
Understanding the Clinical Use of Levosimendan and Perspectives on its Future in Oncology



Abstract
:1. Introduction to Drug Repurposing
2. The Drug Levosimendan
2.1. Pharmacodynamics
2.2. Pharmacokinetics
3. Clinical Use of Levosimendan
3.1. Inotropy
3.2. Vasodilation
3.3. Organ Protection
4. Repurposing Levosimendan for Oncology
4.1. Phosphodiesterase 3 Inhibition
4.2. Nitric Oxide Production
4.3. Reactive Oxygen Species Reduction
Plasma Malondialdehyde
4.4. Drug Combinations
5. Challenges and Perspectives
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, J.; McNamee, C.; et al. Drug repurposing: Progress, challenges and recommendations. Nat. Rev. Drug Discov. 2019, 18, 41–58. [Google Scholar] [CrossRef] [PubMed]
- Khataniar, A.; Pathak, U.; Rajkhowa, S.; Jha, A.N. A Comprehensive Review of Drug Repurposing Strategies against Known Drug Targets of COVID-19. COVID 2022, 2, 148–167. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhou, L.; Xie, N.; Nice, E.C.; Zhang, T.; Cui, Y.; Huang, C. Overcoming cancer therapeutic bottleneck by drug repurposing. Signal Transduct. Target. Ther. 2020, 5, 113. [Google Scholar] [CrossRef]
- Ribeiro, E.; Vale, N. Repurposing of the Drug Tezosentan for Cancer Therapy. Curr. Issues Mol. Biol. 2023, 45, 5118–5131. [Google Scholar] [CrossRef]
- Okuyama, R. Advancements in Drug Repurposing: Examples in Psychiatric Medications. Int. J. Mol. Sci. 2023, 24, 11000. [Google Scholar] [CrossRef]
- Marwick, C. AZT (zidovudine) just a step away from FDA approval for AIDS therapy. JAMA 1987, 257, 1281–1282. [Google Scholar] [CrossRef]
- Haider, M.; Elsherbeny, A.; Pittalà, V.; Fallica, A.N.; Alghamdi, M.A.; Greish, K. The Potential Role of Sildenafil in Cancer Management through EPR Augmentation. J. Pers. Med. 2021, 11, 585. [Google Scholar] [CrossRef]
- 1—Discovery and Development of Doxorubicin. In Medicinal Chemistry; Arcamone, F. (Ed.) Elsevier: Amsterdam, The Netherlands, 1981; Volume 17, pp. 1–47. [Google Scholar]
- Meredith, A.M.; Dass, C.R. Increasing role of the cancer chemotherapeutic doxorubicin in cellular metabolism. J. Pharm. Pharmacol. 2016, 68, 729–741. [Google Scholar] [CrossRef]
- Kim, J.H.; Scialli, A.R. Thalidomide: The tragedy of birth defects and the effective treatment of disease. Toxicol. Sci. 2011, 122, 1–6. [Google Scholar] [CrossRef]
- Zhou, S.; Wang, F.; Hsieh, T.C.; Wu, J.M.; Wu, E. Thalidomide-a notorious sedative to a wonder anticancer drug. Curr. Med. Chem. 2013, 20, 4102–4108. [Google Scholar] [CrossRef]
- Das, A.; Durrant, D.; Mitchell, C.; Mayton, E.; Hoke, N.N.; Salloum, F.N.; Park, M.A.; Qureshi, I.; Lee, R.; Dent, P.; et al. Sildenafil increases chemotherapeutic efficacy of doxorubicin in prostate cancer and ameliorates cardiac dysfunction. Proc. Natl. Acad. Sci. USA 2010, 107, 18202–18207. [Google Scholar] [CrossRef] [PubMed]
- Greish, K.; Fateel, M.; Abdelghany, S.; Rachel, N.; Alimoradi, H.; Bakhiet, M.; Alsaie, A. Sildenafil citrate improves the delivery and anticancer activity of doxorubicin formulations in a mouse model of breast cancer. J. Drug Target 2018, 26, 610–615. [Google Scholar] [CrossRef] [PubMed]
- El-Naa, M.M.; Othman, M.; Younes, S. Sildenafil potentiates the antitumor activity of cisplatin by induction of apoptosis and inhibition of proliferation and angiogenesis. Drug Des. Devel. Ther. 2016, 10, 3661–3672. [Google Scholar] [CrossRef] [PubMed]
- Hsu, J.L.; Leu, W.J.; Hsu, L.C.; Ho, C.H.; Liu, S.P.; Guh, J.H. Phosphodiesterase Type 5 Inhibitors Synergize Vincristine in Killing Castration-Resistant Prostate Cancer Through Amplifying Mitotic Arrest Signaling. Front. Oncol. 2020, 10, 1274. [Google Scholar] [CrossRef]
- Mei, X.L.; Yang, Y.; Zhang, Y.J.; Li, Y.; Zhao, J.M.; Qiu, J.G.; Zhang, W.J.; Jiang, Q.W.; Xue, Y.Q.; Zheng, D.W.; et al. Sildenafil inhibits the growth of human colorectal cancer in vitro and in vivo. Am. J. Cancer Res. 2015, 5, 3311–3324. [Google Scholar]
- Marques, J.G.; Gaspar, V.M.; Markl, D.; Costa, E.C.; Gallardo, E.; Correia, I.J. Co-delivery of Sildenafil (Viagra(®)) and Crizotinib for synergistic and improved anti-tumoral therapy. Pharm. Res. 2014, 31, 2516–2528. [Google Scholar] [CrossRef]
- Hajipour, H.; Ghorbani, M.; Kahroba, H.; Mahmoodzadeh, F.; Emameh, R.Z.; Taheri, R.A. Arginyl-glycyl-aspartic acid (RGD) containing nanostructured lipid carrier co-loaded with doxorubicin and sildenafil citrate enhanced anti-cancer effects and overcomes drug resistance. Process. Biochem. 2019, 84, 172–179. [Google Scholar] [CrossRef]
- Talevi, A.; Bellera, C.L. Challenges and opportunities with drug repurposing: Finding strategies to find alternative uses of therapeutics. Expert Opin. Drug Discov. 2020, 15, 397–401. [Google Scholar] [CrossRef]
- Ribeiro, E.; Costa, B.; Marques, L.; Vasques-Nóvoa, F. Repurposing of Sildenafil, Tezosentan and Levosimendan Drugs against Bladder and Lung Cancer Cell Lines. Unpublished manuscript.
- Pathak, A.; Lebrin, M.; Vaccaro, A.; Senard, J.M.; Despas, F. Pharmacology of levosimendan: Inotropic, vasodilatory and cardioprotective effects. J. Clin. Pharm. Ther. 2013, 38, 341–349. [Google Scholar] [CrossRef]
- Thorvaldsen, T.; Benson, L.; Hagerman, I.; Dahlström, U.; Edner, M.; Lund, L.H. Planned repetitive use of levosimendan for heart failure in cardiology and internal medicine in Sweden. Int. J. Cardiol. 2014, 175, 55–61. [Google Scholar] [CrossRef]
- Kamath, S.R.; Jaykumar, I.; Matha, S. Levosimendan. Indian Pediatr. 2009, 46, 593–596. [Google Scholar] [PubMed]
- Papp, Z.; Agostoni, P.; Alvarez, J.; Bettex, D.; Bouchez, S.; Brito, D.; Černý, V.; Comin-Colet, J.; Crespo-Leiro, M.G.; Delgado, J.F.; et al. Levosimendan Efficacy and Safety: 20 years of SIMDAX in Clinical Use. Card Fail. Rev. 2020, 6, e19. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.; Yang, Y.M.; Zhu, J.Y.; Lu, Y.Q. Multiorgan Drug Action of Levosimendan in Critical Illnesses. Biomed. Res. Int. 2019, 2019, 9731467. [Google Scholar] [CrossRef] [PubMed]
- Grześk, G.; Wołowiec, Ł.; Rogowicz, D.; Gilewski, W.; Kowalkowska, M.; Banach, J.; Hertmanowski, W.; Dobosiewicz, M. The importance of pharmacokinetics, pharmacodynamic and repetitive use of levosimendan. Biomed. Pharmacother. 2022, 153, 113391. [Google Scholar] [CrossRef]
- Põder, P.; Eha, J.; Sundberg, S.; Antila, S.; Heinpalu, M.; Loogna, I.; Planken, Ü.; Rantanen, S.; Lehtonen, L. Pharmacodynamics and Pharmacokinetics of Oral Levosimendan and Its Metabolites in Patients With Severe Congestive Heart Failure: A Dosing Interval Study. J. Clin. Pharmacol. 2004, 44, 1143–1150. [Google Scholar] [CrossRef]
- Antila, S.; Huuskonen, H.; Nevalainen, T.; Kanerva, H.; Vanninen, P.; Lehtonen, L. Site dependent bioavailability and metabolism of levosimendan in dogs. Eur. J. Pharm. Sci. 1999, 9, 85–91. [Google Scholar] [CrossRef]
- Sandell, E.-P.; Häyhä, M.; Antila, S.; Heikkinen, P.; Ottoila, P.; Lehtonen, L.A.; Pentikäinen, P.J. Pharmacokinetics of levosimendan in healthy volunteers and patients with congestive heart failure. J. Cardiovasc. Pharmacol. 1995, 26, 57–62. [Google Scholar] [CrossRef]
- Antila, S.; Honkanen, T.; Lehtonen, L.; Neuvonen, P.J. The CYP3A4 inhibitor intraconazole does not affect the pharmacokinetics of a new calcium-sensitizing drug levosimendan. Int. J. Clin. Pharmacol. Ther. 1998, 36, 446–449. [Google Scholar]
- Papp, Z.; Édes, I.; Fruhwald, S.; De Hert, S.G.; Salmenperä, M.; Leppikangas, H.; Mebazaa, A.; Landoni, G.; Grossini, E.; Caimmi, P. Levosimendan: Molecular mechanisms and clinical implications: Consensus of experts on the mechanisms of action of levosimendan. Int. J. Cardiol. 2012, 159, 82–87. [Google Scholar] [CrossRef]
- Yildiz, O. Vasodilating mechanisms of levosimendan: Involvement of K+ channels. J. Pharmacol. Sci. 2007, 104, 1–5. [Google Scholar] [CrossRef]
- Siegel, G.; Emden, J.; Wenzel, K.; Mironneau, J.; Stock, G. Potassium channel activation in vascular smooth muscle. Excit. Contract. Coupling Skelet. Card. Smooth Muscle 1992, 53–72. [Google Scholar]
- Follath, F.; Cleland, J.G.; Just, H.; Papp, J.; Scholz, H.; Peuhkurinen, K.; Harjola, V.; Mitrovic, V.; Abdalla, M.; Sandell, E. Efficacy and safety of intravenous levosimendan compared with dobutamine in severe low-output heart failure (the LIDO study): A randomised double-blind trial. Lancet 2002, 360, 196–202. [Google Scholar] [CrossRef] [PubMed]
- Rieg, A.D.; Rossaint, R.; Verjans, E.; Maihöfer, N.A.; Uhlig, S.; Martin, C. Levosimendan relaxes pulmonary arteries and veins in precision-cut lung slices-the role of KATP-channels, cAMP and cGMP. PLoS ONE 2013, 8, e66195. [Google Scholar] [CrossRef] [PubMed]
- Rieg, A.D.; Suleiman, S.; Bünting, N.A.; Verjans, E.; Spillner, J.; Schnöring, H.; Kalverkamp, S.; Schröder, T.; von Stillfried, S.; Braunschweig, T. Levosimendan reduces segmental pulmonary vascular resistance in isolated perfused rat lungs and relaxes human pulmonary vessels. PLoS ONE 2020, 15, e0233176. [Google Scholar] [CrossRef] [PubMed]
- Yokoshiki, H.; Katsube, Y.; Sunagawa, M.; Sperelakis, N. Levosimendan, a novel Ca2+ sensitizer, activates the glibenclamide-sensitive K+ channel in rat arterial myocytes. Eur. J. Pharmacol. 1997, 333, 249–259. [Google Scholar] [CrossRef]
- Yokoshiki, H.; Katsube, Y.; Sunagawa, M.; Sperelakis, N. The novel calcium sensitizer levosimendan activates the ATP-sensitive K+ channel in rat ventricular cells. J. Pharmacol. Exp. Ther. 1997, 283, 375–383. [Google Scholar]
- Yildiz, O.; Seyrek, M.; Yildirim, V.; Demirkilic, U.; Nacitarhan, C. Potassium channel-related relaxation by levosimendan in the human internal mammary artery. Ann. Thorac. Sur.g 2006, 81, 1715–1719. [Google Scholar] [CrossRef]
- Kaheinen, P.; Pollesello, P.; Levijoki, J.; Haikala, H. Levosimendan increases diastolic coronary flow in isolated guinea-pig heart by opening ATP-sensitive potassium channels. J. Cardiovasc. Pharmacol. 2001, 37, 367–374. [Google Scholar] [CrossRef]
- Ozdem, S.S.; Yalcin, O.; Meiselman, H.J.; Baskurt, O.K.; Usta, C. The role of potassium channels in relaxant effect of levosimendan in rat small mesenteric arteries. Cardiovasc. Drugs Ther. 2006, 20, 123–127. [Google Scholar] [CrossRef]
- Zager, R.A.; Johnson, A.C.; Lund, S.; Hanson, S.Y.; Abrass, C.K. Levosimendan protects against experimental endotoxemic acute renal failure. Am J Physiol Ren. Physiol 2006, 290, F1453–F1462. [Google Scholar] [CrossRef]
- Pataricza, J.; Krassói, I.; Höhn, J.; Kun, A.; Papp, J.G. Functional role of potassium channels in the vasodilating mechanism of levosimendan in porcine isolated coronary artery. Cardiovasc. Drugs Ther. 2003, 17, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Yildiz, O.; Nacitarhan, C.; Seyrek, M. Potassium channels in the vasodilating action of levosimendan on the human umbilical artery. J. Soc. Gynecol. Investig. 2006, 13, 312–315. [Google Scholar] [CrossRef] [PubMed]
- Usta, C.; Eksert, B.; Gölbasi, I.; Bigat, Z.; Ozdem, S.S. The role of potassium channels in the vasodilatory effect of levosimendan in human internal thoracic arteries. Eur. J. Cardiothorac. Surg. 2006, 30, 329–332. [Google Scholar] [CrossRef] [PubMed]
- Akar, F.; Manavbasi, Y.; Parlar, A.I.; Ulus, A.T.; Katircioglu, S.F. The gender differences in the relaxation to levosimendan of human internal mammary artery. Cardiovasc. Drugs Ther. 2007, 21, 331–338. [Google Scholar] [CrossRef]
- Gruhn, N.; Nielsen-Kudsk, J.E.; Theilgaard, S.; Bang, L.; Olesen, S.P.; Aldershvile, J. Coronary vasorelaxant effect of levosimendan, a new inodilator with calcium-sensitizing properties. J. Cardiovasc. Pharmacol. 1998, 31, 741–749. [Google Scholar] [CrossRef] [PubMed]
- Szilágyi, S.; Pollesello, P.; Levijoki, J.; Kaheinen, P.; Haikala, H.; Edes, I.; Papp, Z. The effects of levosimendan and OR-1896 on isolated hearts, myocyte-sized preparations and phosphodiesterase enzymes of the guinea pig. Eur. J. Pharmacol. 2004, 486, 67–74. [Google Scholar] [CrossRef]
- Grossini, E.; Molinari, C.; Caimmi, P.P.; Uberti, F.; Vacca, G. Levosimendan induces NO production through p38 MAPK, ERK and Akt in porcine coronary endothelial cells: Role for mitochondrial K(ATP) channel. Br. J. Pharmacol. 2009, 156, 250–261. [Google Scholar] [CrossRef]
- Caimmi, P.P.; Molinari, C.; Uberti, F.; Micalizzi, E.; Valente, G.; Mary, D.A.; Vacca, G.; Grossini, E. Intracoronary levosimendan prevents myocardial ischemic damages and activates survival signaling through ATP-sensitive potassium channel and nitric oxide. Eur. J. Cardiothorac. Surg. 2011, 39, e59–e67. [Google Scholar] [CrossRef]
- Erdei, N.; Papp, Z.; Pollesello, P.; Edes, I.; Bagi, Z. The levosimendan metabolite OR-1896 elicits vasodilation by activating the K(ATP) and BK(Ca) channels in rat isolated arterioles. Br. J. Pharmacol. 2006, 148, 696–702. [Google Scholar] [CrossRef]
- Gödény, I.; Pollesello, P.; Edes, I.; Papp, Z.; Bagi, Z. Levosimendan and its metabolite OR-1896 elicit KATP channel-dependent dilation in resistance arteries in vivo. Pharmacol. Rep. 2013, 65, 1304–1310. [Google Scholar] [CrossRef]
- Pataricza, J.; Hõhn, J.; Petri, A.; Balogh, A.; Papp, J.G. Comparison of the vasorelaxing effect of cromakalim and the new inodilator, levosimendan, in human isolated portal vein. J. Pharm. Pharmacol. 2000, 52, 213–217. [Google Scholar] [CrossRef]
- Höhn, J.; Pataricza, J.; Petri, A.; Tóth, G.K.; Balogh, A.; Varró, A.; Papp, J.G. Levosimendan interacts with potassium channel blockers in human saphenous veins. Basic. Clin. Pharmacol. Toxicol. 2004, 94, 271–273. [Google Scholar] [CrossRef] [PubMed]
- De Witt, B.J.; Ibrahim, I.N.; Bayer, E.; Fields, A.M.; Richards, T.A.; Banister, R.E.; Kaye, A.D. An analysis of responses to levosimendan in the pulmonary vascular bed of the cat. Anesth. Analg. 2002, 94, 1427–1433. [Google Scholar] [CrossRef] [PubMed]
- Erb, J.; Beutlhauser, T.; Feldheiser, A.; Schuster, B.; Treskatsch, S.; Grubitzsch, H.; Spies, C. Influence of levosimendan on organ dysfunction in patients with severely reduced left ventricular function undergoing cardiac surgery. J. Int. Med. Res. 2014, 42, 750–764. [Google Scholar] [CrossRef]
- Hausenloy, D.J.; Maddock, H.L.; Baxter, G.F.; Yellon, D.M. Inhibiting mitochondrial permeability transition pore opening: A new paradigm for myocardial preconditioning? Cardiovasc. Res. 2002, 55, 534–543. [Google Scholar] [CrossRef] [PubMed]
- Dolder, M.; Wendt, S.; Wallimann, T. Mitochondrial creatine kinase in contact sites: Interaction with porin and adenine nucleotide translocase, role in permeability transition and sensitivity to oxidative damage. Biol. Signals Recept. 2001, 10, 93–111. [Google Scholar] [CrossRef]
- Zaugg, M.; Schaub, M.C. Signaling and cellular mechanisms in cardiac protection by ischemic and pharmacological preconditioning. J. Muscle Res. Cell Motil. 2003, 24, 219–249. [Google Scholar] [CrossRef] [PubMed]
- Quesada, I.; Rovira, J.M.; Martin, F.; Roche, E.; Nadal, A.; Soria, B. Nuclear KATP channels trigger nuclear Ca(2+) transients that modulate nuclear function. Proc. Natl. Acad. Sci. USA 2002, 99, 9544–9549. [Google Scholar] [CrossRef]
- Soeding, P.E.; Royse, C.F.; Wright, C.E.; Royse, A.G.; Angus, J.A. Inoprotection: The perioperative role of levosimendan. Anaesth. Intensive Care 2007, 35, 845–862. [Google Scholar] [CrossRef]
- Liu, Y.; Sato, T.; O’Rourke, B.; Marban, E. Mitochondrial ATP-dependent potassium channels: Novel effectors of cardioprotection? Circulation 1998, 97, 2463–2469. [Google Scholar] [CrossRef]
- Garlid, K.D.; Dos Santos, P.; Xie, Z.J.; Costa, A.D.; Paucek, P. Mitochondrial potassium transport: The role of the mitochondrial ATP-sensitive K(+) channel in cardiac function and cardioprotection. Biochim. Biophys. Acta 2003, 1606, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Kopustinskiene, D.M.; Pollesello, P.; Saris, N.E. Levosimendan is a mitochondrial K(ATP) channel opener. Eur. J. Pharmacol. 2001, 428, 311–314. [Google Scholar] [CrossRef]
- Sun, B.; Li, H.; Shakur, Y.; Hensley, J.; Hockman, S.; Kambayashi, J.; Manganiello, V.C.; Liu, Y. Role of phosphodiesterase type 3A and 3B in regulating platelet and cardiac function using subtype-selective knockout mice. Cell Signal 2007, 19, 1765–1771. [Google Scholar] [CrossRef]
- Zimmerman, N.P.; Roy, I.; Hauser, A.D.; Wilson, J.M.; Williams, C.L.; Dwinell, M.B. Cyclic AMP regulates the migration and invasion potential of human pancreatic cancer cells. Mol. Carcinog. 2015, 54, 203–215. [Google Scholar] [CrossRef]
- Zhang, H.; Kong, Q.; Wang, J.; Jiang, Y.; Hua, H. Complex roles of cAMP–PKA–CREB signaling in cancer. Exp. Hematol. Oncol. 2020, 9, 32. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Zhang, J.J.; Huang, X.-Y. cAMP Inhibits Cell Migration by Interfering with Rac-induced Lamellipodium Formation. J. Biol. Chem. 2008, 283, 13799–13805. [Google Scholar] [CrossRef] [PubMed]
- Orstavik, O.; Ata, S.H.; Riise, J.; Dahl, C.P.; Andersen, G.; Levy, F.O.; Skomedal, T.; Osnes, J.B.; Qvigstad, E. Inhibition of phosphodiesterase-3 by levosimendan is sufficient to account for its inotropic effect in failing human heart. Br. J. Pharmacol. 2014, 171, 5169–5181. [Google Scholar] [CrossRef]
- Hao, N.; Shen, W.; Du, R.; Jiang, S.; Zhu, J.; Chen, Y.; Huang, C.; Shi, Y.; Xiang, R.; Luo, Y. Phosphodiesterase 3A Represents a Therapeutic Target that Drives Stem Cell-like Property and Metastasis in Breast Cancer. Mol. Cancer Ther. 2020, 19, 868–881. [Google Scholar] [CrossRef] [PubMed]
- Murata, K.; Kameyama, M.; Fukui, F.; Ohigashi, H.; Hiratsuka, M.; Sasaki, Y.; Kabuto, T.; Mukai, M.; Mammoto, T.; Akedo, H.; et al. Phosphodiesterase type III inhibitor, cilostazol, inhibits colon cancer cell motility. Clin. Exp. Metastasis 1999, 17, 525–530. [Google Scholar] [CrossRef]
- Lewis, T.A.; de Waal, L.; Wu, X.; Youngsaye, W.; Wengner, A.; Kopitz, C.; Lange, M.; Gradl, S.; Ellermann, M.; Lienau, P.; et al. Optimization of PDE3A Modulators for SLFN12-Dependent Cancer Cell Killing. ACS Med. Chem. Lett. 2019, 10, 1537–1542. [Google Scholar] [CrossRef]
- Vannini, F.; Kashfi, K.; Nath, N. The dual role of iNOS in cancer. Redox Biol. 2015, 6, 334–343. [Google Scholar] [CrossRef]
- Papapetropoulos, A.; García-Cardeña, G.; Madri, J.A.; Sessa, W.C. Nitric oxide production contributes to the angiogenic properties of vascular endothelial growth factor in human endothelial cells. J. Clin. Investig. 1997, 100, 3131–3139. [Google Scholar] [CrossRef]
- Schenk, M.W.; Humphrey, S.; Hossain, A.M.M.; Revill, M.; Pearsall, S.; Lallo, A.; Brown, S.; Bratt, S.; Galvin, M.; Descamps, T. Soluble guanylate cyclase signalling mediates etoposide resistance in progressing small cell lung cancer. Nat. Commun. 2021, 12, 6652. [Google Scholar] [CrossRef] [PubMed]
- Choudhari, S.K.; Chaudhary, M.; Bagde, S.; Gadbail, A.; Joshi, V. Nitric oxide and cancer: A review, World. J. Surg. Oncol. 2013, 11, 1–11. [Google Scholar]
- Kamm, A.; Przychodzen, P.; Kuban-Jankowska, A.; Jacewicz, D.; Dabrowska, A.M.; Nussberger, S.; Wozniak, M.; Gorska-Ponikowska, M. Nitric oxide and its derivatives in the cancer battlefield. Nitric Oxide 2019, 93, 102–114. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Liu, L.Z.; Loizidou, M.; Ahmed, M.; Charles, I.G. The role of nitric oxide in cancer. Cell Res. 2002, 12, 311–320. [Google Scholar] [CrossRef]
- Sang, J.; Chen, Y.; Tao, Y. Nitric oxide inhibits gastric cancer cell growth through the modulation of the Akt pathway. Mol. Med. Rep. 2011, 4, 1163–1167. [Google Scholar] [CrossRef]
- Pervin, S.; Singh, R.; Chaudhuri, G. Nitric oxide-induced cytostasis and cell cycle arrest of a human breast cancer cell line (MDA-MB-231): Potential role of cyclin D1. Proc. Natl. Acad. Sci. USA 2001, 98, 3583–3588. [Google Scholar] [CrossRef]
- Huguenin, S.; Fleury-Feith, J.; Kheuang, L.; Jaurand, M.C.; Bolla, M.; Riffaud, J.P.; Chopin, D.K.; Vacherot, F. Nitrosulindac (NCX 1102): A new nitric oxide-donating non-steroidal anti-inflammatory drug (NO-NSAID), inhibits proliferation and induces apoptosis in human prostatic epithelial cell lines. Prostate 2004, 61, 132–141. [Google Scholar] [CrossRef]
- Huguenin, S.; Vacherot, F.; Kheuang, L.; Fleury-Feith, J.; Jaurand, M.-C.; Bolla, M.; Riffaud, J.-P.; Chopin, D.K. Antiproliferative effect of nitrosulindac (NCX 1102), a new nitric oxide-donating non-steroidal anti-inflammatory drug, on human bladder carcinoma cell lines. Mol. Cancer Ther. 2004, 3, 291–298. [Google Scholar] [CrossRef]
- Pena-Altamira, E.; Petazzi, P.; Contestabile, A. Nitric oxide control of proliferation in nerve cells and in tumor cells of nervous origin. Curr. Pharm. Des. 2010, 16, 440–450. [Google Scholar] [CrossRef]
- Donia, M.; Mijatovic, S.; Maksimovic-Ivanic, D.; Miljkovic, D.; Mangano, K.; Tumino, S.; Biondi, A.; Basile, F.; Al-Abed, Y.; Stosic-Grujicic, S. The novel NO-donating compound GIT-27NO inhibits in vivo growth of human prostate cancer cells and prevents murine immunoinflammatory hepatitis. Eur. J. Pharmacol. 2009, 615, 228–233. [Google Scholar] [CrossRef] [PubMed]
- Donia, M.; Maksimovic-Ivanic, D.; Mijatovic, S.; Mojic, M.; Miljkovic, D.; Timotijevic, G.; Fagone, P.; Caponnetto, S.; Al-Abed, Y.; McCubrey, J.A. In vitro and in vivo anticancer action of Saquinavir-NO, a novel nitric oxide-derivative of the protease inhibitor saquinavir, on hormone resistant prostate cancer cells. Cell Cycle 2011, 10, 492–499. [Google Scholar] [CrossRef]
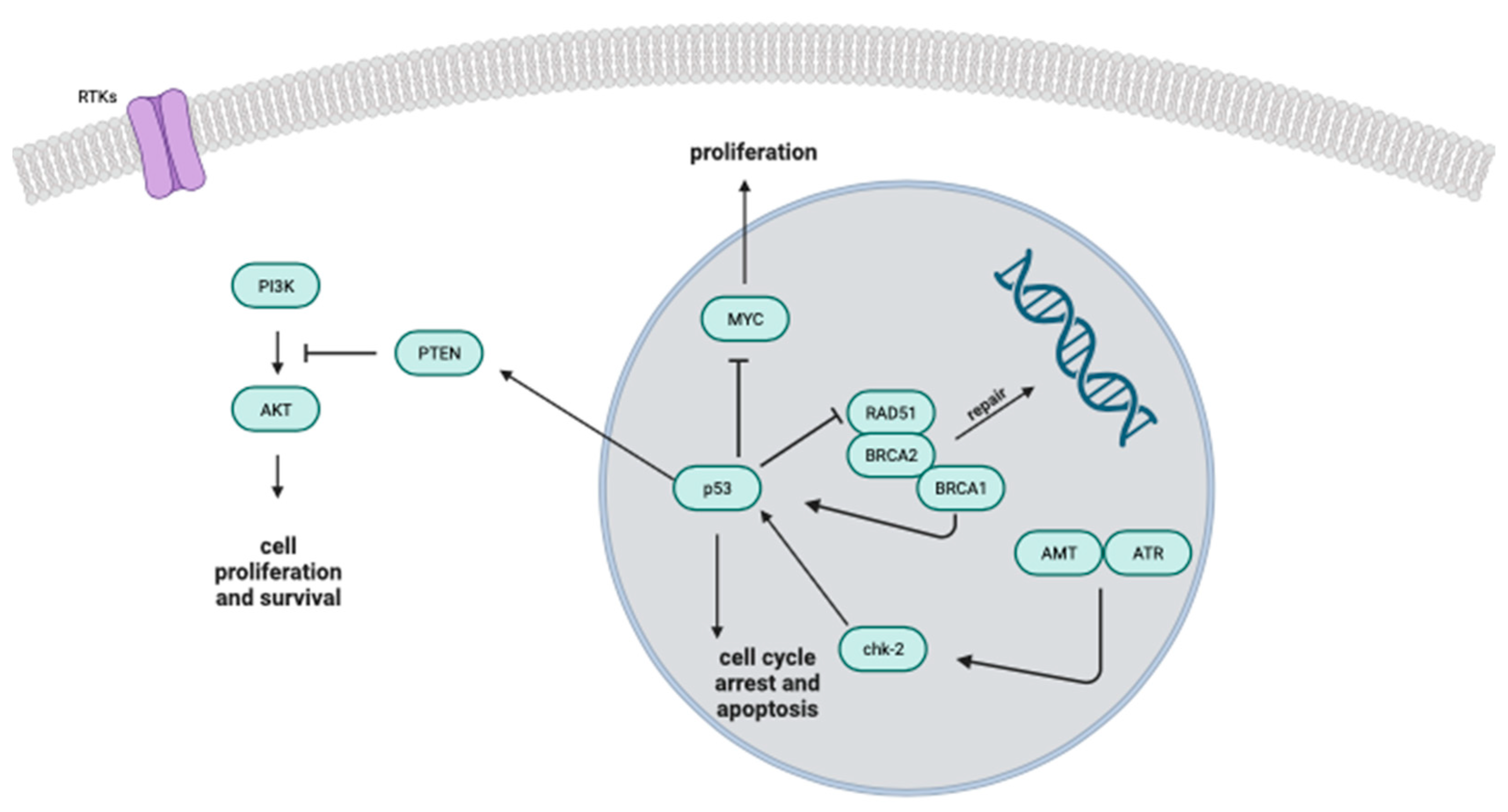
- Van de Wouwer, M.; Couzinié, C.; Serrano-Palero, M.; González-Fernández, Ó.; Galmés-Varela, C.; Menéndez-Antolí, P.; Grau, L.; Villalobo, A. Activation of the BRCA1/Chk1/p53/p21Cip1/Waf1 pathway by nitric oxide and cell cycle arrest in human neuroblastoma NB69 cells. Nitric. Oxide 2012, 26, 182–191. [Google Scholar] [CrossRef] [PubMed]
- Sonveaux, P.; Jordan, B.F.; Gallez, B.; Feron, O. Nitric oxide delivery to cancer: Why and how? Eur. J. Cancer 2009, 45, 1352–1369. [Google Scholar] [CrossRef] [PubMed]
- Cook, T.; Wang, Z.; Alber, S.; Liu, K.; Watkins, S.C.; Vodovotz, Y.; Billiar, T.R.; Blumberg, D. Nitric oxide and ionizing radiation synergistically promote apoptosis and growth inhibition of cancer by activating p53. Cancer Res. 2004, 64, 8015–8021. [Google Scholar] [CrossRef]
- Wang, Z.; Cook, T.; Alber, S.; Liu, K.; Kovesdi, I.; Watkins, S.K.; Vodovotz, Y.; Billiar, T.R.; Blumberg, D. Adenoviral gene transfer of the human inducible nitric oxide synthase gene enhances the radiation response of human colorectal cancer associated with alterations in tumor vascularity. Cancer Res. 2004, 64, 1386–1395. [Google Scholar] [CrossRef]
- Abdal Dayem, A.; Hossain, M.K.; Lee, S.B.; Kim, K.; Saha, S.K.; Yang, G.M.; Choi, H.Y.; Cho, S.G. The Role of Reactive Oxygen Species (ROS) in the Biological Activities of Metallic Nanoparticles. Int J Mol Sci 2017, 18, 120. [Google Scholar] [CrossRef] [PubMed]
- Sies, H.; Jones, D.P. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat. Rev. Mol. Cell Biol. 2020, 21, 363–383. [Google Scholar] [CrossRef]
- Moloney, J.N.; Cotter, T.G. ROS signalling in the biology of cancer. In Seminars in Cell & Developmental Biology; Academic Press: Cambridge, MA, USA, 2018; pp. 50–64. [Google Scholar]
- Trachootham, D.; Alexandre, J.; Huang, P. Targeting cancer cells by ROS-mediated mechanisms: A radical therapeutic approach? Nat. Rev. Drug Discov. 2009, 8, 579–591. [Google Scholar] [CrossRef]
- Liou, G.Y.; Storz, P. Reactive oxygen species in cancer. Free. Radic. Res. 2010, 44, 479–496. [Google Scholar] [CrossRef]
- Perillo, B.; Di Donato, M.; Pezone, A.; Di Zazzo, E.; Giovannelli, P.; Galasso, G.; Castoria, G.; Migliaccio, A. ROS in cancer therapy: The bright side of the moon. Exp. Mol. Med. 2020, 52, 192–203. [Google Scholar] [CrossRef] [PubMed]
- Shah, M.A.; Rogoff, H.A. Implications of reactive oxygen species on cancer formation and its treatment. Semin. Oncol. 2021, 48, 238–245. [Google Scholar] [CrossRef] [PubMed]
- Ushio-Fukai, M.; Nakamura, Y. Reactive oxygen species and angiogenesis: NADPH oxidase as target for cancer therapy. Cancer Lett 2008, 266, 37–52. [Google Scholar] [CrossRef] [PubMed]
- Liao, Z.; Chua, D.; Tan, N.S. Reactive oxygen species: A volatile driver of field cancerization and metastasis. Mol. Cancer 2019, 18, 65. [Google Scholar] [CrossRef] [PubMed]
- Waris, G.; Ahsan, H. Reactive oxygen species: Role in the development of cancer and various chronic conditions. J. Carcinog. 2006, 5, 14. [Google Scholar] [CrossRef] [PubMed]
- El-Kenawi, A.; Ruffell, B. Inflammation, ROS, and mutagenesis. Cancer Cell 2017, 32, 727–729. [Google Scholar] [CrossRef]
- Khan, A.Q.; Kuttikrishnan, S.; Siveen, K.S.; Prabhu, K.S.; Shanmugakonar, M.; Al-Naemi, H.A.; Haris, M.; Dermime, S.; Uddin, S. RAS-mediated oncogenic signaling pathways in human malignancies. In Seminars in Cancer Biology; Academic Press: Cambridge, MA, USA, 2019; pp. 1–13. [Google Scholar]
- Reuter, S.; Gupta, S.C.; Chaturvedi, M.M.; Aggarwal, B.B. Oxidative stress, inflammation, and cancer: How are they linked? Free Radic. Biol. Med. 2010, 49, 1603–1616. [Google Scholar] [CrossRef]
- Hinshaw, D.C.; Shevde, L.A. The tumor microenvironment innately modulates cancer progression. Cancer Res. 2019, 79, 4557–4566. [Google Scholar] [CrossRef]
- Aggarwal, V.; Tuli, H.S.; Varol, A.; Thakral, F.; Yerer, M.B.; Sak, K.; Varol, M.; Jain, A.; Khan, M.A.; Sethi, G. Role of reactive oxygen species in cancer progression: Molecular mechanisms and recent advancements. Biomolecules 2019, 9, 735. [Google Scholar] [CrossRef]
- Weinberg, F.; Ramnath, N.; Nagrath, D. Reactive oxygen species in the tumor microenvironment: An overview. Cancers 2019, 11, 1191. [Google Scholar] [CrossRef] [PubMed]
- Rodic, S.; Vincent, M.D. Reactive oxygen species (ROS) are a key determinant of cancer’s metabolic phenotype. Int. J. Cancer 2018, 142, 440–448. [Google Scholar] [CrossRef]
- Kim, J.; Kim, J.; Bae, J.-S. ROS homeostasis and metabolism: A critical liaison for cancer therapy. Exp. Mol. Med. 2016, 48, e269. [Google Scholar] [CrossRef]
- Muri, J.; Kopf, M. Redox regulation of immunometabolism. Nat. Rev. Immunol. 2021, 21, 363–381. [Google Scholar] [CrossRef] [PubMed]
- Fox, D.B.; Garcia, N.M.G.; McKinney, B.J.; Lupo, R.; Noteware, L.C.; Newcomb, R.; Liu, J.; Locasale, J.W.; Hirschey, M.D.; Alvarez, J.V. NRF2 activation promotes the recurrence of dormant tumour cells through regulation of redox and nucleotide metabolism. Nat. Metab. 2020, 2, 318–334. [Google Scholar] [CrossRef] [PubMed]
- Kipka, H.; Schaflinger, R.; Tomasi, R.; Pogoda, K.; Mannell, H. The Effects of the Levosimendan Metabolites OR-1855 and OR-1896 on Endothelial Pro-Inflammatory Responses. Biomedicines 2023, 11, 918. [Google Scholar] [CrossRef]
- Hasslacher, J.; Bijuklic, K.; Bertocchi, C.; Kountchev, J.; Bellmann, R.; Dunzendorfer, S.; Joannidis, M. Levosimendan inhibits release of reactive oxygen species in polymorphonuclear leukocytes in vitro and in patients with acute heart failure and septic shock: A prospective observational study. Crit. Care 2011, 15, R166. [Google Scholar] [CrossRef]
- Khoubnasabjafari, M.; Ansarin, K.; Jouyban, A. Reliability of malondialdehyde as a biomarker of oxidative stress in psychological disorders. Bioimpacts 2015, 5, 123–127. [Google Scholar] [CrossRef] [PubMed]
- Vona, R.; Pallotta, L.; Cappelletti, M.; Severi, C.; Matarrese, P. The Impact of Oxidative Stress in Human Pathology: Focus on Gastrointestinal Disorders. Antioxidants 2021, 10, 201. [Google Scholar] [CrossRef]
- do Val Carneiro, J.L.; Nixdorf, S.L.; Mantovani, M.S.; da Silva do Amaral Herrera, A.C.; Aoki, M.N.; Amarante, M.K.; Fabris, B.A.; Pelegrinelli Fungaro, M.H.; Ehara Watanabe, M.A. Plasma malondialdehyde levels and CXCR4 expression in peripheral blood cells of breast cancer patients. J. Cancer Res. Clin. Oncol. 2009, 135, 997–1004. [Google Scholar] [CrossRef]
- Gönenç, A.; Özkan, Y.; Torun, M.; Şimşek, B. Plasma malondialdehyde (MDA) levels in breast and lung cancer patients. J. Clin. Pharm. Ther. 2001, 26, 141–144. [Google Scholar] [CrossRef] [PubMed]
- Chole, R.H.; Patil, R.N.; Basak, A.; Palandurkar, K.; Bhowate, R. Estimation of serum malondialdehyde in oral cancer and precancer and its association with healthy individuals, gender, alcohol, and tobacco abuse. J. Cancer Res. Ther. 2010, 6, 487–491. [Google Scholar] [CrossRef] [PubMed]
- Peddireddy, V.; Siva Prasad, B.; Gundimeda, S.D.; Penagaluru, P.R.; Mundluru, H.P. Assessment of 8-oxo-7, 8-dihydro-2′-deoxyguanosine and malondialdehyde levels as oxidative stress markers and antioxidant status in non-small cell lung cancer. Biomarkers 2012, 17, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Rašić, I.; Rašić, A.; Akšamija, G.; Radović, S. The relationship between serum level of malondialdehyde and progression of colorectal cancer. Acta Clin Croat 2018, 57, 411–416. [Google Scholar] [CrossRef]
- Maurya, R.P.; Prajapat, M.K.; Singh, V.P.; Roy, M.; Todi, R.; Bosak, S.; Singh, S.K.; Chaudhary, S.; Kumar, A.; Morekar, S.R. Serum Malondialdehyde as a Biomarker of Oxidative Stress in Patients with Primary Ocular Carcinoma: Impact on Response to Chemotherapy. Clin. Ophthalmol. 2021, 15, 871–879. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Tian, L.; Wang, S.; Yang, W.; Lu, X.; Zhu, C. Levosimendan in rats decreases acute kidney injury after cardiopulmonary resuscitation by improving mitochondrial dysfunction. Transl. Androl. Urol. 2021, 10, 3010–3020. [Google Scholar] [CrossRef]
- Grossini, E.; Farruggio, S.; Pierelli, D.; Bolzani, V.; Rossi, L.; Pollesello, P.; Monaco, C. Levosimendan Improves Oxidative Balance in Cardiogenic Shock/Low Cardiac Output Patients. J. Clin. Med. 2020, 9, 373. [Google Scholar] [CrossRef]
- Adam, M.; Meyer, S.; Knors, H.; Klinke, A.; Radunski, U.K.; Rudolph, T.K.; Rudolph, V.; Spin, J.M.; Tsao, P.S.; Costard-Jäckle, A.; et al. Levosimendan displays anti-inflammatory effects and decreases MPO bioavailability in patients with severe heart failure. Sci. Rep. 2015, 5, 9704. [Google Scholar] [CrossRef]
- Hua, Y.; Dai, X.; Xu, Y.; Xing, G.; Liu, H.; Lu, T.; Chen, Y.; Zhang, Y. Drug repositioning: Progress and challenges in drug discovery for various diseases. Eur. J. Med. Chem. 2022, 234, 114239. [Google Scholar] [CrossRef]
- Yang, F.; Zhang, Q.; Ji, X.; Zhang, Y.; Li, W.; Peng, S.; Xue, F. Machine Learning Applications in Drug Repurposing. Interdiscip. Sci. 2022, 14, 15–21. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug | Effector | Vascular Bed (Species) | Reference |
|---|---|---|---|
| Levosimendan | K(ATP) | Mesenteric artery (rat) | [37] |
| Coronary circulation (guinea pig) | [40] | ||
| Renal circulation (mice) | [42] | ||
| Internal thoracic artery (human) | [39] | ||
| Skeletal muscle arteriole (rat) | [51] | ||
| Portal vein (human) | [53] | ||
| Levosimendan/OR-1896 | K(ATP) | Resistance arteriole | [52] |
| Levosimendan | KV + BKCa | Coronary artery (pig) | [43] |
| Umbilical cord artery (human) | [44] | ||
| OR-1896 | BKCa | Coronary arteriole (rat) | [51] |
| Levosimendan | K(ATP) + BKCa | Internal thoracic artery (human) | [45] |
| Saphenous vein (human) | [54] | ||
| Levosimendan | cAMP | Coronary artery (pig) | [47] |
| Levosimendan | K(ATP) + cAMP + cGMP | Pulmonary circulation (cat) | [55] |
| Pulmonary artery (guinea pig) | [35] | ||
| Levosimendan | K(ATP) + BKCa + cAMP + cGMP | Pulmonary vein (guinea pig) | [35] |
| Levosimendan | K(ATP) + KV + cAMP + cGMP | Pulmonary circulation (human) | [36] |
| Levosimendan | NO | Coronary endothelial cells (pig) | [49] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ribeiro, E.; Vale, N. Understanding the Clinical Use of Levosimendan and Perspectives on its Future in Oncology. Biomolecules 2023, 13, 1296. https://doi.org/10.3390/biom13091296
Ribeiro E, Vale N. Understanding the Clinical Use of Levosimendan and Perspectives on its Future in Oncology. Biomolecules. 2023; 13(9):1296. https://doi.org/10.3390/biom13091296
Chicago/Turabian StyleRibeiro, Eduarda, and Nuno Vale. 2023. "Understanding the Clinical Use of Levosimendan and Perspectives on its Future in Oncology" Biomolecules 13, no. 9: 1296. https://doi.org/10.3390/biom13091296