
Patients with Systemic Juvenile Idiopathic Arthritis (SJIA) Show Differences in Autoantibody Signatures Based on Disease Activity
 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Samples
2.2. IgG Purification
2.3. Protein Microarray Processing
2.4. Protein Microarray Data Analysis
3. Results
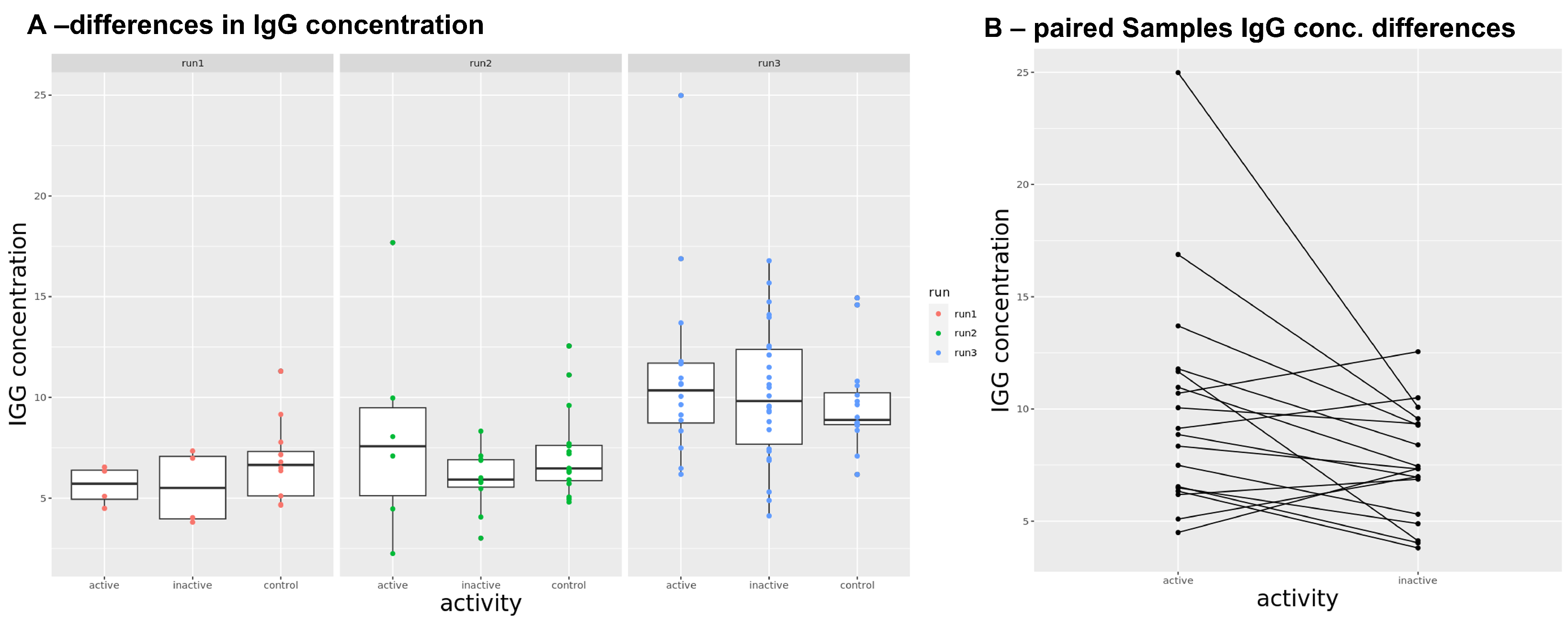
3.1. IgG Concentration in Sera
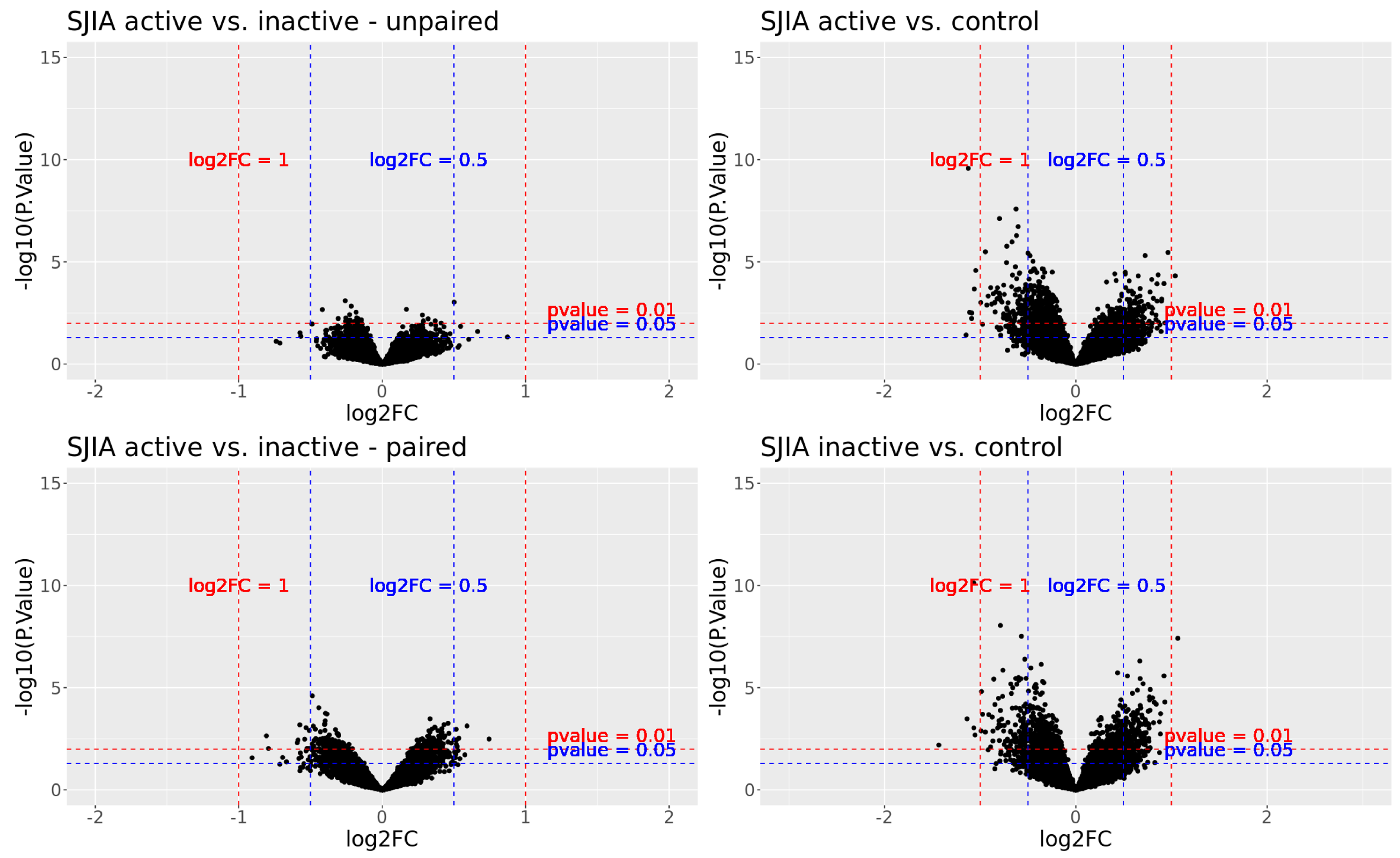
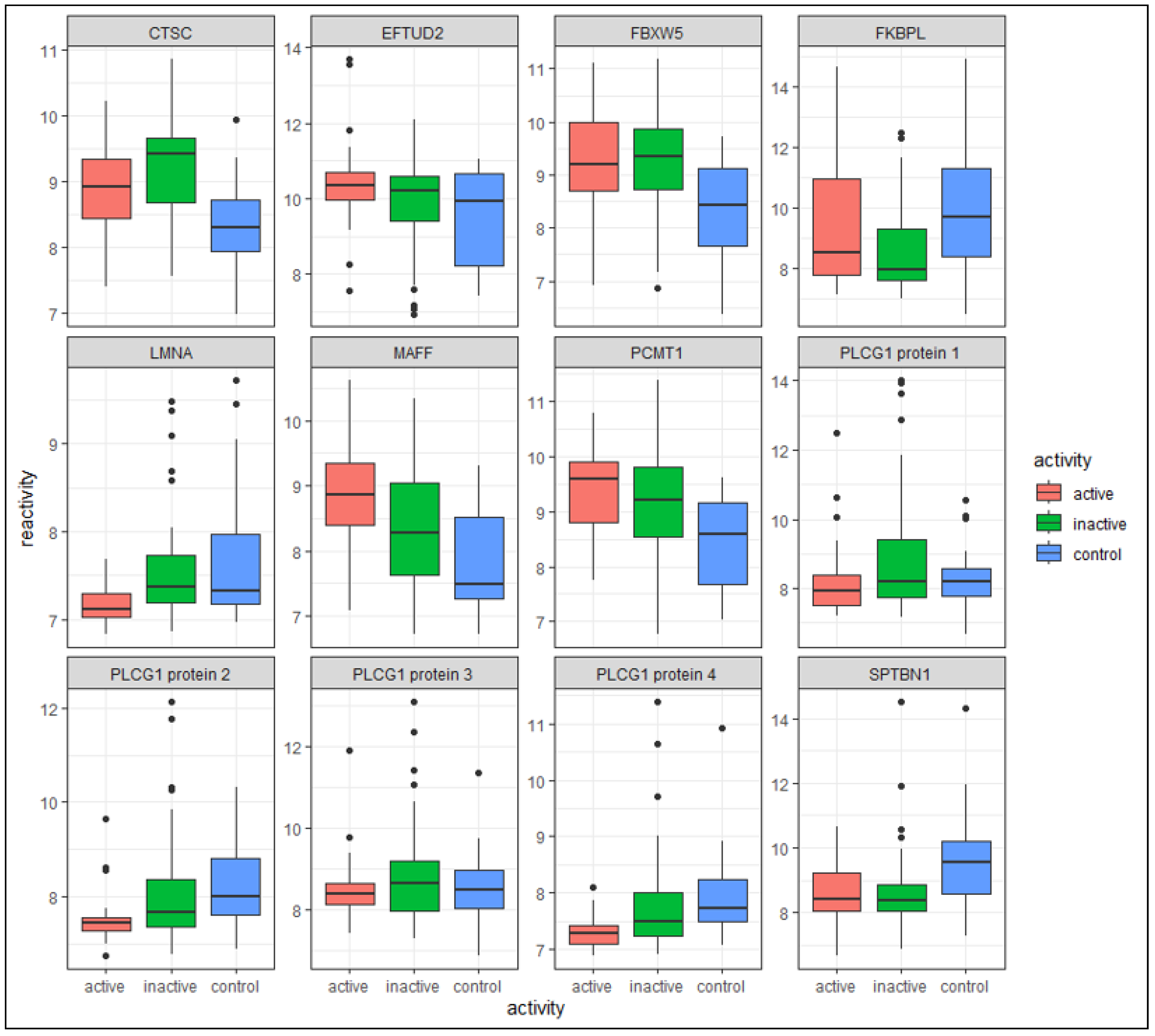
3.2. Differentially Reactive Antigen Analysis and Functional Enrichment
3.2.1. Active SJIA versus Inactive SJIA
3.2.2. Active SJIA versus Healthy Controls
3.2.3. Inactive SJIA versus Healthy Controls
3.2.4. Overlapping DIRAGs in All Three Group Comparisons
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ravelli, A.; Martini, A. Juvenile idiopathic arthritis. Lancet 2007, 369, 767–778. [Google Scholar] [CrossRef] [PubMed]
- Petty, R.E.; Southwood, T.R.; Manners, P.; Baum, J.; Glass, D.N.; Goldenberg, J.; He, X.; Maldonado-Cocco, J.; Orozco-Alcala, J.; Prieur, A.M.; et al. International League of Associations for Rheumatology classification of juvenile idiopathic arthritis: Second revision, Edmonton, 2001. J. Rheumatol. 2004, 31, 390–392. [Google Scholar] [PubMed]
- Ombrello, M.J.; Arthur, V.L.; Remmers, E.F.; Hinks, A.; Tachmazidou, I.; Grom, A.A.; Foell, D.; Martini, A.; Gattorno, M.; Özen, S.; et al. Genetic architecture distinguishes systemic juvenile idiopathic arthritis from other forms of juvenile idiopathic arthritis: Clinical and therapeutic implications. Ann. Rheum. Dis. 2017, 76, 906–913. [Google Scholar] [CrossRef] [PubMed]
- Crayne, C.B.; Albeituni, S.; Nichols, K.E.; Cron, R.Q. The Immunology of Macrophage Activation Syndrome. Front. Immunol. 2019, 10, 119. [Google Scholar] [CrossRef]
- Schulert, G.S.; Grom, A.A. Pathogenesis of Macrophage Activation Syndrome and Potential for Cytokine-Directed Therapies. Annu. Rev. Med. 2015, 66, 145–159. [Google Scholar] [CrossRef]
- Lerkvaleekul, B.; Vilaiyuk, S. Macrophage activation syndrome: Early diagnosis is key. Open Access Rheumatol. Res. Rev. 2018, 10, 117–128. [Google Scholar] [CrossRef]
- Ravelli, A.; Magni-Manzoni, S.; Pistorio, A.; Besana, C.; Foti, T.; Ruperto, N.; Viola, S.; Martini, A. Preliminary diagnostic guidelines for macrophage activation syndrome complicating systemic juvenile idiopathic arthritis. J. Pediatr. 2005, 146, 598–604. [Google Scholar] [CrossRef]
- Arnow, P.M.; Flaherty, J.P. Fever of unknown origin. Lancet 1997, 350, 575–580. [Google Scholar] [CrossRef]
- Wittkowski, H.; Frosch, M.; Wulffraat, N.; Goldbach-Mansky, R.; Kallinich, T.; Kuemmerle-Deschner, J.; Frühwald, M.C.; Dassmann, S.; Pham, T.H.; Roth, J.; et al. S100A12 is a novel molecular marker differentiating systemic-onset juvenile idiopathic arthritis from other causes of fever of unknown origin. Arthritis Rheum. 2008, 58, 3924–3931. [Google Scholar] [CrossRef]
- Pardeo, M.; Bracaglia, C.; De Benedetti, F. Systemic juvenile idiopathic arthritis: New insights into pathogenesis and cytokine directed therapies. Best Pract. Res. Clin. Rheumatol. 2017, 31, 505–516. [Google Scholar] [CrossRef]
- Pardeo, M.; Vastert, S.J.; De Benedetti, F. It is about time: The first validated biomarker for early diagnosis of systemic juvenile idiopathic arthritis. Rheumatology 2022, 61, 2724–2725. [Google Scholar] [CrossRef] [PubMed]
- Swart, J.F.; de Roock, S.; Prakken, B.J. Understanding inflammation in juvenile idiopathic arthritis: How immune biomarkers guide clinical strategies in the systemic onset subtype. Eur. J. Immunol. 2016, 46, 2068–2077. [Google Scholar] [CrossRef] [PubMed]
- Qu, H.; Sundberg, E.; Aulin, C.; Neog, M.; Palmblad, K.; Horne, A.C.; Granath, F.; Ek, A.; Melén, E.; Olsson, M.; et al. Immunoprofiling of active and inactive systemic juvenile idiopathic arthritis reveals distinct biomarkers: A single-center study. Pediatr. Rheumatol. 2021, 19, 173. [Google Scholar] [CrossRef] [PubMed]
- Vastert, S.J.; de Jager, W.; Noordman, B.J.; Holzinger, D.; Kuis, W.; Prakken, B.J.; Wulffraat, N.M. Effectiveness of First-Line Treatment with Recombinant Interleukin-1 Receptor Antagonist in Steroid-Naive Patients with New-Onset Systemic Juvenile Idiopathic Arthritis: Results of a Prospective Cohort Study. Arthritis Rheumatol. 2014, 66, 1034–1043. [Google Scholar] [CrossRef]
- Nigrovic, P.A. Review: Is There a Window of Opportunity for Treatment of Systemic Juvenile Idiopathic Arthritis? Arthritis Rheumatol. 2014, 66, 1405–1413. [Google Scholar] [CrossRef] [PubMed]
- Mannion, M.L.; Cron, R.Q. Therapeutic strategies for treating juvenile idiopathic arthritis. Curr. Opin. Pharmacol. 2022, 64, 102226. [Google Scholar] [CrossRef]
- Vastert, S.J.; Kuis, W.; Grom, A.A. Systemic JIA: New Developments in the Understanding of the Pathophysiology and Therapy. Best Pract. Res. Clin. Rheumatol. 2009, 23, 655–664. [Google Scholar] [CrossRef]
- Autmizguine, J.; Cohen-Wolkowiez, M.; Ilowite, N.; For The Rapport Investigators. Rilonacept pharmacokinetics in children with systemic juvenile idiopathic arthritis: The Journal of Clinical Pharmacology. J. Clin. Pharmacol. 2015, 55, 39–44. [Google Scholar] [CrossRef]
- Arnold, D.D.; Yalamanoglu, A.; Boyman, O. Systematic Review of Safety and Efficacy of IL-1-Targeted Biologics in Treating Immune-Mediated Disorders. Front. Immunol. 2022, 13, 888392. [Google Scholar] [CrossRef]
- Ruperto, N.; Brunner, H.I.; Quartier, P.; Constantin, T.; Wulffraat, N.; Horneff, G.; Brik, R.; McCann, L.; Kasapcopur, O.; Rutkowska-Sak, L.; et al. Two Randomized Trials of Canakinumab in Systemic Juvenile Idiopathic Arthritis. N. Engl. J. Med. 2012, 367, 2396–2406. [Google Scholar] [CrossRef]
- Żuber, Z.; Opoka-Winiarska, V.; Smolewska, E. Recommendations for the therapeutic management of systemic juvenile idiopathic arthritis. Opinion of the Section of Developmental Age Rheumatology of the Polish Society of Rheumatology. Rheumatol. Forum 2022, 8, 6–13. [Google Scholar] [CrossRef]
- Kessel, C.; Hedrich, C.M.; Foell, D. Innately Adaptive or Truly Autoimmune: Is There Something Unique About Systemic Juvenile Idiopathic Arthritis? Arthritis Rheumatol. 2020, 72, 210–219. [Google Scholar] [CrossRef] [PubMed]
- Nigrovic, P.A. Autoinflammation and autoimmunity in systemic juvenile idiopathic arthritis. Proc. Natl. Acad. Sci. USA 2015, 112, 15785–15786. [Google Scholar] [CrossRef]
- Hügle, B.; Hinze, C.; Lainka, E.; Fischer, N.; Haas, J.P. Development of positive antinuclear antibodies and rheumatoid factor in systemic juvenile idiopathic arthritis points toward an autoimmune phenotype later in the disease course. Pediatr. Rheumatol. 2014, 12, 28. [Google Scholar] [CrossRef]
- Kuehn, J.; Schleifenbaum, S.; Hendling, M.; Siebenhandl, S.; Krainer, J.; Fuehner, S.; Hellige, A.; Park, C.; Hinze, C.; Wittkowski, H.; et al. Aberrant Naive CD4–Positive T Cell Differentiation in Systemic Juvenile Idiopathic Arthritis Committed to B Cell Help. Arthritis Rheumatol. 2023, 75, 826–841. [Google Scholar] [CrossRef]
- McGonagle, D.; McDermott, M.F. A Proposed Classification of the Immunological Diseases. PLoS Med. 2006, 3, e297. [Google Scholar] [CrossRef] [PubMed]
- Lleo, A.; Invernizzi, P.; Gao, B.; Podda, M.; Gershwin, M.E. Definition of human autoimmunity—Autoantibodies versus autoimmune disease. Autoimmun. Rev. 2010, 9, A259–A266. [Google Scholar] [CrossRef]
- Bloom, B.J.; Toyoda, M.; Petrosian, A.; Jordan, S. Anti-endothelial cell antibodies are prevalent in juvenile idiopathic arthritis: Implications for clinical disease course and pathogenesis. Rheumatol. Int. 2007, 27, 655–660. [Google Scholar] [CrossRef]
- Serra, C.R.B.; Rodrigues, S.H.; Silva, N.; Sztajnbok, F.; Andrade, L.E.C. Clinical significance of anticardiolipin antibodies in juvenile idiopathic arthritis. Clin. Exp. Rheumatol. 1999, 17, 375–380. [Google Scholar]
- Schulz, C.; Fuehner, S.; Schlüter, B.; Fobker, M.; Sengler, C.; Klotsche, J.; Niewerth, M.; Minden, K.; Foell, D. Prevalence of autoantibodies in patients with juvenile idiopathic arthritis: Results from the German inception cohort ICON-JIA. Pediatr. Rheumatol. 2022, 20, 8. [Google Scholar] [CrossRef]
- Luna Coronell, J.A.; Sergelen, K.; Hofer, P.; Gyurján, I.; Brezina, S.; Hettegger, P.; Leeb, G.; Mach, K.; Gsur, A.; Weinhäusel, A. The Immunome of Colon Cancer: Functional In Silico Analysis of Antigenic Proteins Deduced from IgG Microarray Profiling. Genom. Proteom. Bioinform. 2018, 16, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Gyurján, I.; Rosskopf, S.; Coronell, J.A.L.; Muhr, D.; Singer, C.; Weinhäusel, A. IgG based immunome analyses of breast cancer patients reveal underlying signaling pathways. Oncotarget 2019, 10, 3491–3505. [Google Scholar] [CrossRef] [PubMed]
- Milchram, L.; Fischer, A.; Huber, J.; Soldo, R.; Sieghart, D.; Vierlinger, K.; Blüml, S.; Steiner, G.; Weinhäusel, A. Functional Analysis of Autoantibody Signatures in Rheumatoid Arthritis. Molecules 2022, 27, 1452. [Google Scholar] [CrossRef] [PubMed]
- Jodeleit, H.; Milchram, L.; Soldo, R.; Beikircher, G.; Schönthaler, S.; Al-amodi, O.; Wolf, E.; Beigel, F.; Weinhäusel, A.; Siebeck, M.; et al. Autoantibodies as diagnostic markers and potential drivers of inflammation in ulcerative colitis. PLoS ONE 2020, 15, e0228615. [Google Scholar] [CrossRef]
- Brezina, S.; Soldo, R.; Kreuzhuber, R.; Hofer, P.; Gsur, A.; Weinhaeusel, A. Immune-Signatures for Lung Cancer Diagnostics: Evaluation of Protein Microarray Data Normalization Strategies. Microarrays 2015, 4, 162–187. [Google Scholar] [CrossRef]
- Stempfer, R.; Syed, P.; Vierlinger, K.; Pichler, R.; Meese, E.; Leidinger, P.; Ludwig, N.; Kriegner, A.; Nöhammer, C.; Weinhäusel, A. Tumour auto-antibody screening: Performance of protein microarrays using SEREX derived antigens. BMC Cancer 2010, 10, 627. [Google Scholar] [CrossRef]
- Johnson, W.E.; Li, C.; Rabinovic, A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics 2007, 8, 118–127. [Google Scholar] [CrossRef]
- Leek, J.T.; Johnson, W.E.; Parker, H.S.; Jaffe, A.E.; Storey, J.D. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics 2012, 28, 882–883. [Google Scholar] [CrossRef]
- Gillespie, M.; Jassal, B.; Stephan, R.; Milacic, M.; Rothfels, K.; Senff-Ribeiro, A.; Griss, J.; Sevilla, C.; Matthews, L.; Gong, C.; et al. The reactome pathway knowledgebase 2022. Nucleic Acids Res. 2022, 50, D687–D692. [Google Scholar] [CrossRef]
- Liao, Y.; Wang, J.; Jaehnig, E.J.; Shi, Z.; Zhang, B. WebGestalt 2019: Gene set analysis toolkit with revamped UIs and APIs. Nucleic Acids Res. 2019, 47, W199–W205. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B (Methodol.) 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016. [Google Scholar]
- Yokota, S.; Itoh, Y.; Morio, T.; Origasa, H.; Sumitomo, N.; Tomobe, M.; Tanaka, K.; Minota, S. Tocilizumab in systemic juvenile idiopathic arthritis in a real-world clinical setting: Results from 1 year of postmarketing surveillance follow-up of 417 patients in Japan. Ann. Rheum. Dis. 2016, 75, 1654–1660. [Google Scholar] [CrossRef] [PubMed]
- Aljaberi, N.; Tronconi, E.; Schulert, G.; Grom, A.A.; Lovell, D.J.; Huggins, J.L.; Henrickson, M.; Brunner, H.I. The use of S100 proteins testing in juvenile idiopathic arthritis and autoinflammatory diseases in a pediatric clinical setting: A retrospective analysis. Pediatr. Rheumatol. 2020, 18, 7. [Google Scholar] [CrossRef] [PubMed]
- Rayhan SM, A.M. Comparison of Serum Immunoglobulin (IgG, IgM and IgA) Concentration During Active and Inactive Disease States in Patients with Juvenile Idiopathic Arthritis. Eur. J. Mol. Clin. Med. 2023, 10, 102–110. [Google Scholar]
- Moon, E.J.; Mello, S.S.; Li, C.G.; Chi, J.T.; Thakkar, K.; Kirkland, J.G.; Lagory, E.L.; Lee, I.J.; Diep, A.N.; Miao, Y.; et al. The HIF target MAFF promotes tumor invasion and metastasis through IL11 and STAT3 signaling. Nat. Commun. 2021, 12, 4308. [Google Scholar] [CrossRef] [PubMed]
- Hügle, B.; Schippers, A.; Fischer, N.; Ohl, K.; Denecke, B.; Ticconi, F.; Vastert, B.; Costa, I.G.; Haas, J.P.; Tenbrock, K. Transcription factor motif enrichment in whole transcriptome analysis identifies STAT4 and BCL6 as the most prominent binding motif in systemic juvenile idiopathic arthritis. Arthritis Res. Ther. 2018, 20, 98. [Google Scholar] [CrossRef] [PubMed]
- Milner, J.D.; Vogel, T.P.; Forbes, L.; Ma, C.A.; Stray-Pedersen, A.; Niemela, J.E.; Lyons, J.J.; Engelhardt, K.R.; Zhang, Y.; Topcagic, N.; et al. Early-onset lymphoproliferation and autoimmunity caused by germline STAT3 gain-of-function mutations. Blood 2015, 125, 591–599. [Google Scholar] [CrossRef]
- LI, H.W.; XIE, Y.; LI, F.; SUN, G.C.; CHEN, Z.; ZENG, H.S. Effect of miR-19a and miR-21 on the JAK/STAT signaling pathway in the peripheral blood mononuclear cells of patients with systemic juvenile idiopathic arthritis. Exp. Ther. Med. 2016, 11, 2531–2536. [Google Scholar] [CrossRef]
- Bunney, T.D.; Esposito, D.; Mas-Droux, C.; Lamber, E.; Baxendale, R.W.; Martins, M.; Cole, A.; Svergun, D.; Driscoll, P.C.; Katan, M. Structural and Functional Integration of the PLCγ Interaction Domains Critical for Regulatory Mechanisms and Signaling Deregulation. Structure 2012, 20, 2062–2075. [Google Scholar] [CrossRef]
- Ombrello, M.J.; Remmers, E.F.; Sun, G.; Freeman, A.F.; Datta, S.; Torabi-Parizi, P.; Subramanian, N.; Bunney, T.D.; Baxendale, R.W.; Martins, M.S.; et al. Cold Urticaria, Immunodeficiency, and Autoimmunity Related to PLCG2 Deletions. N. Engl. J. Med. 2012, 366, 330–338. [Google Scholar] [CrossRef]
- Zhou, Q.; Lee, G.S.; Brady, J.; Datta, S.; Katan, M.; Sheikh, A.; Martins, M.S.; Bunney, T.D.; Santich, B.H.; Moir, S.; et al. A Hypermorphic Missense Mutation in PLCG2, Encoding Phospholipase Cγ2, Causes a Dominantly Inherited Autoinflammatory Disease with Immunodeficiency. Am. J. Hum. Genet. 2012, 91, 713–720. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Yang, Z.; Li, H.; Zhu, J.; Wang, Y.; Tang, Q.; Shi, Z. Phospholipase Cγ1 (PLCG1) overexpression is associated with tumor growth and poor survival in IDH wild-type lower-grade gliomas in adult patients. Lab. Investig. 2022, 102, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Duarte, R.; Rebelo de Almeida, C.; Negrão, M.; Fernandes, A.; Borralho, P.; Sobral, D.; Gallego-Paez, L.M.; Machado, D.; Gramaça, J.; Vílchez, J.; et al. Predictive and Therapeutic Implications of a Novel PLCγ1/SHP2-Driven Mechanism of Cetuximab Resistance in Metastatic Colorectal Cancer. Clin. Cancer Res. 2022, 28, 1203–1216. [Google Scholar] [CrossRef]
- Fu, G.; Chen, Y.; Yu, M.; Podd, A.; Schuman, J.; He, Y.; Di, L.; Yassai, M.; Haribhai, D.; North, P.E.; et al. Phospholipase Cγ1 is essential for T cell development, activation, and tolerance. J. Exp. Med. 2010, 207, 309–318. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, G.; Ji, Q.s. Phospholipase C-γ as a Signal-Transducing Element. Exp. Cell Res. 1999, 253, 15–24. [Google Scholar] [CrossRef]
- Alam, S.; Liu, Q.; Liu, S.; Liu, Y.; Zhang, Y.; Yang, X.; Liu, G.; Fan, K.; Ma, J. Up-regulated cathepsin C induces macrophage M1 polarization through FAK-triggered p38 MAPK/NF-κB pathway. Exp. Cell Res. 2019, 382, 111472. [Google Scholar] [CrossRef]
- Srivastava, S.K.; Ramana, K.V. Focus on Molecules: Nuclear Factor-kappaB. Exp. Eye Res. 2009, 88, 2–3. [Google Scholar] [CrossRef]
- Annett, S.; Moore, G.; Short, A.; Marshall, A.; McCrudden, C.; Yakkundi, A.; Das, S.; McCluggage, W.G.; Nelson, L.; Harley, I.; et al. FKBPL-based peptide, ALM201, targets angiogenesis and cancer stem cells in ovarian cancer. Br. J. Cancer 2020, 122, 361–371. [Google Scholar] [CrossRef]
- Annett, S.; Spence, S.; Garciarena, C.; Campbell, C.; Dennehy, M.; Drakeford, C.; Lai, J.; Dowling, J.; Moore, G.; Yakkundi, A.; et al. The immunophilin protein FKBPL and its peptide derivatives are novel regulators of vascular integrity and inflammation via NF-κB signaling. bioRxiv 2021. [Google Scholar] [CrossRef]
- Fan, Y.; Tan, D.; Zhang, X.; Song, D.; Chang, X.; Wang, S.; Yan, H.; Ge, L.; Yang, H.; Bönnemann, C.; et al. Nuclear Factor-κB Pathway Mediates the Molecular Pathogenesis of LMNA-Related Muscular Dystrophies. Biochem. Genet. 2020, 58, 966–980. [Google Scholar] [CrossRef]
- Lavrovsky, Y.; Chatterjee, B.; Clark, R.A.; Roy, A.K. Role of redox-regulated transcription factors in inflammation, aging and age-related diseases. Exp. Gerontol. 2000, 35, 521–532. [Google Scholar] [CrossRef] [PubMed]
- Zhao, P.; Chao, W.; Li, W. FBXW5 reduction alleviates spinal cord injury (SCI) by blocking microglia activity: A mechanism involving p38 and JNK. Biochem. Biophys. Res. Commun. 2019, 514, 558–564. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Chen, S.; Wang, H.; Gao, B.; Kallakury, B.; Bhuvaneshwar, K.; Cahn, K.; Gusev, Y.; Wang, X.; Wu, Y.; et al. SPTBN1 inhibits inflammatory responses and hepatocarcinogenesis via the stabilization of SOCS1 and downregulation of p65 in hepatocellular carcinoma. Theranostics 2021, 11, 4232–4250. [Google Scholar] [CrossRef] [PubMed]
- Dai, L.p.; Xu, X.d.; Yang, T.t.; Yin, Z.h.; Ye, Z.z.; Wei, Y.z. SPTBN1 attenuates rheumatoid arthritis synovial cell proliferation, invasion, migration and inflammatory response by binding to PIK3R2. Immunity Inflamm. Dis. 2022, 10, e724. [Google Scholar] [CrossRef] [PubMed]
- De Arras, L.; Laws, R.; Leach, S.M.; Pontis, K.; Freedman, J.H.; Schwartz, D.A.; Alper, S. Comparative Genomics RNAi Screen Identifies Eftud2 as a Novel Regulator of Innate Immunity. Genetics 2014, 197, 485–496. [Google Scholar] [CrossRef]
- Taylor, J.C.; Bongartz, T.; Massey, J.; Mifsud, B.; Spiliopoulou, A.; Scott, I.C.; Wang, J.; Morgan, M.; Plant, D.; Colombo, M.; et al. Genome-wide association study of response to methotrexate in early rheumatoid arthritis patients. Pharmacogenom. J. 2018, 18, 528–538. [Google Scholar] [CrossRef]
- White, C.R.; Dungan, M.; Carrithers, M.D. Activation of human macrophage sodium channels regulates RNA processing to increase expression of the DNA repair protein PPP1R10. Immunobiology 2019, 224, 80–93. [Google Scholar] [CrossRef]
- Zhang, Z.; Li, F.; Li, Y.; Li, Z.; Jia, G. In vitro Anti-malignant Property of PCMT1 Silencing and Identification of the SNHG16/miR-195/PCMT1 Regulatory Axis in Breast Cancer Cells. Clin. Breast Cancer 2023, 23, 302–316. [Google Scholar] [CrossRef]
- Guo, J.; Du, X.; Li, C. PCMT1 Is a Potential Prognostic Biomarker and Is Correlated with Immune Infiltrates in Breast Cancer. BioMed Res. Int. 2022, 2022, e4434887. [Google Scholar] [CrossRef]
- Belkourchia, F.; Desrosiers, R.R. The Protein L-Isoaspartyl (D-Aspartyl) Methyltransferase Regulates Glial-to-Mesenchymal Transition and Migration Induced by TGF-β1 in Human U-87 MG Glioma Cells. Int. J. Mol. Sci. 2022, 23, 5698. [Google Scholar] [CrossRef]
- Guo, W.; Chen, X.; Zhu, L.; Wang, Q. A six-mRNA signature model for the prognosis of head and neck squamous cell carcinoma. Oncotarget 2017, 8, 94528–94538. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Li, Y.; Liu, H.; Zhang, J.; Wang, J.; Xia, J.; Zhang, Y.; Yu, X.; Ma, J.; Huang, M.; et al. Genome-wide CRISPR/Cas9 library screen identifies PCMT1 as a critical driver of ovarian cancer metastasis. J. Exp. Clin. Cancer Res. CR 2022, 41, 24. [Google Scholar] [CrossRef] [PubMed]
- Dong, L.; Li, Y.; Xue, D.; Liu, Y. PCMT1 is an unfavorable predictor and functions as an oncogene in bladder cancer. IUBMB life 2018, 70, 291–299. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Zhang, G.; Li, X.; Rao, S.; Gong, B.; Jiang, W.; Hao, D.; Wu, P.; Wu, C.; Du, L.; et al. A systematic method for mapping multiple loci: An application to construct a genetic network for rheumatoid arthritis. Gene 2008, 408, 104–111. [Google Scholar] [CrossRef]
- Tong, M.; Jun, T.; Nie, Y.; Hao, J.; Fan, D. The Role of the Slit/Robo Signaling Pathway. J. Cancer 2019, 10, 2694–2705. [Google Scholar] [CrossRef] [PubMed]
- Blockus, H.; Chédotal, A. Slit-Robo signaling. Development 2016, 143, 3037–3044. [Google Scholar] [CrossRef] [PubMed]
- Bros, M.; Haas, K.; Moll, L.; Grabbe, S. RhoA as a Key Regulator of Innate and Adaptive Immunity. Cells 2019, 8, 733. [Google Scholar] [CrossRef]
- Park, Y.H.; Wood, G.; Kastner, D.L.; Chae, J.J. Pyrin inflammasome activation and RhoA signaling in the autoinflammatory diseases FMF and HIDS. Nat. Immunol. 2016, 17, 914–921. [Google Scholar] [CrossRef]
- Supek, F.; Lehner, B.; Lindeboom, R.G.H. To NMD or Not To NMD: Nonsense-Mediated mRNA Decay in Cancer and Other Genetic Diseases. Trends Genet. 2021, 37, 657–668. [Google Scholar] [CrossRef]
- Grandemange, S.; Soler, S.; Touitou, I. Expression of the familial Mediterranean fever gene is regulated by nonsense-mediated decay. Hum. Mol. Genet. 2009, 18, 4746–4755. [Google Scholar] [CrossRef]
- Mino, T.; Murakawa, Y.; Fukao, A.; Vandenbon, A.; Wessels, H.H.; Ori, D.; Uehata, T.; Tartey, S.; Akira, S.; Suzuki, Y.; et al. Regnase-1 and Roquin Regulate a Common Element in Inflammatory mRNAs by Spatiotemporally Distinct Mechanisms. Cell 2015, 161, 1058–1073. [Google Scholar] [CrossRef]
- Johansson, U.; Portinsson, S.; Akesson, A.; Svantesson, H.; Ockerman, P.A.; Akesson, B. Nutritional status in girls with juvenile chronic arthritis. Hum. Nutr. Clin. Nutr. 1986, 40, 57–67. [Google Scholar] [PubMed]
- Ma, Y.; Zhang, X.; Fan, D.; Xia, Q.; Wang, M.; Pan, F. Common trace metals in rheumatoid arthritis: A systematic review and meta-analysis. J. Trace Elem. Med. Biol. 2019, 56, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Barrett, C.W.; Short, S.P.; Williams, C.S. Selenoproteins and oxidative stress-induced inflammatory tumorigenesis in the gut. Cell. Mol. Life Sci. 2017, 74, 607–616. [Google Scholar] [CrossRef]
- Nettleford, S.K.; Prabhu, K.S. Selenium and Selenoproteins in Gut Inflammation—A Review. Antioxidants 2018, 7, 36. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| SJIA | Active | Inactive | Healthy Controls | |
|---|---|---|---|---|
| n | 66 | 26 (39.4%) | 40 (60.6%) | 23 |
| Sex (% male) | 35 (53) | 14 (53.8) | 21 (52.5) | 12 (52.2) |
| Age at diagnosis 1,2 | 7.1 (0.6–17.4) | 6.6 (0.6–17.3) | 7.1 (0.8–17.4) | n/a |
| Age at sampling 1,2 | 11.5 (1.0–20.8) | 10.0 (1.0–20.2) | 12.6 (2.4–20.8) | 14.7 (10.5–15.9) |
| Disease duration 1,2 | 3.5 (<0.1–17.3) | 1.6 (<0.1–15.3) | 3.4 (0.4–17.3) | n/a |
| CRP (mg/dL) 2 | 6.8 (0.5–21.1) % documented: 92.3 | <0.5 % documented: 95 | <0.5 % documented: 95.7 | |
| ESR (mm/hour) 2 | 66 (10–134) % documented: 80.8 | 5.1 (1–30) % documented: 92.5 | 3 (<1–13) % documented: 52.2 | |
| Leukocytes/nL 2 | 15.2 (5–23.8) % documented: 84.6 | 5.9 (3.6–9.4) % documented: 92.5 | 5.5 (<1–7.9) % documented: 95.7 | |
| S100A12 (ng/mL) 2 | 1195 (180–19,410) % documented: 100 | 39 (8–2450) % documented: 100 | n/a | |
| S100A8/A9 (ng/mL) 2 | 7320 (720–220,850) % documented: 34.6 | 560 (100–20,710) % documented: 27.5 | n/a | |
| Steroids 3 | 18 (69.2) | 9 (22.5) | 0 | |
| bDMARDs 3 | 21 (80.8) | 24 (60) | 0 | |
| csDMARDs 3 | 10 (38.4) | 14 (35) | 0 |
| Higher in | log2FC | FC | p Value | Gene | Higher in | log2FC | FC | p Value | Gene | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| a—active vs. control | active | 1.039 | 2.055 | 0.000 | MAFF | b—inactive vs. control | inactive | 1.067 | 2.095 | 0.000 | HPRT1 |
| 0.964 | 1.951 | 0.000 | HPRT1 | 0.931 | 1.907 | 0.000 | LMF2 | ||||
| 0.931 | 1.907 | 0.010 | EFTUD2 | 0.923 | 1.896 | 0.000 | CTSC | ||||
| 0.923 | 1.895 | 0.000 | PSMC4 | 0.887 | 1.849 | 0.000 | RPS11 | ||||
| 0.898 | 1.864 | 0.001 | PCMT1 | 0.885 | 1.847 | 0.002 | FBXW5 | ||||
| 0.895 | 1.860 | 0.001 | ADK | 0.875 | 1.834 | 0.015 | EMC10 | ||||
| 0.892 | 1.855 | 0.025 | EFCC1 | 0.875 | 1.834 | 0.000 | RPS27A | ||||
| 0.880 | 1.841 | 0.012 | FMNL1 | 0.841 | 1.792 | 0.000 | TFAP4 | ||||
| 0.866 | 1.822 | 0.009 | ISYNA1 | 0.827 | 1.774 | 0.046 | BAG5 | ||||
| 0.861 | 1.816 | 0.000 | HDAC2 | 0.820 | 1.765 | 0.000 | AXIN2 | ||||
| control | −1.147 | 0.452 | 0.038 | CFAP36 | control | −1.433 | 0.370 | 0.006 | FKBPL | ||
| −1.125 | 0.458 | 0.000 | SNX1 | −1.137 | 0.455 | 0.000 | PSMD4 | ||||
| −1.111 | 0.463 | 0.003 | GAREM1 | −1.067 | 0.477 | 0.001 | PSMD4 | ||||
| −1.090 | 0.470 | 0.006 | ERCC5 | −1.066 | 0.478 | 0.000 | SNX1 | ||||
| −1.088 | 0.470 | 0.003 | SPTBN1 | −1.054 | 0.482 | 0.002 | GAREM1 | ||||
| −1.062 | 0.479 | 0.000 | MZF1 | −0.990 | 0.503 | 0.001 | OTUB1 | ||||
| −1.047 | 0.484 | 0.000 | ASCC2 | −0.987 | 0.505 | 0.000 | ASCC2 | ||||
| −0.994 | 0.502 | 0.001 | KIF2C | −0.975 | 0.509 | 0.000 | MZF1 | ||||
| −0.975 | 0.509 | 0.011 | MLLT6 | −0.946 | 0.519 | 0.001 | PSMD4 | ||||
| −0.946 | 0.519 | 0.000 | CALB2 | −0.917 | 0.530 | 0.011 | ERCC5 | ||||
| c—active vs. Inactive (paired) | active | 0.745 | 1.676 | 0.003 | IARS2 | d—active vs. Inactive (unpaired) | active | 0.873 | 1.832 | 0.047 | FKBPL |
| 0.591 | 1.507 | 0.001 | CALM3 | 0.665 | 1.586 | 0.025 | ARFIP2 | ||||
| 0.576 | 1.491 | 0.019 | MEIS3 | 0.546 | 1.460 | 0.014 | MAFF | ||||
| 0.542 | 1.456 | 0.030 | ZNF232 | 0.502 | 1.416 | 0.001 | CALM3 | ||||
| 0.535 | 1.449 | 0.003 | MAFF | 0.473 | 1.388 | 0.033 | TMEM183A | ||||
| 0.530 | 1.444 | 0.012 | IGHV3−21 | 0.444 | 1.360 | 0.047 | RTN4 | ||||
| 0.523 | 1.437 | 0.008 | BYSL | 0.443 | 1.359 | 0.034 | APBA2 | ||||
| 0.518 | 1.432 | 0.004 | SDHA | 0.430 | 1.348 | 0.015 | PPP1CA | ||||
| 0.516 | 1.430 | 0.006 | ARID1A | 0.426 | 1.344 | 0.030 | SAP30BP | ||||
| 0.516 | 1.430 | 0.001 | TPM4 | 0.422 | 1.340 | 0.046 | ACLY | ||||
| inactive | −0.907 | 0.533 | 0.027 | PLCG1 | inactive | −0.573 | 0.672 | 0.030 | PLCG1 | ||
| −0.807 | 0.572 | 0.002 | AGAP2 | −0.568 | 0.675 | 0.044 | AGAP2 | ||||
| −0.792 | 0.577 | 0.009 | APBA2 | −0.491 | 0.711 | 0.011 | PLCG1 | ||||
| −0.694 | 0.618 | 0.026 | PLCG1 | −0.487 | 0.713 | 0.011 | CLDN3 | ||||
| −0.667 | 0.630 | 0.042 | PLCG1 | −0.417 | 0.749 | 0.002 | LMNA | ||||
| −0.592 | 0.663 | 0.005 | DOCK2 | −0.394 | 0.761 | 0.023 | BUB1B | ||||
| −0.587 | 0.666 | 0.004 | RSL1D1 | −0.394 | 0.761 | 0.033 | CTSC | ||||
| −0.581 | 0.669 | 0.029 | PLCG1 | −0.388 | 0.764 | 0.046 | ACAD10 | ||||
| −0.574 | 0.672 | 0.001 | RPL10A | −0.387 | 0.765 | 0.022 | RPL36A | ||||
| −0.570 | 0.673 | 0.015 | FLAD1 | −0.372 | 0.773 | 0.048 | SMARCB1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krainer, J.; Hendling, M.; Siebenhandl, S.; Fuehner, S.; Kessel, C.; Verweyen, E.; Vierlinger, K.; Foell, D.; Schönthaler, S.; Weinhäusel, A. Patients with Systemic Juvenile Idiopathic Arthritis (SJIA) Show Differences in Autoantibody Signatures Based on Disease Activity. Biomolecules 2023, 13, 1392. https://doi.org/10.3390/biom13091392
Krainer J, Hendling M, Siebenhandl S, Fuehner S, Kessel C, Verweyen E, Vierlinger K, Foell D, Schönthaler S, Weinhäusel A. Patients with Systemic Juvenile Idiopathic Arthritis (SJIA) Show Differences in Autoantibody Signatures Based on Disease Activity. Biomolecules. 2023; 13(9):1392. https://doi.org/10.3390/biom13091392
Chicago/Turabian StyleKrainer, Julie, Michaela Hendling, Sandra Siebenhandl, Sabrina Fuehner, Christoph Kessel, Emely Verweyen, Klemens Vierlinger, Dirk Foell, Silvia Schönthaler, and Andreas Weinhäusel. 2023. "Patients with Systemic Juvenile Idiopathic Arthritis (SJIA) Show Differences in Autoantibody Signatures Based on Disease Activity" Biomolecules 13, no. 9: 1392. https://doi.org/10.3390/biom13091392