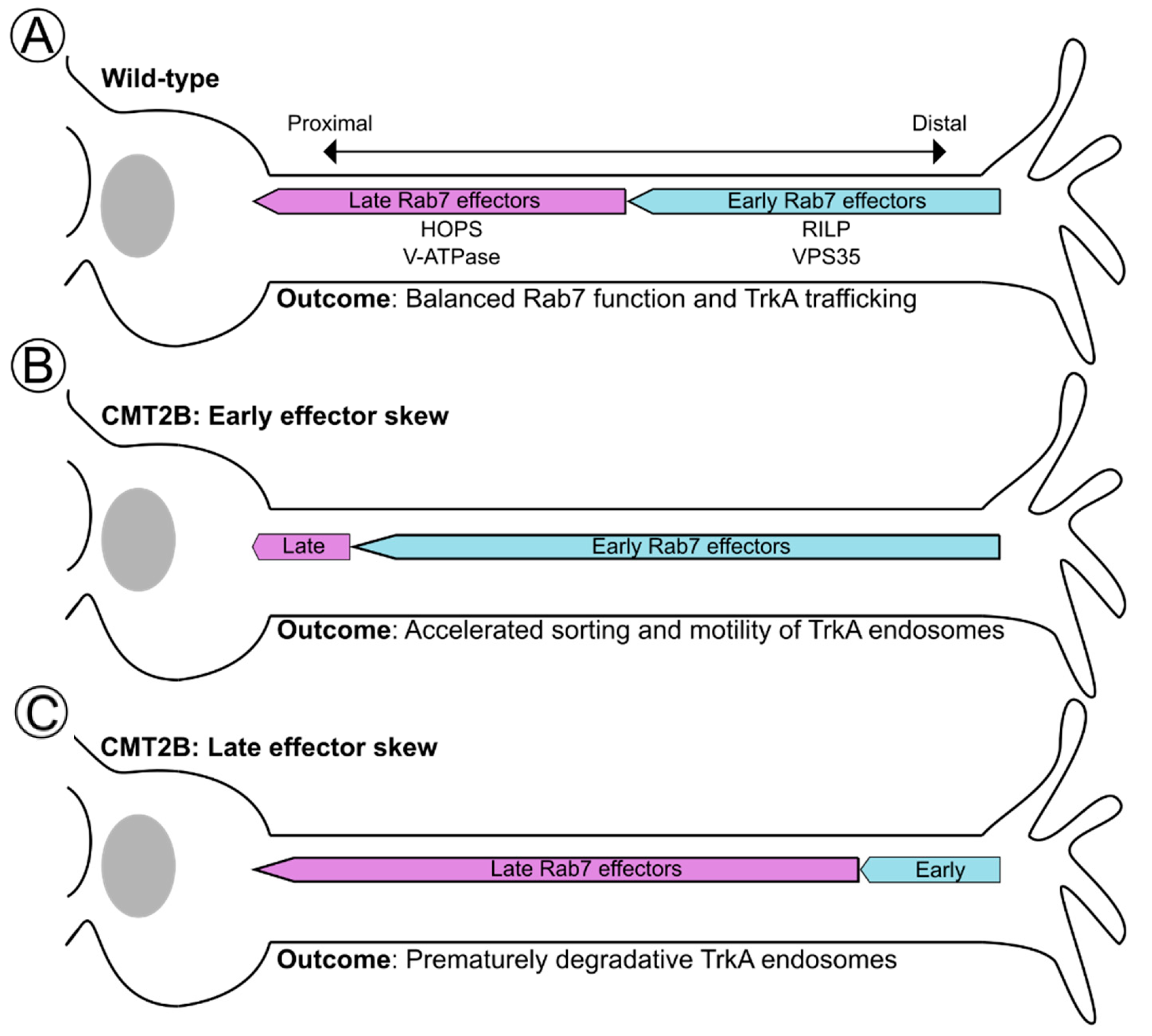
6.3. Spatial Ordering of Rab7 Effectors along the TrkA Trafficking Route
7. Other Rab7 Hypotheses: It Is Not all about TrkA
8. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Azzedine, H.; Senderek, J.; Rivolta, C.; Chrast, R. Molecular genetics of charcot-marie-tooth disease: From genes to genomes. Mol. Syndromol. 2012, 3, 204–214. [Google Scholar] [CrossRef] [PubMed]
- Hoyle, J.C.; Isfort, M.C.; Roggenbuck, J.; Arnold, W.D. The genetics of Charcot-Marie-Tooth disease: Current trends and future implications for diagnosis and management. Appl. Clin. Genet. 2015, 8, 235–243. [Google Scholar] [CrossRef]
- Züchner, S.; Vance, J.M. Mechanisms of disease: A molecular genetic update on hereditary axonal neuropathies. Nat. Clin. Pract. Neurol. 2006, 2, 45–53. [Google Scholar] [CrossRef]
- Harding, A.E.; Thomas, P.K. The clinical features of hereditary motor and sensory neuropathy types I and II. Brain 1980, 103, 259–280. [Google Scholar] [CrossRef]
- Barisic, N.; Claeys, K.G.; Sirotković-Skerlev, M.; Löfgren, A.; Nelis, E.; De Jonghe, P.; Timmerman, V. Charcot-Marie-Tooth disease: A clinico-genetic confrontation. Ann. Hum. Genet. 2008, 72, 416–441. [Google Scholar] [CrossRef] [PubMed]
- Auer-Grumbach, M. Hereditary sensory neuropathies. Drugs Today 2004, 40, 385–394. [Google Scholar] [CrossRef]
- Kwon, J.M.; Elliott, J.L.; Yee, W.C.; Ivanovich, J.; Scavarda, N.J.; Moolsintong, P.J.; Goodfellow, P.J. Assignment of a second Charcot-Marie-Tooth type II locus to chromosome 3q. Am. J. Hum. Genet. 1995, 57, 853–858. [Google Scholar] [PubMed]
- Auer-Grumbach, M.; De Jonghe, P.; Wagner, K.; Verhoeven, K.; Hartung, H.P.; Timmerman, V. Phenotype-genotype correlations in a CMT2B family with refined 3q13-q22 locus. Neurology 2000, 55, 1552–1557. [Google Scholar] [CrossRef] [PubMed]
- De Jonghe, P.; Timmerman, V.; FitzPatrick, D.; Spoelders, P.; Martin, J.J.; Van Broeckhoven, C. Mutilating neuropathic ulcerations in a chromosome 3q13-q22 linked Charcot-Marie-Tooth disease type 2B family. J. Neurol. Neurosurg. Psychiatr. 1997, 62, 570–573. [Google Scholar] [CrossRef] [PubMed]
- Vance, J.M.; Speer, M.C.; Stajich, J.M.; West, S.; Wolpert, C.; Gaskell, P.; Lennon, F.; Tim, R.M.; Rozear, M.; Othmane, K.B. Misclassification and linkage of hereditary sensory and autonomic neuropathy type 1 as Charcot-Marie-Tooth disease, type 2B. Am. J. Hum. Genet. 1996, 59, 258–262. [Google Scholar]
- Verhoeven, K.; De Jonghe, P.; Coen, K.; Verpoorten, N.; Auer-Grumbach, M.; Kwon, J.M.; FitzPatrick, D.; Schmedding, E.; De Vriendt, E.; Jacobs, A.; et al. Mutations in the small GTP-ase late endosomal protein RAB7 cause Charcot-Marie-Tooth type 2B neuropathy. Am. J. Hum. Genet. 2003, 72, 722–727. [Google Scholar] [CrossRef]
- Houlden, H.; King, R.H.M.; Muddle, J.R.; Warner, T.T.; Reilly, M.M.; Orrell, R.W.; Ginsberg, L. A novel RAB7 mutation associated with ulcero-mutilating neuropathy. Ann. Neurol. 2004, 56, 586–590. [Google Scholar] [CrossRef]
- Meggouh, F.; Bienfait, H.M.E.; Weterman, M.A.J.; de Visser, M.; Baas, F. Charcot-Marie-Tooth disease due to a de novo mutation of the RAB7 gene. Neurology 2006, 67, 1476–1478. [Google Scholar] [CrossRef]
- Wang, X.; Han, C.; Liu, W.; Wang, P.; Zhang, X. A novel RAB7 mutation in a Chinese family with Charcot-Marie-Tooth type 2B disease. Gene 2014, 534, 431–434. [Google Scholar] [CrossRef] [PubMed]
- Saveri, P.; De Luca, M.; Nisi, V.; Pisciotta, C.; Romano, R.; Piscosquito, G.; Reilly, M.M.; Polke, J.M.; Cavallaro, T.; Fabrizi, G.M.; et al. Charcot-Marie-Tooth Type 2B: A New Phenotype Associated with a Novel RAB7A Mutation and Inhibited EGFR Degradation. Cells 2020, 9, 1028. [Google Scholar] [CrossRef] [PubMed]
- Homma, Y.; Hiragi, S.; Fukuda, M. Rab family of small GTPases: An updated view on their regulation and functions. FEBS J. 2021, 288, 36–55. [Google Scholar] [CrossRef]
- Grosshans, B.L.; Ortiz, D.; Novick, P. Rabs and their effectors: Achieving specificity in membrane traffic. Proc. Natl. Acad. Sci. USA 2006, 103, 11821–11827. [Google Scholar] [CrossRef]
- Wandinger-Ness, A.; Zerial, M. Rab proteins and the compartmentalization of the endosomal system. Cold Spring Harb. Perspect. Biol. 2014, 6, a022616. [Google Scholar] [CrossRef] [PubMed]
- Cullen, P.J.; Steinberg, F. To degrade or not to degrade: Mechanisms and significance of endocytic recycling. Nat. Rev. Mol. Cell Biol. 2018, 19, 679–696. [Google Scholar] [CrossRef]
- Ceresa, B.P.; Bahr, S.J. rab7 activity affects epidermal growth factor:epidermal growth factor receptor degradation by regulating endocytic trafficking from the late endosome. J. Biol. Chem. 2006, 281, 1099–1106. [Google Scholar] [CrossRef]
- Saxena, S.; Bucci, C.; Weis, J.; Kruttgen, A. The small GTPase Rab7 controls the endosomal trafficking and neuritogenic signaling of the nerve growth factor receptor TrkA. J. Neurosci. 2005, 25, 10930–10940. [Google Scholar] [CrossRef]
- Rink, J.; Ghigo, E.; Kalaidzidis, Y.; Zerial, M. Rab conversion as a mechanism of progression from early to late endosomes. Cell 2005, 122, 735–749. [Google Scholar] [CrossRef] [PubMed]
- Poteryaev, D.; Datta, S.; Ackema, K.; Zerial, M.; Spang, A. Identification of the switch in early-to-late endosome transition. Cell 2010, 141, 497–508. [Google Scholar] [CrossRef] [PubMed]
- Winckler, B.; Faundez, V.; Maday, S.; Cai, Q.; Guimas Almeida, C.; Zhang, H. The endolysosomal system and proteostasis: From development to degeneration. J. Neurosci. 2018, 38, 9364–9374. [Google Scholar] [CrossRef] [PubMed]
- Kawamura, N.; Takaoka, K.; Hamada, H.; Hadjantonakis, A.-K.; Sun-Wada, G.-H.; Wada, Y. Rab7-Mediated Endocytosis Establishes Patterning of Wnt Activity through Inactivation of Dkk Antagonism. Cell Rep. 2020, 31, 107733. [Google Scholar] [CrossRef]
- Gu, Y.; Guerra, F.; Hu, M.; Pope, A.; Sung, K.; Yang, W.; Jetha, S.; Shoff, T.A.; Gunatilake, T.; Dahlkamp, O.; et al. Mitochondria dysfunction in Charcot Marie Tooth 2B Peripheral Sensory Neuropathy. Commun. Biol. 2022, 5, 717. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Wu, C. Charcot marie tooth 2B peripheral sensory neuropathy: How rab7 mutations impact NGF signaling? Int. J. Mol. Sci. 2017, 18, 324. [Google Scholar] [CrossRef]
- Stroupe, C. This is the end: Regulation of rab7 nucleotide binding in endolysosomal trafficking and autophagy. Front. Cell Dev. Biol. 2018, 6, 129. [Google Scholar] [CrossRef] [PubMed]
- Nordmann, M.; Cabrera, M.; Perz, A.; Bröcker, C.; Ostrowicz, C.; Engelbrecht-Vandré, S.; Ungermann, C. The Mon1-Ccz1 complex is the GEF of the late endosomal Rab7 homolog Ypt7. Curr. Biol. 2010, 20, 1654–1659. [Google Scholar] [CrossRef]
- Kinchen, J.M.; Ravichandran, K.S. Identification of two evolutionarily conserved genes regulating processing of engulfed apoptotic cells. Nature 2010, 464, 778–782. [Google Scholar] [CrossRef]
- Cogli, L.; Piro, F.; Bucci, C. Rab7 and the CMT2B disease. Biochem. Soc. Trans. 2009, 37, 1027–1031. [Google Scholar] [CrossRef] [PubMed]
- McCray, B.A.; Skordalakes, E.; Taylor, J.P. Disease mutations in Rab7 result in unregulated nucleotide exchange and inappropriate activation. Hum. Mol. Genet. 2010, 19, 1033–1047. [Google Scholar] [CrossRef] [PubMed]
- Spinosa, M.R.; Progida, C.; De Luca, A.; Colucci, A.M.R.; Alifano, P.; Bucci, C. Functional characterization of Rab7 mutant proteins associated with Charcot-Marie-Tooth type 2B disease. J. Neurosci. 2008, 28, 1640–1648. [Google Scholar] [CrossRef] [PubMed]
- De Luca, A.; Progida, C.; Spinosa, M.R.; Alifano, P.; Bucci, C. Characterization of the Rab7K157N mutant protein associated with Charcot-Marie-Tooth type 2B. Biochem. Biophys. Res. Commun. 2008, 372, 283–287. [Google Scholar] [CrossRef] [PubMed]
- Bucci, C.; De Luca, M. Molecular basis of Charcot-Marie-Tooth type 2B disease. Biochem. Soc. Trans. 2012, 40, 1368–1372. [Google Scholar] [CrossRef]
- Modica, G.; Lefrancois, S. Post-translational modifications: How to modulate Rab7 functions. Small GTPases 2020, 11, 167–173. [Google Scholar] [CrossRef]
- Shinde, S.R.; Maddika, S. PTEN modulates EGFR late endocytic trafficking and degradation by dephosphorylating Rab7. Nat. Commun. 2016, 7, 10689. [Google Scholar] [CrossRef]
- Song, P.; Trajkovic, K.; Tsunemi, T.; Krainc, D. Parkin Modulates Endosomal Organization and Function of the Endo-Lysosomal Pathway. J. Neurosci. 2016, 36, 2425–2437. [Google Scholar] [CrossRef]
- Peng, W.; Schröder, L.F.; Song, P.; Wong, Y.C.; Krainc, D. Parkin regulates amino acid homeostasis at mitochondria-lysosome (M/L) contact sites in Parkinson’s disease. Sci. Adv. 2023, 9, eadh3347. [Google Scholar] [CrossRef]
- Bucci, C.; Thomsen, P.; Nicoziani, P.; McCarthy, J.; van Deurs, B. Rab7: A key to lysosome biogenesis. Mol. Biol. Cell 2000, 11, 467–480. [Google Scholar] [CrossRef]
- Vitelli, R.; Santillo, M.; Lattero, D.; Chiariello, M.; Bifulco, M.; Bruni, C.B.; Bucci, C. Role of the small GTPase Rab7 in the late endocytic pathway. J. Biol. Chem. 1997, 272, 4391–4397. [Google Scholar] [CrossRef]
- Vanlandingham, P.A.; Ceresa, B.P. Rab7 regulates late endocytic trafficking downstream of multivesicular body biogenesis and cargo sequestration. J. Biol. Chem. 2009, 284, 12110–12124. [Google Scholar] [CrossRef]
- Guerra, F.; Bucci, C. Multiple roles of the small gtpase rab7. Cells 2016, 5, 34. [Google Scholar] [CrossRef]
- Langemeyer, L.; Fröhlich, F.; Ungermann, C. Rab gtpase function in endosome and lysosome biogenesis. Trends Cell Biol. 2018, 28, 957–970. [Google Scholar] [CrossRef]
- Borchers, A.-C.; Langemeyer, L.; Ungermann, C. Who’s in control? Principles of Rab GTPase activation in endolysosomal membrane trafficking and beyond. J. Cell Biol. 2021, 220, e202105120. [Google Scholar] [CrossRef]
- Yap, C.C.; Mason, A.J.; Winckler, B. Dynamics and distribution of endosomes and lysosomes in dendrites. Curr. Opin. Neurobiol. 2022, 74, 102537. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Daniszewski, M.; Hao, M.M.; Hernández, D.; Pébay, A.; Gleeson, P.A.; Fourriere, L. Organelle mapping in dendrites of human iPSC-derived neurons reveals dynamic functional dendritic Golgi structures. Cell Rep. 2023, 42, 112709. [Google Scholar] [CrossRef] [PubMed]
- Boecker, C.A.; Olenick, M.A.; Gallagher, E.R.; Ward, M.E.; Holzbaur, E.L.F. ToolBox: Live Imaging of intracellular organelle transport in induced pluripotent stem cell-derived neurons. Traffic 2020, 21, 138–155. [Google Scholar] [CrossRef]
- Yap, C.C.; Digilio, L.; McMahon, L.P.; Garcia, A.D.R.; Winckler, B. Degradation of dendritic cargos requires Rab7-dependent transport to somatic lysosomes. J. Cell Biol. 2018, 217, 3141–3159. [Google Scholar] [CrossRef] [PubMed]
- Yap, C.C.; Winckler, B. Spatial regulation of endosomes in growing dendrites. Dev. Biol. 2022, 486, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Yap, C.C.; Digilio, L.; McMahon, L.P.; Wang, T.; Winckler, B. Dynein Is Required for Rab7-Dependent Endosome Maturation, Retrograde Dendritic Transport, and Degradation. J. Neurosci. 2022, 42, 4415–4434. [Google Scholar] [CrossRef]
- Cheng, X.-T.; Zhou, B.; Lin, M.-Y.; Cai, Q.; Sheng, Z.-H. Axonal autophagosomes recruit dynein for retrograde transport through fusion with late endosomes. J. Cell Biol. 2015, 209, 377–386. [Google Scholar] [CrossRef]
- Cheng, X.-T.; Xie, Y.-X.; Zhou, B.; Huang, N.; Farfel-Becker, T.; Sheng, Z.-H. Characterization of LAMP1-labeled nondegradative lysosomal and endocytic compartments in neurons. J. Cell Biol. 2018, 217, 3127–3139. [Google Scholar] [CrossRef] [PubMed]
- Lie, P.P.Y.; Yang, D.-S.; Stavrides, P.; Goulbourne, C.N.; Zheng, P.; Mohan, P.S.; Cataldo, A.M.; Nixon, R.A. Post-Golgi carriers, not lysosomes, confer lysosomal properties to pre-degradative organelles in normal and dystrophic axons. Cell Rep. 2021, 35, 109034. [Google Scholar] [CrossRef]
- Khobrekar, N.V.; Quintremil, S.; Dantas, T.J.; Vallee, R.B. The dynein adaptor RILP controls neuronal autophagosome biogenesis, transport, and clearance. Dev. Cell 2020, 53, 141–153.e4. [Google Scholar] [CrossRef]
- Lee, S.; Sato, Y.; Nixon, R.A. Lysosomal proteolysis inhibition selectively disrupts axonal transport of degradative organelles and causes an Alzheimer’s-like axonal dystrophy. J. Neurosci. 2011, 31, 7817–7830. [Google Scholar] [CrossRef]
- Kulkarni, V.V.; Stempel, M.H.; Anand, A.; Sidibe, D.K.; Maday, S. Retrograde axonal autophagy and endocytic pathways are parallel and separate in neurons. J. Neurosci. 2022, 42, 8524–8541. [Google Scholar] [CrossRef]
- Nassal, J.P.; Murphy, F.H.; Toonen, R.F.; Verhage, M. Differential axonal trafficking of Neuropeptide Y-, LAMP1-, and RAB7-tagged organelles in vivo. eLife 2022, 11, e81721. [Google Scholar] [CrossRef] [PubMed]
- Krzystek, T.J.; White, J.A.; Rathnayake, R.; Thurston, L.; Hoffmar-Glennon, H.; Li, Y.; Gunawardena, S. HTT (huntingtin) and RAB7 co-migrate retrogradely on a signaling LAMP1-containing late endosome during axonal injury. Autophagy 2023, 19, 1199–1220. [Google Scholar] [CrossRef]
- Zhang, K.; Ben Kenan, R.F.; Osakada, Y.; Xu, W.; Sinit, R.S.; Chen, L.; Zhao, X.; Chen, J.-Y.; Cui, B.; Wu, C. Defective axonal transport of Rab7 GTPase results in dysregulated trophic signaling. J. Neurosci. 2013, 33, 7451–7462. [Google Scholar] [CrossRef] [PubMed]
- Lund, V.K.; Lycas, M.D.; Schack, A.; Andersen, R.C.; Gether, U.; Kjaerulff, O. Rab2 drives axonal transport of dense core vesicles and lysosomal organelles. Cell Rep. 2021, 35, 108973. [Google Scholar] [CrossRef] [PubMed]
- Castle, M.J.; Gershenson, Z.T.; Giles, A.R.; Holzbaur, E.L.F.; Wolfe, J.H. Adeno-associated virus serotypes 1, 8, and 9 share conserved mechanisms for anterograde and retrograde axonal transport. Hum. Gene Ther. 2014, 25, 705–720. [Google Scholar] [CrossRef]
- Deinhardt, K.; Salinas, S.; Verastegui, C.; Watson, R.; Worth, D.; Hanrahan, S.; Bucci, C.; Schiavo, G. Rab5 and Rab7 control endocytic sorting along the axonal retrograde transport pathway. Neuron 2006, 52, 293–305. [Google Scholar] [CrossRef] [PubMed]
- Deppmann, C.D.; Ginty, D.D. Retrograde control of neural circuit formation. Cell 2006, 127, 1306–1307. [Google Scholar] [CrossRef]
- da Silva, S.; Wang, F. Retrograde neural circuit specification by target-derived neurotrophins and growth factors. Curr. Opin. Neurobiol. 2011, 21, 61–67. [Google Scholar] [CrossRef]
- Scott-Solomon, E.; Kuruvilla, R. Mechanisms of neurotrophin trafficking via Trk receptors. Mol. Cell. Neurosci. 2018, 91, 25–33. [Google Scholar] [CrossRef]
- Howe, C.L.; Mobley, W.C. Long-distance retrograde neurotrophic signaling. Curr. Opin. Neurobiol. 2005, 15, 40–48. [Google Scholar] [CrossRef]
- Ye, M.; Lehigh, K.M.; Ginty, D.D. Multivesicular bodies mediate long-range retrograde NGF-TrkA signaling. eLife 2018, 7, 33012. [Google Scholar] [CrossRef]
- Delcroix, J.-D.; Valletta, J.S.; Wu, C.; Hunt, S.J.; Kowal, A.S.; Mobley, W.C. NGF signaling in sensory neurons: Evidence that early endosomes carry NGF retrograde signals. Neuron 2003, 39, 69–84. [Google Scholar] [CrossRef] [PubMed]
- Barford, K.; Keeler, A.; McMahon, L.; McDaniel, K.; Yap, C.C.; Deppmann, C.D.; Winckler, B. Transcytosis of TrkA leads to diversification of dendritic signaling endosomes. Sci. Rep. 2018, 8, 4715. [Google Scholar] [CrossRef]
- Barford, K.; Deppmann, C.; Winckler, B. The neurotrophin receptor signaling endosome: Where trafficking meets signaling. Dev. Neurobiol. 2017, 77, 405–418. [Google Scholar] [CrossRef]
- BasuRay, S.; Mukherjee, S.; Romero, E.G.; Seaman, M.N.J.; Wandinger-Ness, A. Rab7 mutants associated with Charcot-Marie-Tooth disease cause delayed growth factor receptor transport and altered endosomal and nuclear signaling. J. Biol. Chem. 2013, 288, 1135–1149. [Google Scholar] [CrossRef] [PubMed]
- Romano, R.; Rivellini, C.; De Luca, M.; Tonlorenzi, R.; Beli, R.; Manganelli, F.; Nolano, M.; Santoro, L.; Eskelinen, E.-L.; Previtali, S.C.; et al. Alteration of the late endocytic pathway in Charcot-Marie-Tooth type 2B disease. Cell. Mol. Life Sci. 2021, 78, 351–372. [Google Scholar] [CrossRef]
- BasuRay, S.; Mukherjee, S.; Romero, E.; Wilson, M.C.; Wandinger-Ness, A. Rab7 mutants associated with Charcot-Marie-Tooth disease exhibit enhanced NGF-stimulated signaling. PLoS ONE 2010, 5, e15351. [Google Scholar] [CrossRef]
- Cogli, L.; Progida, C.; Lecci, R.; Bramato, R.; Krüttgen, A.; Bucci, C. CMT2B-associated Rab7 mutants inhibit neurite outgrowth. Acta Neuropathol. 2010, 120, 491–501. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, J.; Torii, T.; Kusakawa, S.; Sanbe, A.; Nakamura, K.; Takashima, S.; Hamasaki, H.; Kawaguchi, S.; Miyamoto, Y.; Tanoue, A. The mood stabilizer valproic acid improves defective neurite formation caused by Charcot-Marie-Tooth disease-associated mutant Rab7 through the JNK signaling pathway. J. Neurosci. Res. 2010, 88, 3189–3197. [Google Scholar] [CrossRef]
- Markworth, R.; Dambeck, V.; Steinbeck, L.M.; Koufali, A.; Bues, B.; Dankovich, T.M.; Wichmann, C.; Burk, K. Tubular microdomains of Rab7-endosomes retrieve TrkA, a mechanism disrupted in Charcot-Marie-Tooth 2B. J. Cell Sci. 2021, 134, 258559. [Google Scholar] [CrossRef]
- Ponomareva, O.Y.; Eliceiri, K.W.; Halloran, M.C. Charcot-Marie-Tooth 2b associated Rab7 mutations cause axon growth and guidance defects during vertebrate sensory neuron development. Neural Dev. 2016, 11, 2. [Google Scholar] [CrossRef]
- Cioni, J.-M.; Lin, J.Q.; Holtermann, A.V.; Koppers, M.; Jakobs, M.A.H.; Azizi, A.; Turner-Bridger, B.; Shigeoka, T.; Franze, K.; Harris, W.A.; et al. Late Endosomes Act as mRNA Translation Platforms and Sustain Mitochondria in Axons. Cell 2019, 176, 56–72.e15. [Google Scholar] [CrossRef] [PubMed]
- Jordens, I.; Fernandez-Borja, M.; Marsman, M.; Dusseljee, S.; Janssen, L.; Calafat, J.; Janssen, H.; Wubbolts, R.; Neefjes, J. The Rab7 effector protein RILP controls lysosomal transport by inducing the recruitment of dynein-dynactin motors. Curr. Biol. 2001, 11, 1680–1685. [Google Scholar] [CrossRef]
- Cantalupo, G.; Alifano, P.; Roberti, V.; Bruni, C.B.; Bucci, C. Rab-interacting lysosomal protein (RILP): The Rab7 effector required for transport to lysosomes. EMBO J. 2001, 20, 683–693. [Google Scholar] [CrossRef] [PubMed]
- Yap, C.C.; Digilio, L.; McMahon, L.; Winckler, B. Disruption of Golgi markers by two RILP-directed shRNAs in neurons: A new role for RILP or a neuron-specific off-target phenotype? J. Biol. Chem. 2023, 299, 104916. [Google Scholar] [CrossRef] [PubMed]
- Cason, S.E.; Carman, P.J.; Van Duyne, C.; Goldsmith, J.; Dominguez, R.; Holzbaur, E.L.F. Sequential dynein effectors regulate axonal autophagosome motility in a maturation-dependent pathway. J. Cell Biol. 2021, 220, e202010179. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, E.M.; Elfmark, L.A.; Stenmark, H.; Raiborg, C. ER as master regulator of membrane trafficking and organelle function. J. Cell Biol. 2022, 221, e202205135. [Google Scholar] [CrossRef] [PubMed]
- Nakatsu, F.; Kawasaki, A. Functions of Oxysterol-Binding Proteins at Membrane Contact Sites and Their Control by Phosphoinositide Metabolism. Front. Cell Dev. Biol. 2021, 9, 664788. [Google Scholar] [CrossRef]
- Johansson, M.; Lehto, M.; Tanhuanpää, K.; Cover, T.L.; Olkkonen, V.M. The oxysterol-binding protein homologue ORP1L interacts with Rab7 and alters functional properties of late endocytic compartments. Mol. Biol. Cell 2005, 16, 5480–5492. [Google Scholar] [CrossRef]
- Johansson, M.; Rocha, N.; Zwart, W.; Jordens, I.; Janssen, L.; Kuijl, C.; Olkkonen, V.M.; Neefjes, J. Activation of endosomal dynein motors by stepwise assembly of Rab7-RILP-p150Glued, ORP1L, and the receptor betalll spectrin. J. Cell Biol. 2007, 176, 459–471. [Google Scholar] [CrossRef]
- Rocha, N.; Kuijl, C.; van der Kant, R.; Janssen, L.; Houben, D.; Janssen, H.; Zwart, W.; Neefjes, J. Cholesterol sensor ORP1L contacts the ER protein VAP to control Rab7-RILP-p150 Glued and late endosome positioning. J. Cell Biol. 2009, 185, 1209–1225. [Google Scholar] [CrossRef]
- Ma, X.; Liu, K.; Li, J.; Li, H.; Li, J.; Liu, Y.; Yang, C.; Liang, H. A non-canonical GTPase interaction enables ORP1L-Rab7-RILP complex formation and late endosome positioning. J. Biol. Chem. 2018, 293, 14155–14164. [Google Scholar] [CrossRef]
- Mukherjee, S.; Maxfield, F.R. Lipid and cholesterol trafficking in NPC. Biochim. Biophys. Acta 2004, 1685, 28–37. [Google Scholar] [CrossRef]
- Sugii, S.; Lin, S.; Ohgami, N.; Ohashi, M.; Chang, C.C.Y.; Chang, T.-Y. Roles of endogenously synthesized sterols in the endocytic pathway. J. Biol. Chem. 2006, 281, 23191–23206. [Google Scholar] [CrossRef] [PubMed]
- Levin-Konigsberg, R.; Montaño-Rendón, F.; Keren-Kaplan, T.; Li, R.; Ego, B.; Mylvaganam, S.; DiCiccio, J.E.; Trimble, W.S.; Bassik, M.C.; Bonifacino, J.S.; et al. Phagolysosome resolution requires contacts with the endoplasmic reticulum and phosphatidylinositol-4-phosphate signalling. Nat. Cell Biol. 2019, 21, 1234–1247. [Google Scholar] [CrossRef] [PubMed]
- Collins, M.P.; Forgac, M. Regulation and function of V-ATPases in physiology and disease. Biochim. Biophys. Acta Biomembr. 2020, 1862, 183341. [Google Scholar] [CrossRef] [PubMed]
- Chadwick, S.R.; Grinstein, S.; Freeman, S.A. From the inside out: Ion fluxes at the centre of endocytic traffic. Curr. Opin. Cell Biol. 2021, 71, 77–86. [Google Scholar] [CrossRef]
- Freeman, S.A.; Grinstein, S.; Orlowski, J. Determinants, maintenance, and function of organellar pH. Physiol. Rev. 2023, 103, 515–606. [Google Scholar] [CrossRef]
- Nakanishi-Matsui, M.; Matsumoto, N. V-ATPase a3 Subunit in Secretory Lysosome Trafficking in Osteoclasts. Biol. Pharm. Bull. 2022, 45, 1426–1431. [Google Scholar] [CrossRef]
- De Luca, M.; Cogli, L.; Progida, C.; Nisi, V.; Pascolutti, R.; Sigismund, S.; Di Fiore, P.P.; Bucci, C. RILP regulates vacuolar ATPase through interaction with the V1G1 subunit. J. Cell Sci. 2014, 127, 2697–2708. [Google Scholar] [CrossRef]
- Johnson, D.E.; Ostrowski, P.; Jaumouillé, V.; Grinstein, S. The position of lysosomes within the cell determines their luminal pH. J. Cell Biol. 2016, 212, 677–692. [Google Scholar] [CrossRef]
- Diering, G.H.; Numata, Y.; Fan, S.; Church, J.; Numata, M. Endosomal acidification by Na+/H+ exchanger NHE5 regulates TrkA cell-surface targeting and NGF-induced PI3K signaling. Mol. Biol. Cell 2013, 24, 3435–3448. [Google Scholar] [CrossRef]
- Ouyang, Q.; Lizarraga, S.B.; Schmidt, M.; Yang, U.; Gong, J.; Ellisor, D.; Kauer, J.A.; Morrow, E.M. Christianson syndrome protein NHE6 modulates TrkB endosomal signaling required for neuronal circuit development. Neuron 2013, 80, 97–112. [Google Scholar] [CrossRef]
- Ghosh, P.; Dahms, N.M.; Kornfeld, S. Mannose 6-phosphate receptors: New twists in the tale. Nat. Rev. Mol. Cell Biol. 2003, 4, 202–212. [Google Scholar] [CrossRef]
- Seaman, M.N.J. Cargo-selective endosomal sorting for retrieval to the Golgi requires retromer. J. Cell Biol. 2004, 165, 111–122. [Google Scholar] [CrossRef]
- Arighi, C.N.; Hartnell, L.M.; Aguilar, R.C.; Haft, C.R.; Bonifacino, J.S. Role of the mammalian retromer in sorting of the cation-independent mannose 6-phosphate receptor. J. Cell Biol. 2004, 165, 123–133. [Google Scholar] [CrossRef]
- Seaman, M.N.J. Retromer and the cation-independent mannose 6-phosphate receptor-Time for a trial separation? Traffic 2018, 19, 150–152. [Google Scholar] [CrossRef] [PubMed]
- McNally, K.E.; Cullen, P.J. Endosomal retrieval of cargo: Retromer is not alone. Trends Cell Biol. 2018, 28, 807–822. [Google Scholar] [CrossRef] [PubMed]
- Rojas, R.; van Vlijmen, T.; Mardones, G.A.; Prabhu, Y.; Rojas, A.L.; Mohammed, S.; Heck, A.J.R.; Raposo, G.; van der Sluijs, P.; Bonifacino, J.S. Regulation of retromer recruitment to endosomes by sequential action of Rab5 and Rab7. J. Cell Biol. 2008, 183, 513–526. [Google Scholar] [CrossRef] [PubMed]
- Seaman, M.N.J.; Harbour, M.E.; Tattersall, D.; Read, E.; Bright, N. Membrane recruitment of the cargo-selective retromer subcomplex is catalysed by the small GTPase Rab7 and inhibited by the Rab-GAP TBC1D5. J. Cell Sci. 2009, 122, 2371–2382. [Google Scholar] [CrossRef]
- Priya, A.; Kalaidzidis, I.V.; Kalaidzidis, Y.; Lambright, D.; Datta, S. Molecular insights into Rab7-mediated endosomal recruitment of core retromer: Deciphering the role of Vps26 and Vps35. Traffic 2015, 16, 68–84. [Google Scholar] [CrossRef]
- Seaman, M.N.J.; Mukadam, A.S.; Breusegem, S.Y. Inhibition of TBC1D5 activates Rab7a and can enhance the function of the retromer cargo-selective complex. J. Cell Sci. 2018, 131, jcs217398. [Google Scholar] [CrossRef]
- Jimenez-Orgaz, A.; Kvainickas, A.; Nägele, H.; Denner, J.; Eimer, S.; Dengjel, J.; Steinberg, F. Control of RAB7 activity and localization through the retromer-TBC1D5 complex enables RAB7-dependent mitophagy. EMBO J. 2018, 37, 235–254. [Google Scholar] [CrossRef]
- Maruzs, T.; Lőrincz, P.; Szatmári, Z.; Széplaki, S.; Sándor, Z.; Lakatos, Z.; Puska, G.; Juhász, G.; Sass, M. Retromer ensures the degradation of autophagic cargo by maintaining lysosome function in drosophila. Traffic 2015, 16, 1088–1107. [Google Scholar] [CrossRef]
- Cui, Y.; Carosi, J.M.; Yang, Z.; Ariotti, N.; Kerr, M.C.; Parton, R.G.; Sargeant, T.J.; Teasdale, R.D. Retromer has a selective function in cargo sorting via endosome transport carriers. J. Cell Biol. 2019, 218, 615–631. [Google Scholar] [CrossRef]
- Rahman, A.A.; Morrison, B.E. Contributions of VPS35 mutations to parkinson’s disease. Neuroscience 2019, 401, 1–10. [Google Scholar] [CrossRef]
- Sassone, J.; Reale, C.; Dati, G.; Regoni, M.; Pellecchia, M.T.; Garavaglia, B. The role of VPS35 in the pathobiology of parkinson’s disease. Cell. Mol. Neurobiol. 2021, 41, 199–227. [Google Scholar] [CrossRef]
- Muraleedharan, A.; Vanderperre, B. The Endo-lysosomal System in Parkinson’s Disease: Expanding the Horizon. J. Mol. Biol. 2023, 435, 168140. [Google Scholar] [CrossRef] [PubMed]
- Daly, J.L.; Danson, C.M.; Lewis, P.A.; Zhao, L.; Riccardo, S.; Di Filippo, L.; Cacchiarelli, D.; Lee, D.; Cross, S.J.; Heesom, K.J.; et al. Multi-omic approach characterises the neuroprotective role of retromer in regulating lysosomal health. Nat. Commun. 2023, 14, 3086. [Google Scholar] [CrossRef] [PubMed]
- Spang, A. Membrane tethering complexes in the endosomal system. Front. Cell Dev. Biol. 2016, 4, 35. [Google Scholar] [CrossRef] [PubMed]
- Seals, D.F.; Eitzen, G.; Margolis, N.; Wickner, W.T.; Price, A. A Ypt/Rab effector complex containing the Sec1 homolog Vps33p is required for homotypic vacuole fusion. Proc. Natl. Acad. Sci. USA 2000, 97, 9402–9407. [Google Scholar] [CrossRef]
- Wurmser, A.E.; Sato, T.K.; Emr, S.D. New component of the vacuolar class C-Vps complex couples nucleotide exchange on the Ypt7 GTPase to SNARE-dependent docking and fusion. J. Cell Biol. 2000, 151, 551–562. [Google Scholar] [CrossRef]
- Plemel, R.L.; Lobingier, B.T.; Brett, C.L.; Angers, C.G.; Nickerson, D.P.; Paulsel, A.; Sprague, D.; Merz, A.J. Subunit organization and Rab interactions of Vps-C protein complexes that control endolysosomal membrane traffic. Mol. Biol. Cell 2011, 22, 1353–1363. [Google Scholar] [CrossRef]
- Ho, R.; Stroupe, C. The HOPS/class C Vps complex tethers membranes by binding to one Rab GTPase in each apposed membrane. Mol. Biol. Cell 2015, 26, 2655–2663. [Google Scholar] [CrossRef] [PubMed]
- Khatter, D.; Raina, V.B.; Dwivedi, D.; Sindhwani, A.; Bahl, S.; Sharma, M. The small GTPase Arl8b regulates assembly of the mammalian HOPS complex on lysosomes. J. Cell Sci. 2015, 128, 1746–1761. [Google Scholar] [CrossRef]
- Schleinitz, A.; Pöttgen, L.-A.; Keren-Kaplan, T.; Pu, J.; Saftig, P.; Bonifacino, J.S.; Haas, A.; Jeschke, A. Consecutive functions of small GTPases guide HOPS-mediated tethering of late endosomes and lysosomes. Cell Rep. 2023, 42, 111969. [Google Scholar] [CrossRef] [PubMed]
- Jongsma, M.L.; Bakker, J.; Cabukusta, B.; Liv, N.; van Elsland, D.; Fermie, J.; Akkermans, J.L.; Kuijl, C.; van der Zanden, S.Y.; Janssen, L.; et al. SKIP-HOPS recruits TBC1D15 for a Rab7-to-Arl8b identity switch to control late endosome transport. EMBO J. 2020, 39, e102301. [Google Scholar] [CrossRef]
- Jiang, P.; Nishimura, T.; Sakamaki, Y.; Itakura, E.; Hatta, T.; Natsume, T.; Mizushima, N. The HOPS complex mediates autophagosome-lysosome fusion through interaction with syntaxin 17. Mol. Biol. Cell 2014, 25, 1327–1337. [Google Scholar] [CrossRef] [PubMed]
- Takáts, S.; Pircs, K.; Nagy, P.; Varga, Á.; Kárpáti, M.; Hegedűs, K.; Kramer, H.; Kovács, A.L.; Sass, M.; Juhász, G. Interaction of the HOPS complex with Syntaxin 17 mediates autophagosome clearance in Drosophila. Mol. Biol. Cell 2014, 25, 1338–1354. [Google Scholar] [CrossRef]
- Lin, X.; Yang, T.; Wang, S.; Wang, Z.; Yun, Y.; Sun, L.; Zhou, Y.; Xu, X.; Akazawa, C.; Hong, W.; et al. RILP interacts with HOPS complex via VPS41 subunit to regulate endocytic trafficking. Sci. Rep. 2014, 4, 7282. [Google Scholar] [CrossRef]
- van der Kant, R.; Fish, A.; Janssen, L.; Janssen, H.; Krom, S.; Ho, N.; Brummelkamp, T.; Carette, J.; Rocha, N.; Neefjes, J. Late endosomal transport and tethering are coupled processes controlled by RILP and the cholesterol sensor ORP1L. J. Cell Sci. 2013, 126, 3462–3474. [Google Scholar] [CrossRef]
- Janssens, K.; Goethals, S.; Atkinson, D.; Ermanoska, B.; Fransen, E.; Jordanova, A.; Auer-Grumbach, M.; Asselbergh, B.; Timmerman, V. Human Rab7 mutation mimics features of Charcot-Marie-Tooth neuropathy type 2B in Drosophila. Neurobiol. Dis. 2014, 65, 211–219. [Google Scholar] [CrossRef]
- Harrison, M.S.; Hung, C.-S.; Liu, T.; Christiano, R.; Walther, T.C.; Burd, C.G. A mechanism for retromer endosomal coat complex assembly with cargo. Proc. Natl. Acad. Sci. USA 2014, 111, 267–272. [Google Scholar] [CrossRef]
- Romano, R.; Del Fiore, V.S.; Saveri, P.; Palamà, I.E.; Pisciotta, C.; Pareyson, D.; Bucci, C.; Guerra, F. Autophagy and lysosomal functionality in CMT2B fibroblasts carrying the RAB7K126R mutation. Cells 2022, 11, 496. [Google Scholar] [CrossRef] [PubMed]
- Harrington, A.W.; St Hillaire, C.; Zweifel, L.S.; Glebova, N.O.; Philippidou, P.; Halegoua, S.; Ginty, D.D. Recruitment of actin modifiers to TrkA endosomes governs retrograde NGF signaling and survival. Cell 2011, 146, 421–434. [Google Scholar] [CrossRef] [PubMed]
- Overly, C.C.; Hollenbeck, P.J. Dynamic organization of endocytic pathways in axons of cultured sympathetic neurons. J. Neurosci. 1996, 16, 6056–6064. [Google Scholar] [CrossRef] [PubMed]
- Farfel-Becker, T.; Roney, J.C.; Cheng, X.-T.; Li, S.; Cuddy, S.R.; Sheng, Z.-H. Neuronal Soma-Derived Degradative Lysosomes Are Continuously Delivered to Distal Axons to Maintain Local Degradation Capacity. Cell Rep. 2019, 28, 51–64.e4. [Google Scholar] [CrossRef]
- Colecchia, D.; Stasi, M.; Leonardi, M.; Manganelli, F.; Nolano, M.; Veneziani, B.M.; Santoro, L.; Eskelinen, E.-L.; Chiariello, M.; Bucci, C. Alterations of autophagy in the peripheral neuropathy Charcot-Marie-Tooth type 2B. Autophagy 2018, 14, 930–941. [Google Scholar] [CrossRef] [PubMed]
- Giudetti, A.M.; Guerra, F.; Longo, S.; Beli, R.; Romano, R.; Manganelli, F.; Nolano, M.; Mangini, V.; Santoro, L.; Bucci, C. An altered lipid metabolism characterizes Charcot-Marie-Tooth type 2B peripheral neuropathy. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2020, 1865, 158805. [Google Scholar] [CrossRef]
- Kuchitsu, Y.; Fukuda, M. Revisiting Rab7 functions in mammalian autophagy: Rab7 knockout studies. Cells 2018, 7, 215. [Google Scholar] [CrossRef]
- Hancock-Cerutti, W.; Wu, Z.; Xu, P.; Yadavalli, N.; Leonzino, M.; Tharkeshwar, A.K.; Ferguson, S.M.; Shadel, G.S.; De Camilli, P. ER-lysosome lipid transfer protein VPS13C/PARK23 prevents aberrant mtDNA-dependent STING signaling. J. Cell Biol. 2022, 221, e202106046. [Google Scholar] [CrossRef]
- Özkan, N.; Koppers, M.; van Soest, I.; van Harten, A.; Jurriens, D.; Liv, N.; Klumperman, J.; Kapitein, L.C.; Hoogenraad, C.C.; Farías, G.G. ER-lysosome contacts at a pre-axonal region regulate axonal lysosome availability. Nat. Commun. 2021, 12, 4493. [Google Scholar] [CrossRef]
- Gao, Y.; Xiong, J.; Chu, Q.-Z.; Ji, W.-K. PDZD8-mediated lipid transfer at contacts between the ER and late endosomes/lysosomes is required for neurite outgrowth. J. Cell Sci. 2022, 135, jcs255026. [Google Scholar] [CrossRef]
- Kuijpers, M.; Nguyen, P.T.; Haucke, V. The endoplasmic reticulum and its contacts: Emerging roles in axon development, neurotransmission, and degeneration. Neuroscientist 2023, 10738584231162810. [Google Scholar] [CrossRef] [PubMed]
- Wijdeven, R.H.; Janssen, H.; Nahidiazar, L.; Janssen, L.; Jalink, K.; Berlin, I.; Neefjes, J. Cholesterol and ORP1L-mediated ER contact sites control autophagosome transport and fusion with the endocytic pathway. Nat. Commun. 2016, 7, 11808. [Google Scholar] [CrossRef] [PubMed]
- Cogli, L.; Progida, C.; Thomas, C.L.; Spencer-Dene, B.; Donno, C.; Schiavo, G.; Bucci, C. Charcot-Marie-Tooth type 2B disease-causing RAB7A mutant proteins show altered interaction with the neuronal intermediate filament peripherin. Acta Neuropathol. 2013, 125, 257–272. [Google Scholar] [CrossRef] [PubMed]
- Wong, Y.C.; Kim, S.; Peng, W.; Krainc, D. Regulation and Function of Mitochondria-Lysosome Membrane Contact Sites in Cellular Homeostasis. Trends Cell Biol. 2019, 29, 500–513. [Google Scholar] [CrossRef] [PubMed]
- Wong, Y.C.; Ysselstein, D.; Krainc, D. Mitochondria-lysosome contacts regulate mitochondrial fission via RAB7 GTP hydrolysis. Nature 2018, 554, 382–386. [Google Scholar] [CrossRef] [PubMed]
- Wong, Y.C.; Peng, W.; Krainc, D. Lysosomal Regulation of Inter-mitochondrial Contact Fate and Motility in Charcot-Marie-Tooth Type 2. Dev. Cell 2019, 50, 339–354.e4. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Effector | Function | Rab7CMT2B Interaction vs. Rab7WT | Abundance | Observed Outcomes for CMT2B Alleles Attributable to Effector |
|---|---|---|---|---|
| RILP | Dynein motor adaptor protein; late endosomal positioning and motility | Equal, in overexpression followed by immunoprecipitation experiments [32,33] | Decreased in N161T and K126R patient tissues [12,15] | -Increased anterograde motility [60] -Increased retrograde motility [79] -Decreased stationary and pausing time [78,79,129] -Increased endosome speeds [60,79] -Decreased endosome speeds [78] |
| ORP1L | Late endosome positioning and motility (w/RILP); lipid exchange and ER contacts | Increased by IP-MS [L129F, V162M] [32] | Undetermined | -Positioning and motility same as RILP -ER contact sites undetermined |
| VPS13C | Lipid exchange and ER contact sites | Increased by IP-MS [L129F, V162M] [32] | Undetermined | -Increased lipid droplet abundance [131,136] -Increased cholesterol ester:cholesterol ratios, increased monounsaturated fatty acids, free fatty acids, and total neutral lipids [136] |
| VPS35 | Core retromer complex component; sorting and recycling of membrane receptors | -Preserved [L129F, N161T] -Decreased [K157N] -Undetermined [V162M, K126R] [105] | Undetermined | -Increased M6PR levels [V162M] [73] -Increased mature cathepsins [V162M/K126R] [73,131] -Increased TrkA tubule behavior [L129F/N161T]; no change in TrkA tubule behavior [K157N/V162M] [77] |
| V-ATPase Subunits | Endosomal and lysosomal acidification (w/RILP) | Undetermined | Undetermined | Undetermined |
| HOPS Subunits | Endosomal tethering and fusion (w/RILP) | Undetermined (possibly decreased [27]) | Undetermined | Undetermined |
| Peripherin | Peripheral nervous system specific intermediate filament | Increased [15,143] | Increased [15] | -Increased soluble: insoluble peripherin [143] -Undetermined impact on endosome behavior |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mulligan, R.J.; Winckler, B. Regulation of Endosomal Trafficking by Rab7 and Its Effectors in Neurons: Clues from Charcot–Marie–Tooth 2B Disease. Biomolecules 2023, 13, 1399. https://doi.org/10.3390/biom13091399
Mulligan RJ, Winckler B. Regulation of Endosomal Trafficking by Rab7 and Its Effectors in Neurons: Clues from Charcot–Marie–Tooth 2B Disease. Biomolecules. 2023; 13(9):1399. https://doi.org/10.3390/biom13091399
Chicago/Turabian StyleMulligan, Ryan J., and Bettina Winckler. 2023. "Regulation of Endosomal Trafficking by Rab7 and Its Effectors in Neurons: Clues from Charcot–Marie–Tooth 2B Disease" Biomolecules 13, no. 9: 1399. https://doi.org/10.3390/biom13091399