
Cyclophilin D in Mitochondrial Dysfunction: A Key Player in Neurodegeneration?

Abstract
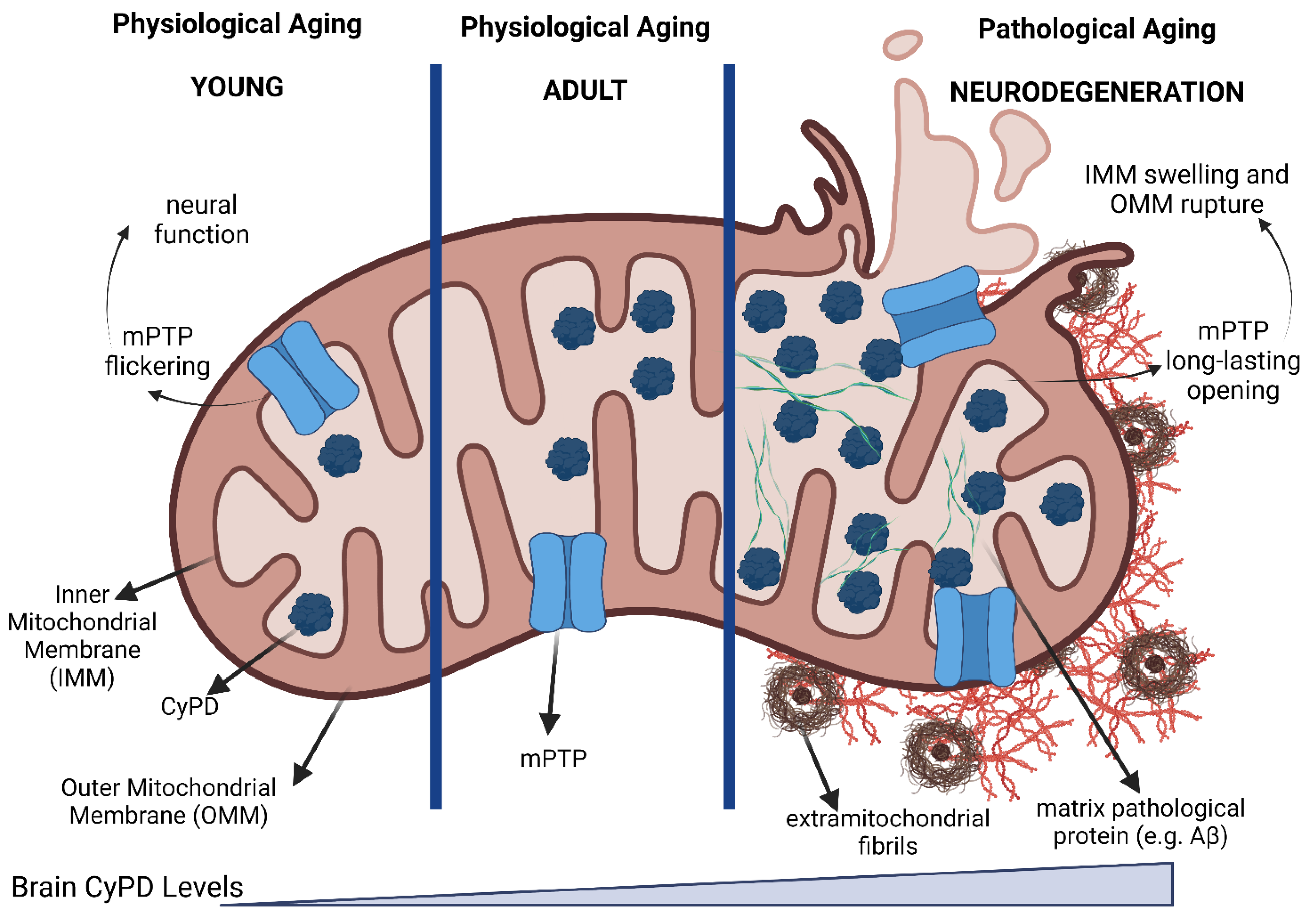
:1. Introduction
2. The Superfamily of Peptidylprolyl cis-trans Isomerases
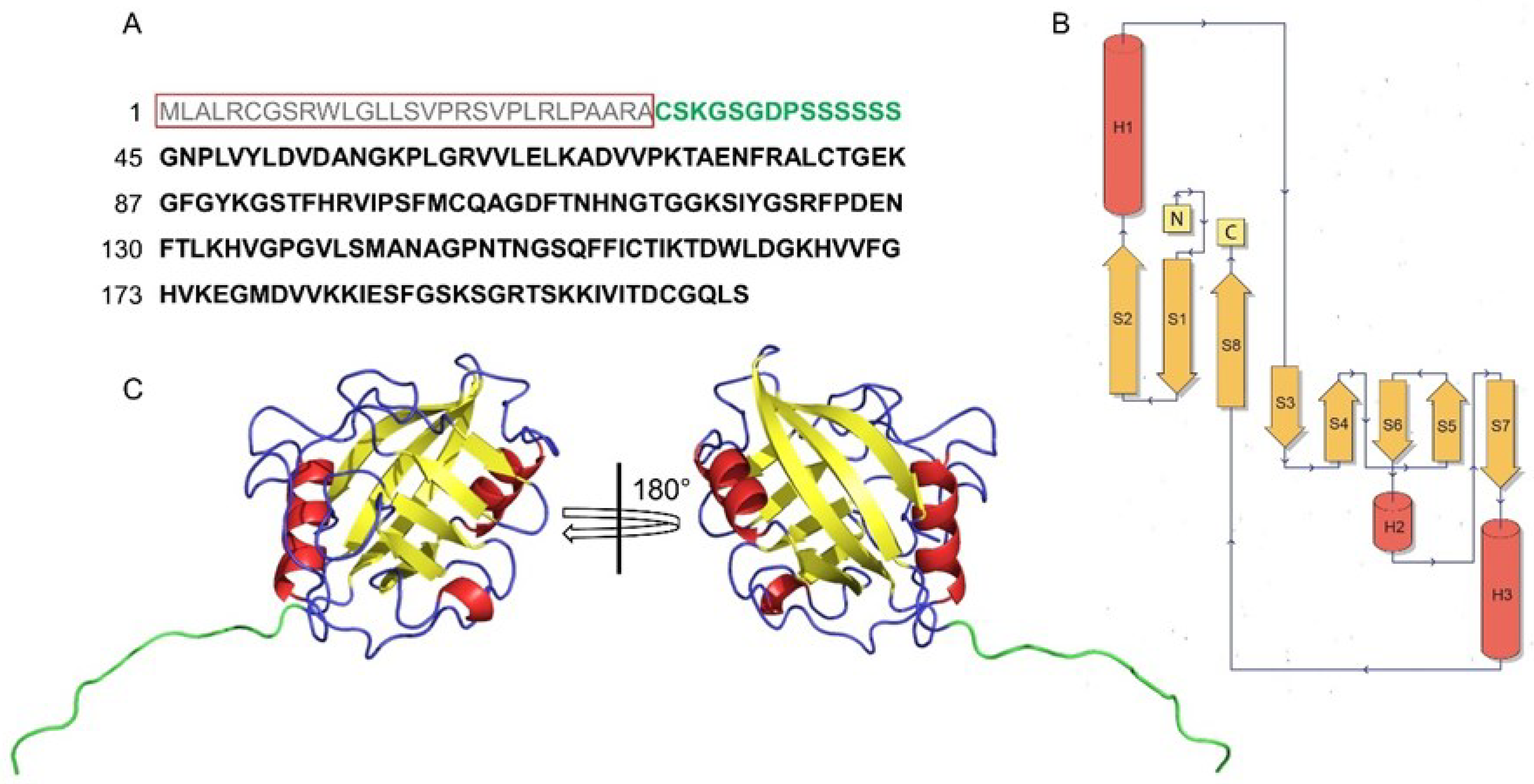
3. Generalities of CyPD

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Chromosome | Protein Name(s) | Length (AA) | Subcellular Localization |
|---|---|---|---|---|
| ppia | 7p13 | CyPA | 165 | Cytoplasm, Nucleus, ECM |
| ppib | 15q22.31 | CyPB | 216 | ER Lumen |
| ppic | 5q23.2 | CyPC | 212 | Cytoplasm |
| ppid | 4q32.1 | CyP-40, CyPD | 370 | Cytoplasm, Nucleus |
| ppie | 1p34.2 | CyP-33, CyPE | 301 | Nucleus |
| ppif | 10q22.3 | CyPD, CyP3, CyP-M, CyPF | 207 | Mitochondrial Matrix |
| ppig | 2q31.1 | SRCyP, SCAF10 | 754 | Nucleus, Nuclear Speckles |
| ppih | 1p34.2 | CyP-20, CyPH, USA-CyP | 177 | Cytoplasm, Nuclear Speckles |
| ppil1 | 6p21.2 | CYPL1, hCyPX, CGI-124 | 166 | Nucleus |
| ppil2 | 22q11.21 | CyP-60, CyC4, UBOX7 | 520 | Nucleus |
| ppil3 | 2q33.1 | CyPJ | 161 | Nucleus |
| ppil4 | 6q25.1 | PPIL4, HDCME13P | 492 | Nucleus |
| ppil6 | 6q21 | PPIL6, RSPH12 | 311 | Cytoplasm |
| ppwd1 | 5q12.3 | PPWD1 | 646 | Nucleus |
| ranbp2 | 2q13 | RANBP2, NUP358 | 3224 | Nucleus |
| cwc27 | 5q12.3 | CWC27, SDCCAG10 | 472 | Nucleus |
| nktr | 3p22.1 | CyPNK, P104 | 1462 | Cell Membrane |
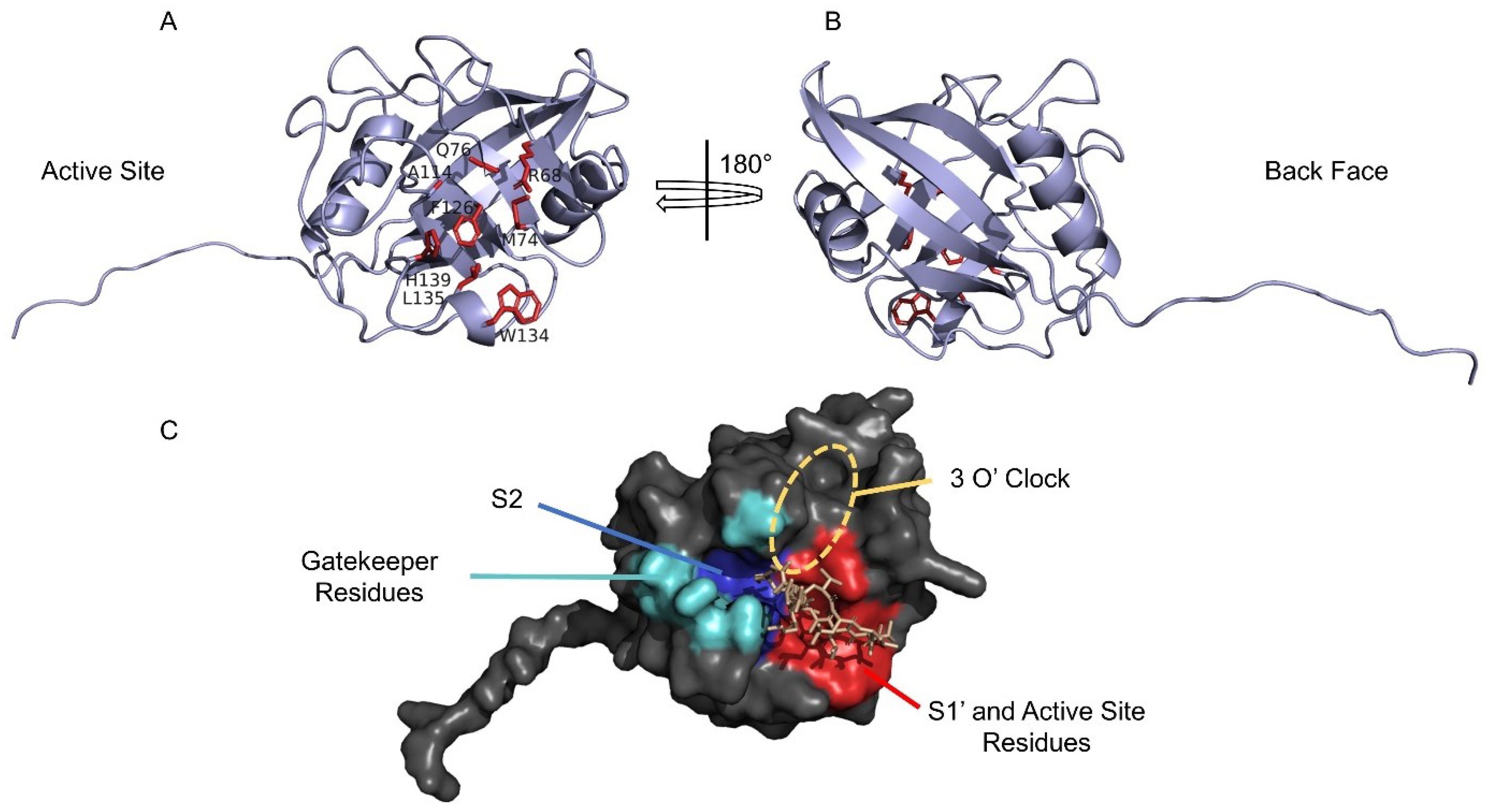
4. Structural Features of CyPD

5. Cyclosporine A
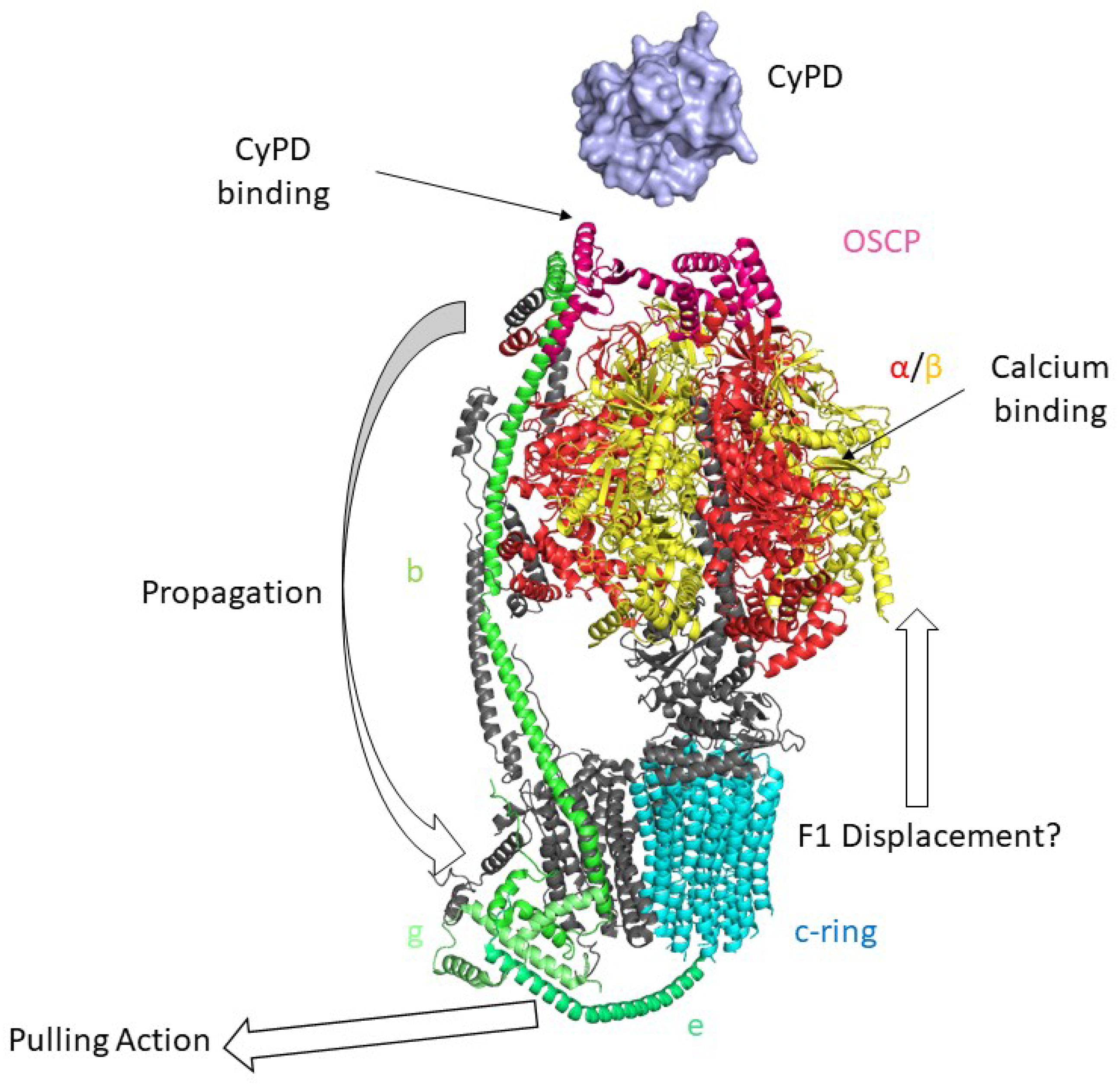
6. The Permeability Transition Pore

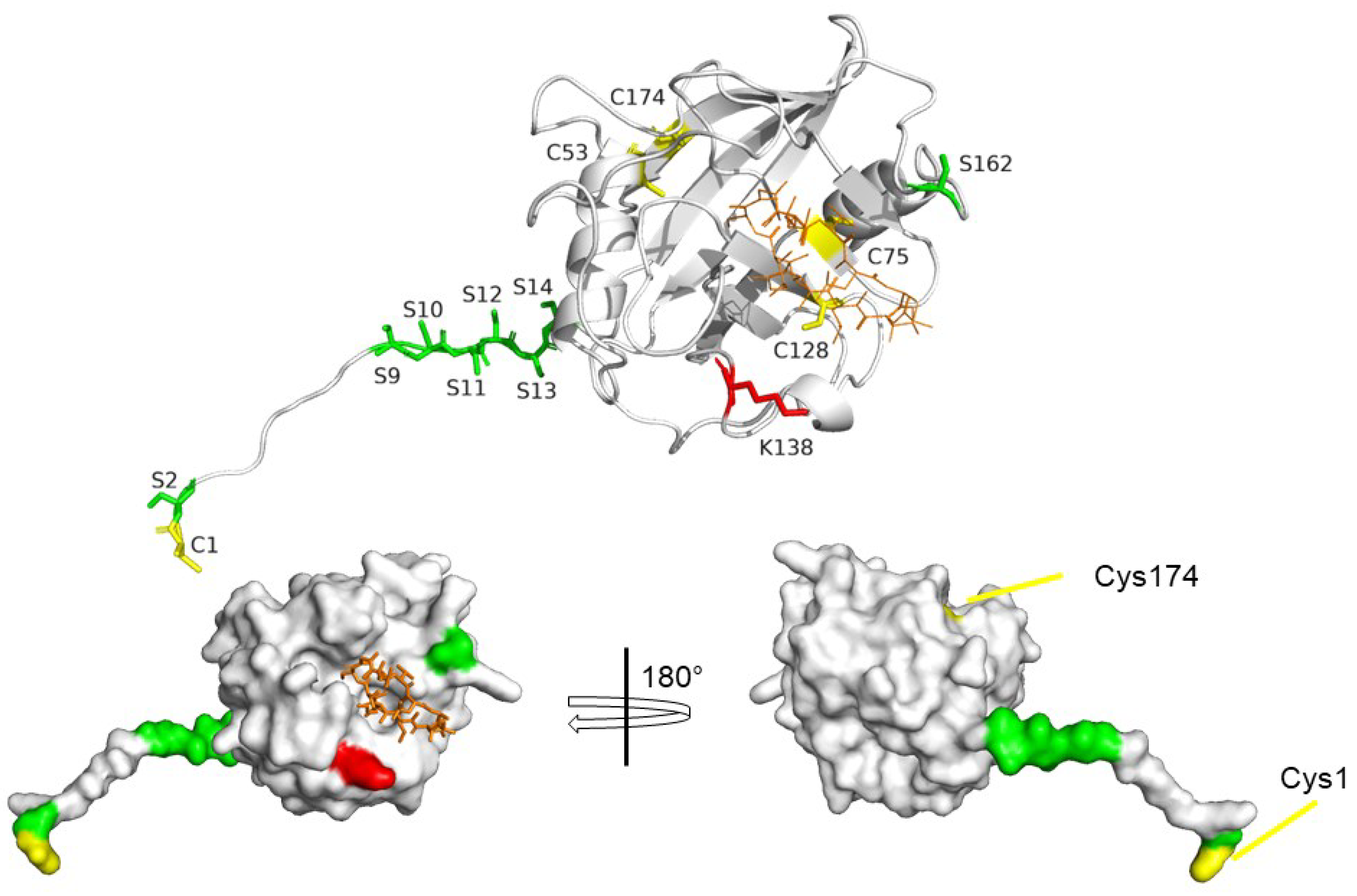
7. Post-Translational Modifications of CyPD
7.1. CyPD Acetylation
7.2. CyPD Phosphorylation
7.3. Oxidative Modifications of CyPD
8. CyPD in Brain
9. The Involvement of CyPD in Alzheimer’s Disease
10. The Involvement of CyPD in Parkinson’s Disease
11. Pharmacological Inhibition of CyPD
- (1)
- N-4-Aminobenzyl-N′-(2-(2-arylpyrrolidin)-2-oxoethyl)urea-based compounds,
- (2)
- 2-(Benzyloxy)arylurea-based compounds
- (3)
- Other small-molecule inhibitors
12. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hou, Y.; Dan, X.; Babbar, M.; Wei, Y.; Hasselbalch, S.G.; Croteau, D.L.; Bohr, V.A. Ageing as a Risk Factor for Neurodegenerative Disease. Nat. Rev. Neurol. 2019, 15, 565–581. [Google Scholar] [CrossRef] [PubMed]
- Coleman, C.; Martin, I. Unraveling Parkinson’s Disease Neurodegeneration: Does Aging Hold the Clues? J. Park. Dis. 2022, 12, 2321–2338. [Google Scholar] [CrossRef] [PubMed]
- Lang, K.; Schmid, F.X.; Fischer, G. Catalysis of Protein Folding by Prolyl Isomerase. Nature 1987, 329, 268–270. [Google Scholar] [CrossRef] [PubMed]
- Fischer, G.; Bang, H.; Mech, C. Determination of enzymatic catalysis for the cis-trans-isomerization of peptide binding in proline-containing peptides. Biomed. Biochim. Acta 1984, 43, 1101–1111. [Google Scholar]
- Handschumacher, R.E.; Harding, M.W.; Rice, J.; Drugge, R.J.; Speicher, D.W. Cyclophilin: A Specific Cytosolic Binding Protein for Cyclosporin A. Science 1984, 226, 544–547. [Google Scholar] [CrossRef]
- Fischer, G.; Wittmann-Liebold, B.; Lang, K.; Kiefhaber, T.; Schmid, F.X. Cyclophilin and Peptidyl-Prolyl Cis-Trans Isomerase Are Probably Identical Proteins. Nature 1989, 337, 476–478. [Google Scholar] [CrossRef]
- Harikishore, A.; Sup Yoon, H. Immunophilins: Structures, Mechanisms and Ligands. CMP 2015, 9, 37–47. [Google Scholar] [CrossRef]
- Matena, A.; Rehic, E.; Hönig, D.; Kamba, B.; Bayer, P. Structure and Function of the Human Parvulins Pin1 and Par14/17. Biol. Chem. 2018, 399, 101–125. [Google Scholar] [CrossRef]
- Fanghänel, J.; Fischer, G. Insights into the Catalytic Mechanism of Peptidyl Prolyl Cis/Trans Isomerases. Front. Biosci. 2004, 9, 3453. [Google Scholar] [CrossRef]
- Rein, T. Peptidylprolylisomerases, Protein Folders, or Scaffolders? The Example of FKBP51 and FKBP52. BioEssays 2020, 42, 1900250. [Google Scholar] [CrossRef]
- Stewart, D.E.; Sarkar, A.; Wampler, J.E. Occurrence and Role of Cis Peptide Bonds in Protein Structures. J. Mol. Biol. 1990, 214, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Galat, A. Peptidylproline Cis-Trans-Isomerases: Immunophilins. Eur. J. Biochem. 1993, 216, 689–707. [Google Scholar] [CrossRef] [PubMed]
- Hanes, S.D. Prolyl Isomerases in Gene Transcription. Biochim. Biophys. Acta BBA Gen. Subj. 2015, 1850, 2017–2034. [Google Scholar] [CrossRef] [PubMed]
- Horowitz, D.S.; Lee, E.J.; Mabon, S.A.; Misteli, T. A Cyclophilin Functions in Pre-MRNA Splicing. EMBO J. 2002, 21, 470–480. [Google Scholar] [CrossRef]
- Mamatis, J.E.; Pellizzari-Delano, I.E.; Gallardo-Flores, C.E.; Colpitts, C.C. Emerging Roles of Cyclophilin A in Regulating Viral Cloaking. Front. Microbiol. 2022, 13, 828078. [Google Scholar] [CrossRef]
- Xu, Q.; Leiva, M.C.; Fischkoff, S.A.; Handschumacher, R.E.; Lyttle, C.R. Leukocyte Chemotactic Activity of Cyclophilin. J. Biol. Chem. 1992, 267, 11968–11971. [Google Scholar] [CrossRef]
- Bergsma, D.J.; Eder, C.; Gross, M.; Kersten, H.; Sylvester, D.; Appelbaum, E.; Cusimano, D.; Livi, G.P.; McLaughlin, M.M.; Kasyan, K. The Cyclophilin Multigene Family of Peptidyl-Prolyl Isomerases. Characterization of Three Separate Human Isoforms. J. Biol. Chem. 1991, 266, 23204–23214. [Google Scholar] [CrossRef]
- Woodfield, K.-Y.; Price, N.T.; Halestrap, A.P. CDNA Cloning of Rat Mitochondrial Cyclophilin. Biochim. Biophys. Acta BBA Gene Struct. Expr. 1997, 1351, 27–30. [Google Scholar] [CrossRef]
- Gutiérrez-Aguilar, M.; Baines, C.P. Structural Mechanisms of Cyclophilin D-Dependent Control of the Mitochondrial Permeability Transition Pore. Biochim. Biophys. Acta BBA Gen. Subj. 2015, 1850, 2041–2047. [Google Scholar] [CrossRef]
- Elrod, J.W.; Molkentin, J.D. Physiologic Functions of Cyclophilin D and the Mitochondrial Permeability Transition Pore. Circ. J. 2013, 77, 1111–1122. [Google Scholar] [CrossRef]
- Stelzer, G.; Rosen, N.; Plaschkes, I.; Zimmerman, S.; Twik, M.; Fishilevich, S.; Stein, T.I.; Nudel, R.; Lieder, I.; Mazor, Y.; et al. The GeneCards Suite: From Gene Data Mining to Disease Genome Sequence Analyses. Curr. Protoc. Bioinform. 2016, 54, 1.30.1–1.30.33. [Google Scholar] [CrossRef] [PubMed]
- The UniProt Consortium; Bateman, A.; Martin, M.-J.; Orchard, S.; Magrane, M.; Ahmad, S.; Alpi, E.; Bowler-Barnett, E.H.; Britto, R.; Bye-A-Jee, H.; et al. UniProt: The Universal Protein Knowledgebase in 2023. Nucleic Acids Res. 2023, 51, D523–D531. [Google Scholar] [CrossRef]
- Johnson, N.; Khan, A.; Virji, S.; Ward, J.M.; Crompton, M. Import and Processing of Heart Mitochondrial Cyclophilin D. Eur. J. Biochem. 1999, 263, 353–359. [Google Scholar] [CrossRef]
- Connern, C.P.; Halestrap, A.P. Purification and N-Terminal Sequencing of Peptidyl-Prolyl Cis-Trans-Isomerase from Rat Liver Mitochondrial Matrix Reveals the Existence of a Distinct Mitochondrial Cyclophilin. Biochem. J. 1992, 284, 381–385. [Google Scholar] [CrossRef]
- Porter, G.A.; Beutner, G. Cyclophilin D, Somehow a Master Regulator of Mitochondrial Function. Biomolecules 2018, 8, 176. [Google Scholar] [CrossRef] [PubMed]
- Giorgio, V.; Bisetto, E.; Soriano, M.E.; Dabbeni-Sala, F.; Basso, E.; Petronilli, V.; Forte, M.A.; Bernardi, P.; Lippe, G. Cyclophilin D Modulates Mitochondrial F0F1-ATP Synthase by Interacting with the Lateral Stalk of the Complex. J. Biol. Chem. 2009, 284, 33982–33988. [Google Scholar] [CrossRef] [PubMed]
- Giorgio, V.; von Stockum, S.; Antoniel, M.; Fabbro, A.; Fogolari, F.; Forte, M.; Glick, G.D.; Petronilli, V.; Zoratti, M.; Szabó, I.; et al. Dimers of Mitochondrial ATP Synthase Form the Permeability Transition Pore. Proc. Natl. Acad. Sci. USA 2013, 110, 5887–5892. [Google Scholar] [CrossRef]
- Urbani, A.; Giorgio, V.; Carrer, A.; Franchin, C.; Arrigoni, G.; Jiko, C.; Abe, K.; Maeda, S.; Shinzawa-Itoh, K.; Bogers, J.F.M.; et al. Purified F-ATP Synthase Forms a Ca2+-Dependent High-Conductance Channel Matching the Mitochondrial Permeability Transition Pore. Nat. Commun. 2019, 10, 4341. [Google Scholar] [CrossRef]
- Mnatsakanyan, N.; Llaguno, M.C.; Yang, Y.; Yan, Y.; Weber, J.; Sigworth, F.J.; Jonas, E.A. A Mitochondrial Megachannel Resides in Monomeric F1FO ATP Synthase. Nat. Commun. 2019, 10, 5823. [Google Scholar] [CrossRef]
- Crompton, M.; Virji, S.; Ward, J.M. Cyclophilin-D Binds Strongly to Complexes of the Voltage-Dependent Anion Channel and the Adenine Nucleotide Translocase to Form the Permeability Transition Pore. Eur. J. Biochem. 1998, 258, 729–735. [Google Scholar] [CrossRef]
- Rasola, A.; Sciacovelli, M.; Chiara, F.; Pantic, B.; Brusilow, W.S.; Bernardi, P. Activation of Mitochondrial ERK Protects Cancer Cells from Death through Inhibition of the Permeability Transition. Proc. Natl. Acad. Sci. USA 2010, 107, 726–731. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, J.C.; Siegelin, M.D.; Vaira, V.; Faversani, A.; Tavecchio, M.; Chae, Y.C.; Lisanti, S.; Rampini, P.; Giroda, M.; Caino, M.C.; et al. Adaptive Mitochondrial Reprogramming and Resistance to PI3K Therapy. JNCI J. Natl. Cancer Inst. 2015, 107, dju502. [Google Scholar] [CrossRef] [PubMed]
- Vaseva, A.V.; Marchenko, N.D.; Ji, K.; Tsirka, S.E.; Holzmann, S.; Moll, U.M. P53 Opens the Mitochondrial Permeability Transition Pore to Trigger Necrosis. Cell 2012, 149, 1536–1548. [Google Scholar] [CrossRef] [PubMed]
- Kumutima, J.; Yao, X.-Q.; Hamelberg, D. P53 Is Potentially Regulated by Cyclophilin D in the Triple-Proline Loop of the DNA Binding Domain. Biochemistry 2021, 60, 597–606. [Google Scholar] [CrossRef]
- Zhao, J.; Liu, X.; Blayney, A.; Zhang, Y.; Gandy, L.; Mirsky, P.O.; Smith, N.; Zhang, F.; Linhardt, R.J.; Chen, J.; et al. Intrinsically Disordered N-Terminal Domain (NTD) of P53 Interacts with Mitochondrial PTP Regulator Cyclophilin D. J. Mol. Biol. 2022, 434, 167552. [Google Scholar] [CrossRef]
- Cannino, G.; Urbani, A.; Gaspari, M.; Varano, M.; Negro, A.; Filippi, A.; Ciscato, F.; Masgras, I.; Gerle, C.; Tibaldi, E.; et al. The Mitochondrial Chaperone TRAP1 Regulates F-ATP Synthase Channel Formation. Cell Death Differ. 2022, 29, 2335–2346. [Google Scholar] [CrossRef]
- Etzler, J.C.; Bollo, M.; Holstein, D.; Deng, J.J.; Perez, V.; Lin, D.; Richardson, A.; Bai, Y.; Lechleiter, J.D. Cyclophilin D Over-Expression Increases Mitochondrial Complex III Activity and Accelerates Supercomplex Formation. Arch. Biochem. Biophys. 2017, 613, 61–68. [Google Scholar] [CrossRef]
- Eliseev, R.A.; Malecki, J.; Lester, T.; Zhang, Y.; Humphrey, J.; Gunter, T.E. Cyclophilin D Interacts with Bcl2 and Exerts an Anti-Apoptotic Effect. J. Biol. Chem. 2009, 284, 9692–9699. [Google Scholar] [CrossRef]
- Beutner, G.; Alanzalon, R.E.; Porter, G.A. Cyclophilin D Regulates the Dynamic Assembly of Mitochondrial ATP Synthase into Synthasomes. Sci. Rep. 2017, 7, 14488. [Google Scholar] [CrossRef]
- Ruprecht, J.J.; Kunji, E.R. Structural Changes in the Transport Cycle of the Mitochondrial ADP/ATP Carrier. Curr. Opin. Struct. Biol. 2019, 57, 135–144. [Google Scholar] [CrossRef]
- Ruprecht, J.J.; Kunji, E.R.S. Structural Mechanism of Transport of Mitochondrial Carriers. Annu. Rev. Biochem. 2021, 90, 535–558. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.T.M.; Wong, R.; Menazza, S.; Sun, J.; Chen, Y.; Wang, G.; Gucek, M.; Steenbergen, C.; Sack, M.N.; Murphy, E. Cyclophilin D Modulates Mitochondrial Acetylome. Circ. Res. 2013, 113, 1308–1319. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, P.; Carraro, M.; Lippe, G. The Mitochondrial Permeability Transition: Recent Progress and Open Questions. FEBS J. 2022, 289, 7051–7074. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, E.J.; Halestrap, A.P. Protection by Cyclosporin A of Ischemia/Reperfusion-Induced Damage in Isolated Rat Hearts. J. Mol. Cell. Cardiol. 1993, 25, 1461–1469. [Google Scholar] [CrossRef] [PubMed]
- Broekemeier, K.M.; Dempsey, M.E.; Pfeiffer, D.R. Cyclosporin A Is a Potent Inhibitor of the Inner Membrane Permeability Transition in Liver Mitochondria. J. Biol. Chem. 1989, 264, 7826–7830. [Google Scholar] [CrossRef] [PubMed]
- Halestrap, A.P.; Davidson, A.M. Inhibition of Ca2+-Induced Large-Amplitude Swelling of Liver and Heart Mitochondria by Cyclosporin Is Probably Caused by the Inhibitor Binding to Mitochondrial-Matrix Peptidyl-Prolyl Cis-Trans Isomerase and Preventing It Interacting with the Adenine Nucleotide Translocase. Biochem. J. 1990, 268, 153–160. [Google Scholar] [CrossRef]
- Basso, E.; Fante, L.; Fowlkes, J.; Petronilli, V.; Forte, M.A.; Bernardi, P. Properties of the Permeability Transition Pore in Mitochondria Devoid of Cyclophilin D. J. Biol. Chem. 2005, 280, 18558–18561. [Google Scholar] [CrossRef]
- Ke, H.M.; Zydowsky, L.D.; Liu, J.; Walsh, C.T. Crystal Structure of Recombinant Human T-Cell Cyclophilin A at 2.5 A Resolution. Proc. Natl. Acad. Sci. USA 1991, 88, 9483–9487. [Google Scholar] [CrossRef]
- Kallen, J.; Spitzfaden, C.; Zurini, M.G.M.; Wider, G.; Widmer, H.; Wüthrich, K.; Walkinshaw, M.D. Structure of Human Cyclophilin and Its Binding Site for Cyclosporin a Determined by X-Ray Crystallography and NMR Spectroscopy. Nature 1991, 353, 276–279. [Google Scholar] [CrossRef]
- Spitzfaden, C.; Braun, W.; Wider, G.; Widmer, H.; Wüthrich, K. Determination of the NMR Solution Structure of the Cyclophilin A-Cyclosporin A Complex. J. Biomol. NMR 1994, 4, 463–482. [Google Scholar] [CrossRef]
- Wang, P.; Heitman, J. The Cyclophilins. Genome Biol. 2005, 6, 226. [Google Scholar] [CrossRef] [PubMed]
- Davis, T.L.; Walker, J.R.; Campagna-Slater, V.; Finerty, P.J.; Paramanathan, R.; Bernstein, G.; MacKenzie, F.; Tempel, W.; Ouyang, H.; Lee, W.H.; et al. Structural and Biochemical Characterization of the Human Cyclophilin Family of Peptidyl-Prolyl Isomerases. PLoS Biol. 2010, 8, e1000439. [Google Scholar] [CrossRef] [PubMed]
- Schlatter, D.; Thoma, R.; Küng, E.; Stihle, M.; Müller, F.; Borroni, E.; Cesura, A.; Hennig, M. Crystal Engineering Yields Crystals of Cyclophilin D Diffracting to 1.7 Å Resolution. Acta Crystallogr. D Biol. Crystallogr. 2005, 61, 513–519. [Google Scholar] [CrossRef] [PubMed]
- Kajitani, K.; Fujihashi, M.; Kobayashi, Y.; Shimizu, S.; Tsujimoto, Y.; Miki, K. Crystal Structure of Human Cyclophilin D in Complex with Its Inhibitor, Cyclosporin A at 0.96-Å Resolution. Proteins 2007, 70, 1635–1639. [Google Scholar] [CrossRef]
- wwPDB Consortium. Protein Data Bank: The Single Global Archive for 3D Macromolecular Structure Data. Nucleic Acids Res. 2019, 47, D520–D528. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly Accurate Protein Structure Prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A.; et al. AlphaFold Protein Structure Database: Massively Expanding the Structural Coverage of Protein-Sequence Space with High-Accuracy Models. Nucleic Acids Res. 2022, 50, D439–D444. [Google Scholar] [CrossRef]
- De Simone, A.; Georgiou, C.; Ioannidis, H.; Gupta, A.A.; Juárez-Jiménez, J.; Doughty-Shenton, D.; Blackburn, E.A.; Wear, M.A.; Richards, J.P.; Barlow, P.N.; et al. A Computationally Designed Binding Mode Flip Leads to a Novel Class of Potent Tri-Vector Cyclophilin Inhibitors. Chem. Sci. 2019, 10, 542–547. [Google Scholar] [CrossRef]
- Borel, J.F.; Feurer, C.; Gubler, H.U.; Stähelin, H. Biological Effects of Cyclosporin A: A New Antilymphocytic Agent. Agents Actions 1976, 6, 468–475. [Google Scholar] [CrossRef]
- Matsuda, S.; Koyasu, S. Mechanisms of Action of Cyclosporine. Immunopharmacology 2000, 47, 119–125. [Google Scholar] [CrossRef]
- Wüthrich, K.; Von Freyberg, B.; Weber, C.; Wider, G.; Traber, R.; Widmer, H.; Braun, W. Receptor-Induced Conformation Change of the Immunosuppressant Cyclosporin A. Science 1991, 254, 953–954. [Google Scholar] [CrossRef] [PubMed]
- Loosli, H.-R.; Kessler, H.; Oschkinat, H.; Weber, H.-P.; Petcher, T.J.; Widmer, A. Peptide conformations. Part 31. The conformation of cyclosporin a in the crystal and in solution. Helv. Chim. Acta 1985, 68, 682–704. [Google Scholar] [CrossRef]
- Ko, S.Y.; Dalvit, C. Conformation of Cyclosporin A in Polar Solvents. Int. J. Pept. Protein Res. 1992, 40, 380–382. [Google Scholar] [CrossRef] [PubMed]
- Fesik, S.W.; Gampe, R.T.; Eaton, H.L.; Gemmecker, G.; Olejniczak, E.T.; Neri, P.; Holzman, T.F.; Egan, D.A.; Edalji, R.; Simmer, R. NMR Studies of [U-13C]Cyclosporin A Bound to Cyclophilin: Bound Conformation and Portions of Cyclosporin Involved in Binding. Biochemistry 1991, 30, 6574–6583. [Google Scholar] [CrossRef]
- Thériault, Y.; Logan, T.M.; Meadows, R.; Yu, L.; Olejniczak, E.T.; Holzman, T.F.; Simmer, R.L.; Fesik, S.W. Solution Structure of the Cyclosporin A/Cyclophilin Complex by NMR. Nature 1993, 361, 88–91. [Google Scholar] [CrossRef]
- Peterson, A.A.; Rangwala, A.M.; Thakur, M.K.; Ward, P.S.; Hung, C.; Outhwaite, I.R.; Chan, A.I.; Usanov, D.L.; Mootha, V.K.; Seeliger, M.A.; et al. Discovery and Molecular Basis of Subtype-Selective Cyclophilin Inhibitors. Nat. Chem. Biol. 2022, 18, 1184–1195. [Google Scholar] [CrossRef]
- Daum, S.; Schumann, M.; Mathea, S.; Aumüller, T.; Balsley, M.A.; Constant, S.L.; De Lacroix, B.F.; Kruska, F.; Braun, M.; Schiene-Fischer, C. Isoform-Specific Inhibition of Cyclophilins. Biochemistry 2009, 48, 6268–6277. [Google Scholar] [CrossRef]
- Bernardi, P.; Rasola, A.; Forte, M.; Lippe, G. The Mitochondrial Permeability Transition Pore: Channel Formation by F-ATP Synthase, Integration in Signal Transduction, and Role in Pathophysiology. Physiol. Rev. 2015, 95, 1111–1155. [Google Scholar] [CrossRef]
- Massari, S.; Frigeri, L.; Azzone, G.F. A Quantitative Correlation between the Kinetics of Solutes and Water Translocation in Liver Mitochondria. J. Membr. Biol. 1972, 9, 71–82. [Google Scholar] [CrossRef]
- Nakagawa, T.; Shimizu, S.; Watanabe, T.; Yamaguchi, O.; Otsu, K.; Yamagata, H.; Inohara, H.; Kubo, T.; Tsujimoto, Y. Cyclophilin D-Dependent Mitochondrial Permeability Transition Regulates Some Necrotic but Not Apoptotic Cell Death. Nature 2005, 434, 652–658. [Google Scholar] [CrossRef]
- Rasola, A.; Bernardi, P. Mitochondrial Permeability Transition in Ca2+-Dependent Apoptosis and Necrosis. Cell Calcium 2011, 50, 222–233. [Google Scholar] [CrossRef]
- Baines, C.P.; Kaiser, R.A.; Purcell, N.H.; Blair, N.S.; Osinska, H.; Hambleton, M.A.; Brunskill, E.W.; Sayen, M.R.; Gottlieb, R.A.; Dorn, G.W.; et al. Loss of Cyclophilin D Reveals a Critical Role for Mitochondrial Permeability Transition in Cell Death. Nature 2005, 434, 658–662. [Google Scholar] [CrossRef] [PubMed]
- Forte, M.; Gold, B.G.; Marracci, G.; Chaudhary, P.; Basso, E.; Johnsen, D.; Yu, X.; Fowlkes, J.; Rahder, M.; Stem, K.; et al. Cyclophilin D Inactivation Protects Axons in Experimental Autoimmune Encephalomyelitis, an Animal Model of Multiple Sclerosis. Proc. Natl. Acad. Sci. USA 2007, 104, 7558–7563. [Google Scholar] [CrossRef]
- Fujimoto, K.; Chen, Y.; Polonsky, K.S.; Dorn, G.W. Targeting Cyclophilin D and the Mitochondrial Permeability Transition Enhances Beta-Cell Survival and Prevents Diabetes in Pdx1 Deficiency. Proc. Natl. Acad. Sci. USA 2010, 107, 10214–10219. [Google Scholar] [CrossRef] [PubMed]
- Martin, L.J.; Gertz, B.; Pan, Y.; Price, A.C.; Molkentin, J.D.; Chang, Q. The Mitochondrial Permeability Transition Pore in Motor Neurons: Involvement in the Pathobiology of ALS Mice. Exp. Neurol. 2009, 218, 333–346. [Google Scholar] [CrossRef] [PubMed]
- Millay, D.P.; Sargent, M.A.; Osinska, H.; Baines, C.P.; Barton, E.R.; Vuagniaux, G.; Sweeney, H.L.; Robbins, J.; Molkentin, J.D. Genetic and Pharmacologic Inhibition of Mitochondrial-Dependent Necrosis Attenuates Muscular Dystrophy. Nat. Med. 2008, 14, 442–447. [Google Scholar] [CrossRef] [PubMed]
- Parone, P.A.; Da Cruz, S.; Han, J.S.; McAlonis-Downes, M.; Vetto, A.P.; Lee, S.K.; Tseng, E.; Cleveland, D.W. Enhancing Mitochondrial Calcium Buffering Capacity Reduces Aggregation of Misfolded SOD1 and Motor Neuron Cell Death without Extending Survival in Mouse Models of Inherited Amyotrophic Lateral Sclerosis. J. Neurosci. 2013, 33, 4657–4671. [Google Scholar] [CrossRef] [PubMed]
- Schinzel, A.C.; Takeuchi, O.; Huang, Z.; Fisher, J.K.; Zhou, Z.; Rubens, J.; Hetz, C.; Danial, N.N.; Moskowitz, M.A.; Korsmeyer, S.J. Cyclophilin D Is a Component of Mitochondrial Permeability Transition and Mediates Neuronal Cell Death after Focal Cerebral Ischemia. Proc. Natl. Acad. Sci. USA 2005, 102, 12005–12010. [Google Scholar] [CrossRef]
- Wang, X.; Carlsson, Y.; Basso, E.; Zhu, C.; Rousset, C.I.; Rasola, A.; Johansson, B.R.; Blomgren, K.; Mallard, C.; Bernardi, P.; et al. Developmental Shift of Cyclophilin D Contribution to Hypoxic-Ischemic Brain Injury. J. Neurosci. 2009, 29, 2588–2596. [Google Scholar] [CrossRef]
- Palma, E.; Tiepolo, T.; Angelin, A.; Sabatelli, P.; Maraldi, N.M.; Basso, E.; Forte, M.A.; Bernardi, P.; Bonaldo, P. Genetic Ablation of Cyclophilin D Rescues Mitochondrial Defects and Prevents Muscle Apoptosis in Collagen VI Myopathic Mice. Hum. Mol. Genet. 2009, 18, 2024–2031. [Google Scholar] [CrossRef]
- Hunter, D.R.; Haworth, R.A. The Ca2+-Induced Membrane Transition in Mitochondria: I. The Protective Mechanisms. Arch. Biochem. Biophys. 1979, 195, 453–459. [Google Scholar] [CrossRef] [PubMed]
- Beutner, G.; Rück, A.; Riede, B.; Brdiczka, D. Complexes between Porin, Hexokinase, Mitochondrial Creatine Kinase and Adenylate Translocator Display Properties of the Permeability Transition Pore. Implication for Regulation of Permeability Transition by the Kinases. Biochim. Biophys. Acta 1998, 1368, 7–18. [Google Scholar] [CrossRef]
- Bernardi, P.; Di Lisa, F.; Fogolari, F.; Lippe, G. From ATP to PTP and Back: A Dual Function for the Mitochondrial ATP Synthase. Circ. Res. 2015, 116, 1850–1862. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Ford, H.C.; Carroll, J.; Ding, S.; Fearnley, I.M.; Walker, J.E. Persistence of the Mitochondrial Permeability Transition in the Absence of Subunit c of Human ATP Synthase. Proc. Natl. Acad. Sci. USA 2017, 114, 3409–3414. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Carroll, J.; Ding, S.; Fearnley, I.M.; Walker, J.E. Permeability Transition in Human Mitochondria Persists in the Absence of Peripheral Stalk Subunits of ATP Synthase. Proc. Natl. Acad. Sci. USA 2017, 114, 9086–9091. [Google Scholar] [CrossRef] [PubMed]
- Carroll, J.; He, J.; Ding, S.; Fearnley, I.M.; Walker, J.E. Persistence of the Permeability Transition Pore in Human Mitochondria Devoid of an Assembled ATP Synthase. Proc. Natl. Acad. Sci. USA 2019, 116, 12816–12821. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, P. Mechanisms for Ca2+-Dependent Permeability Transition in Mitochondria. Proc. Natl. Acad. Sci. USA 2020, 117, 2743–2744. [Google Scholar] [CrossRef]
- Neginskaya, M.A.; Solesio, M.E.; Berezhnaya, E.V.; Amodeo, G.F.; Mnatsakanyan, N.; Jonas, E.A.; Pavlov, E.V. ATP Synthase C-Subunit-Deficient Mitochondria Have a Small Cyclosporine A-Sensitive Channel, but Lack the Permeability Transition Pore. Cell Rep. 2019, 26, 11–17.e2. [Google Scholar] [CrossRef]
- Kühlbrandt, W. Structure and Mechanisms of F-Type ATP Synthases. Annu. Rev. Biochem. 2019, 88, 515–549. [Google Scholar] [CrossRef]
- Spikes, T.E.; Montgomery, M.G.; Walker, J.E. Structure of the Dimeric ATP Synthase from Bovine Mitochondria. Proc. Natl. Acad. Sci. USA 2020, 117, 23519–23526. [Google Scholar] [CrossRef]
- Gu, J.; Zhang, L.; Zong, S.; Guo, R.; Liu, T.; Yi, J.; Wang, P.; Zhuo, W.; Yang, M. Cryo-EM Structure of the Mammalian ATP Synthase Tetramer Bound with Inhibitory Protein IF1. Science 2019, 364, 1068–1075. [Google Scholar] [CrossRef] [PubMed]
- Paumard, P.; Vaillier, J.; Coulary, B.; Schaeffer, J.; Soubannier, V.; Mueller, D.M.; Brèthes, D.; di Rago, J.-P.; Velours, J. The ATP Synthase Is Involved in Generating Mitochondrial Cristae Morphology. EMBO J. 2002, 21, 221–230. [Google Scholar] [CrossRef] [PubMed]
- Boyer, P.D. The ATP Synthase—A Splendid Molecular Machine. Annu. Rev. Biochem. 1997, 66, 717–749. [Google Scholar] [CrossRef] [PubMed]
- Stock, D.; Leslie, A.G.; Walker, J.E. Molecular Architecture of the Rotary Motor in ATP Synthase. Science 1999, 286, 1700–1705. [Google Scholar] [CrossRef]
- Kobayashi, R.; Ueno, H.; Li, C.-B.; Noji, H. Rotary Catalysis of Bovine Mitochondrial F1-ATPase Studied by Single-Molecule Experiments. Proc. Natl. Acad. Sci. USA 2020, 117, 1447–1456. [Google Scholar] [CrossRef]
- Guo, H.; Rubinstein, J.L. Structure of ATP Synthase under Strain during Catalysis. Nat. Commun. 2022, 13, 2232. [Google Scholar] [CrossRef]
- Papageorgiou, S.; Melandri, A.B.; Solaini, G. Relevance of Divalent Cations to ATP-Driven Proton Pumping in Beef Heart Mitochondrial F0F1-ATPase. J. Bioenerg. Biomembr. 1998, 30, 533–541. [Google Scholar] [CrossRef]
- Nathanson, L.; Gromet-Elhanan, Z. Mutations in the Beta-Subunit Thr(159) and Glu(184) of the Rhodospirillum Rubrum F(0)F(1) ATP Synthase Reveal Differences in Ligands for the Coupled Mg2+- and Decoupled Ca2+-Dependent F(0)F(1) Activities. J. Biol. Chem. 2000, 275, 901–905. [Google Scholar] [CrossRef]
- Nesci, S.; Trombetti, F.; Ventrella, V.; Pirini, M.; Pagliarani, A. Kinetic Properties of the Mitochondrial F1FO-ATPase Activity Elicited by Ca2+ in Replacement of Mg2+. Biochimie 2017, 140, 73–81. [Google Scholar] [CrossRef]
- Lai, Y.; Zhang, Y.; Zhou, S.; Xu, J.; Du, Z.; Feng, Z.; Yu, L.; Zhao, Z.; Wang, W.; Tang, Y.; et al. Structure of the Human ATP Synthase. Mol. Cell 2023, 83, 2137–2147.e4. [Google Scholar] [CrossRef]
- Pinke, G.; Zhou, L.; Sazanov, L.A. Cryo-EM Structure of the Entire Mammalian F-Type ATP Synthase. Nat. Struct. Mol. Biol. 2020, 27, 1077–1085. [Google Scholar] [CrossRef]
- Colina-Tenorio, L.; Dautant, A.; Miranda-Astudillo, H.; Giraud, M.-F.; González-Halphen, D. The Peripheral Stalk of Rotary ATPases. Front. Physiol. 2018, 9, 1243. [Google Scholar] [CrossRef] [PubMed]
- Giorgio, V.; Fogolari, F.; Lippe, G.; Bernardi, P. OSCP Subunit of Mitochondrial ATP Synthase: Role in Regulation of Enzyme Function and of Its Transition to a Pore. Br. J. Pharmacol. 2019, 176, 4247–4257. [Google Scholar] [CrossRef] [PubMed]
- Symersky, J.; Osowski, D.; Walters, D.E.; Mueller, D.M. Oligomycin Frames a Common Drug-Binding Site in the ATP Synthase. Proc. Natl. Acad. Sci. USA 2012, 109, 13961–13965. [Google Scholar] [CrossRef] [PubMed]
- Alavian, K.N.; Beutner, G.; Lazrove, E.; Sacchetti, S.; Park, H.-A.; Licznerski, P.; Li, H.; Nabili, P.; Hockensmith, K.; Graham, M.; et al. An Uncoupling Channel within the C-Subunit Ring of the F1FO ATP Synthase Is the Mitochondrial Permeability Transition Pore. Proc. Natl. Acad. Sci. USA 2014, 111, 10580–10585. [Google Scholar] [CrossRef] [PubMed]
- Giorgio, V.; Burchell, V.; Schiavone, M.; Bassot, C.; Minervini, G.; Petronilli, V.; Argenton, F.; Forte, M.; Tosatto, S.; Lippe, G.; et al. Ca2+ Binding to F-ATP Synthase β Subunit Triggers the Mitochondrial Permeability Transition. EMBO Rep. 2017, 18, 1065–1076. [Google Scholar] [CrossRef] [PubMed]
- Antoniel, M.; Jones, K.; Antonucci, S.; Spolaore, B.; Fogolari, F.; Petronilli, V.; Giorgio, V.; Carraro, M.; Di Lisa, F.; Forte, M.; et al. The Unique Histidine in OSCP Subunit of F-ATP Synthase Mediates Inhibition of the Permeability Transition Pore by Acidic PH. EMBO Rep. 2018, 19, 257–268. [Google Scholar] [CrossRef]
- Guo, L.; Carraro, M.; Sartori, G.; Minervini, G.; Eriksson, O.; Petronilli, V.; Bernardi, P. Arginine 107 of Yeast ATP Synthase Subunit g Mediates Sensitivity of the Mitochondrial Permeability Transition to Phenylglyoxal. J. Biol. Chem. 2018, 293, 14632–14645. [Google Scholar] [CrossRef]
- Guo, L.; Carraro, M.; Carrer, A.; Minervini, G.; Urbani, A.; Masgras, I.; Tosatto, S.C.E.; Szabò, I.; Bernardi, P.; Lippe, G. Arg-8 of Yeast Subunit e Contributes to the Stability of F-ATP Synthase Dimers and to the Generation of the Full-Conductance Mitochondrial Megachannel. J. Biol. Chem. 2019, 294, 10987–10997. [Google Scholar] [CrossRef]
- Carrer, A.; Tommasin, L.; Šileikytė, J.; Ciscato, F.; Filadi, R.; Urbani, A.; Forte, M.; Rasola, A.; Szabò, I.; Carraro, M.; et al. Defining the Molecular Mechanisms of the Mitochondrial Permeability Transition through Genetic Manipulation of F-ATP Synthase. Nat. Commun. 2021, 12, 4835. [Google Scholar] [CrossRef]
- Morciano, G.; Pedriali, G.; Bonora, M.; Pavasini, R.; Mikus, E.; Calvi, S.; Bovolenta, M.; Lebiedzinska-Arciszewska, M.; Pinotti, M.; Albertini, A.; et al. A Naturally Occurring Mutation in ATP Synthase Subunit c Is Associated with Increased Damage Following Hypoxia/Reoxygenation in STEMI Patients. Cell Rep. 2021, 35, 108983. [Google Scholar] [CrossRef] [PubMed]
- Carraro, M.; Jones, K.; Sartori, G.; Schiavone, M.; Antonucci, S.; Kucharczyk, R.; di Rago, J.-P.; Franchin, C.; Arrigoni, G.; Forte, M.; et al. The Unique Cysteine of F-ATP Synthase OSCP Subunit Participates in Modulation of the Permeability Transition Pore. Cell Rep. 2020, 32, 108095. [Google Scholar] [CrossRef] [PubMed]
- Gerle, C. Mitochondrial F-ATP Synthase as the Permeability Transition Pore. Pharmacol. Res. 2020, 160, 105081. [Google Scholar] [CrossRef] [PubMed]
- Mnatsakanyan, N.; Park, H.-A.; Wu, J.; He, X.; Llaguno, M.C.; Latta, M.; Miranda, P.; Murtishi, B.; Graham, M.; Weber, J.; et al. Mitochondrial ATP Synthase C-Subunit Leak Channel Triggers Cell Death upon Loss of Its F1 Subcomplex. Cell Death Differ. 2022, 29, 1874–1887. [Google Scholar] [CrossRef]
- Amodeo, G.F.; Krilyuk, N.; Pavlov, E.V. Formation of High-Conductive C Subunit Channels upon Interaction with Cyclophilin D. Int. J. Mol. Sci. 2021, 22, 11022. [Google Scholar] [CrossRef]
- Amodeo, G.F.; Lee, B.Y.; Krilyuk, N.; Filice, C.T.; Valyuk, D.; Otzen, D.E.; Noskov, S.; Leonenko, Z.; Pavlov, E.V. C Subunit of the ATP Synthase Is an Amyloidogenic Calcium Dependent Channel-Forming Peptide with Possible Implications in Mitochondrial Permeability Transition. Sci. Rep. 2021, 11, 8744. [Google Scholar] [CrossRef]
- Amodeo, G.F.; Pavlov, E.V. Amyloid β, α-Synuclein and the c Subunit of the ATP Synthase: Can These Peptides Reveal an Amyloidogenic Pathway of the Permeability Transition Pore? Biochim. Biophys. Acta 2021, 1863, 183531. [Google Scholar] [CrossRef]
- Scorrano, L.; Nicolli, A.; Basso, E.; Petronilli, V.; Bernardi, P. Two Modes of Activation of the Permeability Transition Pore: The Role of Mitochondrial Cyclophilin. Mol. Cell Biochem. 1997, 174, 181–184. [Google Scholar] [CrossRef]
- Frigo, E.; Tommasin, L.; Lippe, G.; Carraro, M.; Bernardi, P. The Haves and Have-Nots: The Mitochondrial Permeability Transition Pore across Species. Cells 2023, 12, 1409. [Google Scholar] [CrossRef]
- Khoury, G.A.; Baliban, R.C.; Floudas, C.A. Proteome-Wide Post-Translational Modification Statistics: Frequency Analysis and Curation of the Swiss-Prot Database. Sci. Rep. 2011, 1, 90. [Google Scholar] [CrossRef]
- Hafner, A.V.; Dai, J.; Gomes, A.P.; Xiao, C.-Y.; Palmeira, C.M.; Rosenzweig, A.; Sinclair, D.A. Regulation of the MPTP by SIRT3-Mediated Deacetylation of CypD at Lysine 166 Suppresses Age-Related Cardiac Hypertrophy. Aging 2010, 2, 914–923. [Google Scholar] [CrossRef] [PubMed]
- Bochaton, T.; Crola-Da-Silva, C.; Pillot, B.; Villedieu, C.; Ferreras, L.; Alam, M.R.; Thibault, H.; Strina, M.; Gharib, A.; Ovize, M.; et al. Inhibition of Myocardial Reperfusion Injury by Ischemic Postconditioning Requires Sirtuin 3-Mediated Deacetylation of Cyclophilin D. J. Mol. Cell. Cardiol. 2015, 84, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Castillo, E.C.; Morales, J.A.; Chapoy-Villanueva, H.; Silva-Platas, C.; Trevino-Saldana, N.; Guerrero-Beltrán, C.E.; Bernal-Ramìrez, J.; Torres-Quintanilla, A.; Garcìa, N.; Youker, K.; et al. Mitochondrial Hyperacetylation in the Failing Hearts of Obese Patients Mediated Partly by a Reduction in SIRT3: The Involvement of the Mitochondrial Permeability Transition Pore. Cell Physiol. Biochem. 2019, 53, 465–479. [Google Scholar] [CrossRef] [PubMed]
- Hurst, S.; Gonnot, F.; Dia, M.; Crola Da Silva, C.; Gomez, L.; Sheu, S.-S. Phosphorylation of Cyclophilin D at Serine 191 Regulates Mitochondrial Permeability Transition Pore Opening and Cell Death after Ischemia-Reperfusion. Cell Death Dis. 2020, 11, 661. [Google Scholar] [CrossRef]
- Parks, R.J.; Menazza, S.; Holmström, K.M.; Amanakis, G.; Fergusson, M.; Ma, H.; Aponte, A.M.; Bernardi, P.; Finkel, T.; Murphy, E. Cyclophilin D-Mediated Regulation of the Permeability Transition Pore Is Altered in Mice Lacking the Mitochondrial Calcium Uniporter. Cardiovasc. Res. 2019, 115, 385–394. [Google Scholar] [CrossRef]
- Amanakis, G.; Sun, J.; Fergusson, M.M.; McGinty, S.; Liu, C.; Molkentin, J.D.; Murphy, E. Cysteine 202 of Cyclophilin D Is a Site of Multiple Post-Translational Modifications and Plays a Role in Cardioprotection. Cardiovasc. Res. 2021, 117, 212–223. [Google Scholar] [CrossRef]
- Teodoro, J.S.; Varela, A.T.; Duarte, F.V.; Gomes, A.P.; Palmeira, C.M.; Rolo, A.P. Indirubin and NAD+ Prevent Mitochondrial Ischaemia/Reperfusion Damage in Fatty Livers. Eur. J. Clin. Invest. 2018, 48, e12932. [Google Scholar] [CrossRef]
- Nguyen, T.T.; Stevens, M.V.; Kohr, M.; Steenbergen, C.; Sack, M.N.; Murphy, E. Cysteine 203 of Cyclophilin D Is Critical for Cyclophilin D Activation of the Mitochondrial Permeability Transition Pore. J. Biol. Chem. 2011, 286, 40184–40192. [Google Scholar] [CrossRef]
- Sambri, I.; Massa, F.; Gullo, F.; Meneghini, S.; Cassina, L.; Carraro, M.; Dina, G.; Quattrini, A.; Patanella, L.; Carissimo, A.; et al. Impaired Flickering of the Permeability Transition Pore Causes SPG7 Spastic Paraplegia. eBioMedicine 2020, 61, 103050. [Google Scholar] [CrossRef]
- Hu, M.; He, F.; Thompson, E.W.; Ostrikov, K.; Dai, X. Lysine Acetylation, Cancer Hallmarks and Emerging Onco-Therapeutic Opportunities. Cancers 2022, 14, 346. [Google Scholar] [CrossRef]
- Ziętara, P.; Dziewięcka, M.; Augustyniak, M. Why Is Longevity Still a Scientific Mystery? Sirtuins—Past, Present and Future. Int. J. Mol. Sci. 2022, 24, 728. [Google Scholar] [CrossRef] [PubMed]
- Baeza, J.; Smallegan, M.J.; Denu, J.M. Mechanisms and Dynamics of Protein Acetylation in Mitochondria. Trends Biochem. Sci. 2016, 41, 231–244. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.C.; Sprung, R.; Chen, Y.; Xu, Y.; Ball, H.; Pei, J.; Cheng, T.; Kho, Y.; Xiao, H.; Xiao, L.; et al. Substrate and Functional Diversity of Lysine Acetylation Revealed by a Proteomics Survey. Mol. Cell 2006, 23, 607–618. [Google Scholar] [CrossRef]
- Boreikaite, V.; Wicky, B.I.M.; Watt, I.N.; Clarke, J.; Walker, J.E. Extrinsic Conditions Influence the Self-Association and Structure of IF1, the Regulatory Protein of Mitochondrial ATP Synthase. Proc. Natl. Acad. Sci. USA 2019, 116, 10354–10359. [Google Scholar] [CrossRef]
- Bilbrough, T.; Piemontese, E.; Seitz, O. Dissecting the Role of Protein Phosphorylation: A Chemical Biology Toolbox. Chem. Soc. Rev. 2022, 51, 5691–5730. [Google Scholar] [CrossRef] [PubMed]
- Manning, G.; Whyte, D.B.; Martinez, R.; Hunter, T.; Sudarsanam, S. The Protein Kinase Complement of the Human Genome. Science 2002, 298, 1912–1934. [Google Scholar] [CrossRef]
- Barford, D. Protein Phosphatases. Curr. Opin. Struct. Biol. 1995, 5, 728–734. [Google Scholar] [CrossRef]
- Juhaszova, M.; Zorov, D.B.; Kim, S.H.; Pepe, S.; Fu, Q.; Fishbein, K.W.; Ziman, B.D.; Wang, S.; Ytrehus, K.; Antos, C.L.; et al. Glycogen Synthase Kinase-3beta Mediates Convergence of Protection Signaling to Inhibit the Mitochondrial Permeability Transition Pore. J. Clin. Investig. 2004, 113, 1535–1549. [Google Scholar] [CrossRef]
- Kumutima, J.; Yao, X.-Q.; Hamelberg, D. Post-Translational Modifications of Cyclophilin D Fine-Tune Its Conformational Dynamics and Activity: Implications for Its Mitochondrial Function. J. Phys. Chem. B 2022, 126, 10844–10853. [Google Scholar] [CrossRef]
- Rinalducci, S.; Murgiano, L.; Zolla, L. Redox Proteomics: Basic Principles and Future Perspectives for the Detection of Protein Oxidation in Plants. J. Exp. Bot. 2008, 59, 3781–3801. [Google Scholar] [CrossRef]
- Di Meo, S.; Reed, T.T.; Venditti, P.; Victor, V.M. Role of ROS and RNS Sources in Physiological and Pathological Conditions. Oxid. Med. Cell Longev. 2016, 2016, 1245049. [Google Scholar] [CrossRef] [PubMed]
- Balaban, R.S.; Nemoto, S.; Finkel, T. Mitochondria, Oxidants, and Aging. Cell 2005, 120, 483–495. [Google Scholar] [CrossRef] [PubMed]
- Radi, R.; Cassina, A.; Hodara, R.; Quijano, C.; Castro, L. Peroxynitrite Reactions and Formation in Mitochondria. Free Radic. Biol. Med. 2002, 33, 1451–1464. [Google Scholar] [CrossRef] [PubMed]
- Linard, D.; Kandlbinder, A.; Degand, H.; Morsomme, P.; Dietz, K.-J.; Knoops, B. Redox Characterization of Human Cyclophilin D: Identification of a New Mammalian Mitochondrial Redox Sensor? Arch. Biochem. Biophys. 2009, 491, 39–45. [Google Scholar] [CrossRef]
- Folda, A.; Citta, A.; Scalcon, V.; Calì, T.; Zonta, F.; Scutari, G.; Bindoli, A.; Rigobello, M.P. Mitochondrial Thioredoxin System as a Modulator of Cyclophilin D Redox State. Sci. Rep. 2016, 6, 23071. [Google Scholar] [CrossRef]
- Kohr, M.J.; Sun, J.; Aponte, A.; Wang, G.; Gucek, M.; Murphy, E.; Steenbergen, C. Simultaneous Measurement of Protein Oxidation and S-Nitrosylation during Preconditioning and Ischemia/Reperfusion Injury with Resin-Assisted Capture. Circ. Res. 2011, 108, 418–426. [Google Scholar] [CrossRef]
- Andreyev, A.; Fiskum, G. Calcium Induced Release of Mitochondrial Cytochrome c by Different Mechanisms Selective for Brain versus Liver. Cell Death Differ. 1999, 6, 825–832. [Google Scholar] [CrossRef]
- Berman, S.B.; Watkins, S.C.; Hastings, T.G. Quantitative Biochemical and Ultrastructural Comparison of Mitochondrial Permeability Transition in Isolated Brain and Liver Mitochondria: Evidence for Reduced Sensitivity of Brain Mitochondria. Exp. Neurol. 2000, 164, 415–425. [Google Scholar] [CrossRef]
- Eliseev, R.A.; Filippov, G.; Velos, J.; VanWinkle, B.; Goldman, A.; Rosier, R.N.; Gunter, T.E. Role of Cyclophilin D in the Resistance of Brain Mitochondria to the Permeability Transition. Neurobiol. Aging 2007, 28, 1532–1542. [Google Scholar] [CrossRef]
- Du, H.; Guo, L.; Fang, F.; Chen, D.; Sosunov, A.A.; McKhann, G.M.; Yan, Y.; Wang, C.; Zhang, H.; Molkentin, J.D.; et al. Cyclophilin D Deficiency Attenuates Mitochondrial and Neuronal Perturbation and Ameliorates Learning and Memory in Alzheimer’s Disease. Nat. Med. 2008, 14, 1097–1105. [Google Scholar] [CrossRef]
- Du, H.; Guo, L.; Zhang, W.; Rydzewska, M.; Yan, S. Cyclophilin D Deficiency Improves Mitochondrial Function and Learning/Memory in Aging Alzheimer Disease Mouse Model. Neurobiol. Aging 2011, 32, 398–406. [Google Scholar] [CrossRef]
- Gauba, E.; Guo, L.; Du, H. Cyclophilin D Promotes Brain Mitochondrial F1FO ATP Synthase Dysfunction in Aging Mice. J. Alzheimer’s Dis. 2016, 55, 1351–1362. [Google Scholar] [CrossRef]
- Luvisetto, S.; Basso, E.; Petronilli, V.; Bernardi, P.; Forte, M. Enhancement of Anxiety, Facilitation of Avoidance Behavior, and Occurrence of Adult-Onset Obesity in Mice Lacking Mitochondrial Cyclophilin D. Neuroscience 2008, 155, 585–596. [Google Scholar] [CrossRef]
- Rebolledo-Solleiro, D.; Roldán-Roldán, G.; Díaz, D.; Velasco, M.; Larqué, C.; Rico-Rosillo, G.; Vega-Robledo, G.B.; Zambrano, E.; Hiriart, M.; Pérez de la Mora, M. Increased Anxiety-like Behavior Is Associated with the Metabolic Syndrome in Non-Stressed Rats. PLoS ONE 2017, 12, e0176554. [Google Scholar] [CrossRef]
- Tzioras, M.; McGeachan, R.I.; Durrant, C.S.; Spires-Jones, T.L. Synaptic Degeneration in Alzheimer Disease. Nat. Rev. Neurol. 2023, 19, 19–38. [Google Scholar] [CrossRef]
- O’Brien, R.J.; Wong, P.C. Amyloid Precursor Protein Processing and Alzheimer’s Disease. Annu. Rev. Neurosci. 2011, 34, 185–204. [Google Scholar] [CrossRef]
- Friedrich, R.P.; Tepper, K.; Rönicke, R.; Soom, M.; Westermann, M.; Reymann, K.; Kaether, C.; Fändrich, M. Mechanism of Amyloid Plaque Formation Suggests an Intracellular Basis of Abeta Pathogenicity. Proc. Natl. Acad. Sci. USA 2010, 107, 1942–1947. [Google Scholar] [CrossRef]
- Hardy, J.A.; Higgins, G.A. Alzheimer’s Disease: The Amyloid Cascade Hypothesis. Science 1992, 256, 184–185. [Google Scholar] [CrossRef]
- Caspersen, C.; Wang, N.; Yao, J.; Sosunov, A.; Chen, X.; Lustbader, J.W.; Xu, H.W.; Stern, D.; McKhann, G.; Du Yan, S. Mitochondrial Aβ: A Potential Focal Point for Neuronal Metabolic Dysfunction in Alzheimer’s Disease. FASEB J. 2005, 19, 2040–2041. [Google Scholar] [CrossRef]
- Casley, C.S.; Canevari, L.; Land, J.M.; Clark, J.B.; Sharpe, M.A. β-Amyloid Inhibits Integrated Mitochondrial Respiration and Key Enzyme Activities. J. Neurochem. 2002, 80, 91–100. [Google Scholar] [CrossRef]
- Abramov, A.Y.; Canevari, L.; Duchen, M.R. Beta-Amyloid Peptides Induce Mitochondrial Dysfunction and Oxidative Stress in Astrocytes and Death of Neurons through Activation of NADPH Oxidase. J. Neurosci. 2004, 24, 565–575. [Google Scholar] [CrossRef]
- Wang, S.; Mims, P.N.; Roman, R.J.; Fan, F. Is Beta-Amyloid Accumulation a Cause or Consequence of Alzheimer’s Disease? J. Alzheimers Park. Dement. 2016, 1, 007. [Google Scholar]
- Imbimbo, B.P.; Balducci, C.; Ippati, S.; Watling, M. Initial Failures of Anti-Tau Antibodies in Alzheimer’s Disease Are Reminiscent of the Amyloid-β Story. Neural Regen. Res. 2022, 18, 117–118. [Google Scholar] [CrossRef]
- Price, J.L.; McKeel, D.W.; Buckles, V.D.; Roe, C.M.; Xiong, C.; Grundman, M.; Hansen, L.A.; Petersen, R.C.; Parisi, J.E.; Dickson, D.W.; et al. Neuropathology of Nondemented Aging: Presumptive Evidence for Preclinical Alzheimer Disease. Neurobiol. Aging 2009, 30, 1026–1036. [Google Scholar] [CrossRef]
- Kolarova, M.; García-Sierra, F.; Bartos, A.; Ricny, J.; Ripova, D. Structure and Pathology of Tau Protein in Alzheimer Disease. Int. J. Alzheimer’s Dis. 2012, 2012, e731526. [Google Scholar] [CrossRef]
- Du, H.; Yan, S.S. Mitochondrial Permeability Transition Pore in Alzheimer’s Disease: Cyclophilin D and Amyloid Beta. Biochim. Biophys. Acta 2010, 1802, 198–204. [Google Scholar] [CrossRef]
- Singh, P.; Suman, S.; Chandna, S.; Das, T.K. Possible Role of Amyloid-Beta, Adenine Nucleotide Translocase and Cyclophilin-D Interaction in Mitochondrial Dysfunction of Alzheimer’s Disease. Bioinformation 2009, 3, 440–445. [Google Scholar] [CrossRef]
- Drummond, E.; Wisniewski, T. Alzheimer’s Disease: Experimental Models and Reality. Acta Neuropathol. 2017, 133, 155–175. [Google Scholar] [CrossRef]
- Waltereit, R.; Weller, M. Signaling from CAMP/PKA to MAPK and Synaptic Plasticity. Mol. Neurobiol. 2003, 27, 99–106. [Google Scholar] [CrossRef]
- Du, H.; Guo, L.; Wu, X.; Sosunov, A.A.; McKhann, G.M.; Chen, J.X.; Yan, S.S. Cyclophilin D Deficiency Rescues Aβ-Impaired PKA/CREB Signaling and Alleviates Synaptic Degeneration. Biochim. Biophys. Acta 2014, 1842, 2517–2527. [Google Scholar] [CrossRef]
- Jara, C.; Cerpa, W.; Tapia-Rojas, C.; Quintanilla, R.A. Tau Deletion Prevents Cognitive Impairment and Mitochondrial Dysfunction Age Associated by a Mechanism Dependent on Cyclophilin-D. Front. Neurosci. 2021, 14, 586710. [Google Scholar] [CrossRef] [PubMed]
- Beck, S.J.; Guo, L.; Phensy, A.; Tian, J.; Wang, L.; Tandon, N.; Gauba, E.; Lu, L.; Pascual, J.M.; Kroener, S.; et al. Deregulation of Mitochondrial F1FO-ATP Synthase via OSCP in Alzheimer’s Disease. Nat. Commun. 2016, 7, 11483. [Google Scholar] [CrossRef] [PubMed]
- Pérez, M.J.; Ponce, D.P.; Aranguiz, A.; Behrens, M.I.; Quintanilla, R.A. Mitochondrial Permeability Transition Pore Contributes to Mitochondrial Dysfunction in Fibroblasts of Patients with Sporadic Alzheimer’s Disease. Redox Biol. 2018, 19, 290–300. [Google Scholar] [CrossRef]
- Gauba, E.; Chen, H.; Guo, L.; Du, H. Cyclophilin D Deficiency Attenuates Mitochondrial F1Fo ATP Synthase Dysfunction via OSCP in Alzheimer’s Disease. Neurobiol. Dis. 2019, 121, 138–147. [Google Scholar] [CrossRef]
- Sui, S.; Tian, J.; Gauba, E.; Wang, Q.; Guo, L.; Du, H. Cyclophilin D Regulates Neuronal Activity-Induced Filopodiagenesis by Fine-Tuning Dendritic Mitochondrial Calcium Dynamics. J. Neurochem. 2018, 146, 403–415. [Google Scholar] [CrossRef]
- Hamilton, J.; Brustovetsky, T.; Brustovetsky, N. The Effect of Mitochondrial Calcium Uniporter and Cyclophilin D Knockout on Resistance of Brain Mitochondria to Ca2+-Induced Damage. J. Biol. Chem. 2021, 296, 100669. [Google Scholar] [CrossRef]
- Misrani, A.; Tabassum, S.; Yang, L. Mitochondrial Dysfunction and Oxidative Stress in Alzheimer’s Disease. Front. Aging Neurosci. 2021, 13, 57. [Google Scholar] [CrossRef]
- Covarrubias, A.J.; Perrone, R.; Grozio, A.; Verdin, E. NAD+ Metabolism and Its Roles in Cellular Processes during Ageing. Nat. Rev. Mol. Cell Biol. 2021, 22, 119–141. [Google Scholar] [CrossRef]
- Sun, L.; Bhawal, R.; Xu, H.; Chen, H.; Anderson, E.T.; Haroutunian, V.; Cross, A.C.; Zhang, S.; Gibson, G.E. The Human Brain Acetylome Reveals That Decreased Acetylation of Mitochondrial Proteins Associates with Alzheimer’s Disease. J. Neurochem. 2021, 158, 282–296. [Google Scholar] [CrossRef]
- Ping, L.; Kundinger, S.R.; Duong, D.M.; Yin, L.; Gearing, M.; Lah, J.J.; Levey, A.I.; Seyfried, N.T. Global Quantitative Analysis of the Human Brain Proteome and Phosphoproteome in Alzheimer’s Disease. Sci. Data 2020, 7, 315. [Google Scholar] [CrossRef]
- Rottenberg, H.; Hoek, J.B. The Mitochondrial Permeability Transition: Nexus of Aging, Disease and Longevity. Cells 2021, 10, 79. [Google Scholar] [CrossRef] [PubMed]
- Rani, L.; Sahu, M.R.; Mondal, A.C. Age-Related Mitochondrial Dysfunction in Parkinson’s Disease: New Insights into the Disease Pathology. Neuroscience 2022, 499, 152–169. [Google Scholar] [CrossRef] [PubMed]
- Surmeier, D.J. Determinants of Dopaminergic Neuron Loss in Parkinson’s Disease. FEBS J. 2018, 285, 3657–3668. [Google Scholar] [CrossRef]
- Roberts, H.L.; Brown, D.R. Seeking a Mechanism for the Toxicity of Oligomeric α-Synuclein. Biomolecules 2015, 5, 282–305. [Google Scholar] [CrossRef] [PubMed]
- Iljina, M.; Garcia, G.A.; Horrocks, M.H.; Tosatto, L.; Choi, M.L.; Ganzinger, K.A.; Abramov, A.Y.; Gandhi, S.; Wood, N.W.; Cremades, N.; et al. Kinetic Model of the Aggregation of Alpha-Synuclein Provides Insights into Prion-like Spreading. Proc. Natl. Acad. Sci. USA 2016, 113, E1206–E1215. [Google Scholar] [CrossRef] [PubMed]
- Luk, K.C.; Kehm, V.; Carroll, J.; Zhang, B.; O’Brien, P.; Trojanowski, J.Q.; Lee, V.M.-Y. Pathological α-Synuclein Transmission Initiates Parkinson-like Neurodegeneration in Nontransgenic Mice. Science 2012, 338, 949–953. [Google Scholar] [CrossRef]
- Mao, X.; Ou, M.T.; Karuppagounder, S.S.; Kam, T.-I.; Yin, X.; Xiong, Y.; Ge, P.; Umanah, G.E.; Brahmachari, S.; Shin, J.-H.; et al. Pathological α-Synuclein Transmission Initiated by Binding Lymphocyte-Activation Gene 3. Science 2016, 353, aah3374. [Google Scholar] [CrossRef]
- Chartier-Harlin, M.-C.; Kachergus, J.; Roumier, C.; Mouroux, V.; Douay, X.; Lincoln, S.; Levecque, C.; Larvor, L.; Andrieux, J.; Hulihan, M.; et al. Alpha-Synuclein Locus Duplication as a Cause of Familial Parkinson’s Disease. Lancet 2004, 364, 1167–1169. [Google Scholar] [CrossRef]
- Singleton, A.B.; Farrer, M.; Johnson, J.; Singleton, A.; Hague, S.; Kachergus, J.; Hulihan, M.; Peuralinna, T.; Dutra, A.; Nussbaum, R.; et al. Alpha-Synuclein Locus Triplication Causes Parkinson’s Disease. Science 2003, 302, 841. [Google Scholar] [CrossRef]
- Schapira, A.H.; Cooper, J.M.; Dexter, D.; Clark, J.B.; Jenner, P.; Marsden, C.D. Mitochondrial Complex I Deficiency in Parkinson’s Disease. J. Neurochem. 1990, 54, 823–827. [Google Scholar] [CrossRef]
- Bindoff, L.A.; Birch-Machin, M.; Cartlidge, N.E.; Parker, W.D.; Turnbull, D.M. Mitochondrial Function in Parkinson’s Disease. Lancet 1989, 2, 49. [Google Scholar] [CrossRef] [PubMed]
- Ramsay, R.R.; Salach, J.I.; Dadgar, J.; Singer, T.P. Inhibition of Mitochondrial NADH Dehydrogenase by Pyridine Derivatives and Its Possible Relation to Experimental and Idiopathic Parkinsonism. Biochem. Biophys. Res. Commun. 1986, 135, 269–275. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.; Hwang, I.; Park, S.; Hong, S.; Hwang, B.; Cho, Y.; Son, J.; Yu, J.-W. MPTP-Driven NLRP3 Inflammasome Activation in Microglia Plays a Central Role in Dopaminergic Neurodegeneration. Cell Death Differ. 2019, 26, 213–228. [Google Scholar] [CrossRef] [PubMed]
- González-Rodríguez, P.; Zampese, E.; Stout, K.A.; Guzman, J.N.; Ilijic, E.; Yang, B.; Tkatch, T.; Stavarache, M.A.; Wokosin, D.L.; Gao, L.; et al. Disruption of Mitochondrial Complex I Induces Progressive Parkinsonism. Nature 2021, 599, 650–656. [Google Scholar] [CrossRef]
- Burbulla, L.F.; Song, P.; Mazzulli, J.R.; Zampese, E.; Wong, Y.C.; Jeon, S.; Santos, D.P.; Blanz, J.; Obermaier, C.D.; Strojny, C.; et al. Dopamine Oxidation Mediates Mitochondrial and Lysosomal Dysfunction in Parkinson’s Disease. Science 2017, 357, 1255–1261. [Google Scholar] [CrossRef]
- Nakamura, K.; Nemani, V.M.; Azarbal, F.; Skibinski, G.; Levy, J.M.; Egami, K.; Munishkina, L.; Zhang, J.; Gardner, B.; Wakabayashi, J.; et al. Direct Membrane Association Drives Mitochondrial Fission by the Parkinson Disease-Associated Protein Alpha-Synuclein. J. Biol. Chem. 2011, 286, 20710–20726. [Google Scholar] [CrossRef]
- Ludtmann, M.H.R.; Angelova, P.R.; Horrocks, M.H.; Choi, M.L.; Rodrigues, M.; Baev, A.Y.; Berezhnov, A.V.; Yao, Z.; Little, D.; Banushi, B.; et al. α-Synuclein Oligomers Interact with ATP Synthase and Open the Permeability Transition Pore in Parkinson’s Disease. Nat. Commun. 2018, 9, 2293. [Google Scholar] [CrossRef]
- Schöndorf, D.C.; Ivanyuk, D.; Baden, P.; Sanchez-Martinez, A.; De Cicco, S.; Yu, C.; Giunta, I.; Schwarz, L.K.; Di Napoli, G.; Panagiotakopoulou, V.; et al. The NAD+ Precursor Nicotinamide Riboside Rescues Mitochondrial Defects and Neuronal Loss in IPSC and Fly Models of Parkinson’s Disease. Cell Rep. 2018, 23, 2976–2988. [Google Scholar] [CrossRef]
- Bonello, F.; Hassoun, S.-M.; Mouton-Liger, F.; Shin, Y.S.; Muscat, A.; Tesson, C.; Lesage, S.; Beart, P.M.; Brice, A.; Krupp, J.; et al. LRRK2 Impairs PINK1/Parkin-Dependent Mitophagy via Its Kinase Activity: Pathologic Insights into Parkinson’s Disease. Hum. Mol. Genet. 2019, 28, 1645–1660. [Google Scholar] [CrossRef]
- Yang, M.; Li, C.; Yang, S.; Xiao, Y.; Xiong, X.; Chen, W.; Zhao, H.; Zhang, Q.; Han, Y.; Sun, L. Mitochondria-Associated ER Membranes—The Origin Site of Autophagy. Front. Cell Dev. Biol. 2020, 8, 595. [Google Scholar] [CrossRef]
- Wong, E.; Cuervo, A.M. Autophagy Gone Awry in Neurodegenerative Diseases. Nat. Neurosci. 2010, 13, 805–811. [Google Scholar] [CrossRef] [PubMed]
- Surmeier, D.J.; Nguyen, J.T.; Lancki, N.; Venuto, C.S.; Oakes, D.; Simuni, T.; Wyse, R.K. Re-Analysis of the STEADY-PD II Trial-Evidence for Slowing the Progression of Parkinson’s Disease. Mov. Disord. 2022, 37, 334–342. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Verma, P.; Balaji, G.; Samantaray, S.; Mohanakumar, K.P. Nimodipine, an L-Type Calcium Channel Blocker Attenuates Mitochondrial Dysfunctions to Protect against 1-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine-Induced Parkinsonism in Mice. Neurochem. Int. 2016, 99, 221–232. [Google Scholar] [CrossRef] [PubMed]
- Guzman, J.N.; Ilijic, E.; Yang, B.; Sanchez-Padilla, J.; Wokosin, D.; Galtieri, D.; Kondapalli, J.; Schumacker, P.T.; Surmeier, D.J. Systemic Isradipine Treatment Diminishes Calcium-Dependent Mitochondrial Oxidant Stress. J. Clin. Invest. 2018, 128, 2266–2280. [Google Scholar] [CrossRef] [PubMed]
- Costantini, P.; Petronilli, V.; Colonna, R.; Bernardi, P. On the Effects of Paraquat on Isolated Mitochondria. Evidence That Paraquat Causes Opening of the Cyclosporin A-Sensitive Permeability Transition Pore Synergistically with Nitric Oxide. Toxicology 1995, 99, 77–88. [Google Scholar] [CrossRef]
- Seaton, T.A.; Cooper, J.M.; Schapira, A.H. Cyclosporin Inhibition of Apoptosis Induced by Mitochondrial Complex I Toxins. Brain Res. 1998, 809, 12–17. [Google Scholar] [CrossRef]
- Thomas, B.; Banerjee, R.; Starkova, N.N.; Zhang, S.F.; Calingasan, N.Y.; Yang, L.; Wille, E.; Lorenzo, B.J.; Ho, D.J.; Beal, M.F.; et al. Mitochondrial Permeability Transition Pore Component Cyclophilin D Distinguishes Nigrostriatal Dopaminergic Death Paradigms in the MPTP Mouse Model of Parkinson’s Disease. Antioxid. Redox Signal. 2012, 16, 855–868. [Google Scholar] [CrossRef]
- Martin, L.J.; Semenkow, S.; Hanaford, A.; Wong, M. The Mitochondrial Permeability Transition Pore Regulates Parkinson’s Disease Development in Mutant α-Synuclein Transgenic Mice. Neurobiol. Aging 2014, 35, 1132–1152. [Google Scholar] [CrossRef]
- Sison, S.L.; Ebert, A.D. Decreased NAD+ in Dopaminergic Neurons. Aging 2018, 10, 526–527. [Google Scholar] [CrossRef]
- Murata, H.; Khine, C.C.; Nishikawa, A.; Yamamoto, K.-I.; Kinoshita, R.; Sakaguchi, M. C-Jun N-Terminal Kinase (JNK)-Mediated Phosphorylation of SARM1 Regulates NAD+ Cleavage Activity to Inhibit Mitochondrial Respiration. J. Biol. Chem. 2018, 293, 18933–18943. [Google Scholar] [CrossRef]
- Shi, H.; Deng, H.-X.; Gius, D.; Schumacker, P.T.; Surmeier, D.J.; Ma, Y.-C. Sirt3 Protects Dopaminergic Neurons from Mitochondrial Oxidative Stress. Hum. Mol. Genet. 2017, 26, 1915–1926. [Google Scholar] [CrossRef] [PubMed]
- Torpey, J.; Madine, J.; Wood, A.; Lian, L.-Y. Cyclophilin D Binds to the Acidic C-Terminus Region of α-Synuclein and Affects Its Aggregation Characteristics. Sci. Rep. 2020, 10, 10159. [Google Scholar] [CrossRef] [PubMed]
- Grassi, D.; Howard, S.; Zhou, M.; Diaz-Perez, N.; Urban, N.T.; Guerrero-Given, D.; Kamasawa, N.; Volpicelli-Daley, L.A.; LoGrasso, P.; Lasmézas, C.I. Identification of a Highly Neurotoxic α-Synuclein Species Inducing Mitochondrial Damage and Mitophagy in Parkinson’s Disease. Proc. Natl. Acad. Sci. USA 2018, 115, E2634–E2643. [Google Scholar] [CrossRef] [PubMed]
- Ludtmann, M.H.R.; Angelova, P.R.; Ninkina, N.N.; Gandhi, S.; Buchman, V.L.; Abramov, A.Y. Monomeric Alpha-Synuclein Exerts a Physiological Role on Brain ATP Synthase. J. Neurosci. 2016, 36, 10510–10521. [Google Scholar] [CrossRef] [PubMed]
- Barsukova, A.G.; Bourdette, D.; Forte, M. Mitochondrial Calcium and Its Regulation in Neurodegeneration Induced by Oxidative Stress: Neuronal Calcium during Oxidative Stress. Eur. J. Neurosci. 2011, 34, 437–447. [Google Scholar] [CrossRef]
- Qadri, R.; Namdeo, M.; Behari, M.; Goyal, V.; Sharma, S.; Mukhopadhyay, A.K. Alterations in Mitochondrial Membrane Potential in Peripheral Blood Mononuclear Cells in Parkinson’s Disease: Potential for a Novel Biomarker. Restor. Neurol. Neurosci. 2018, 36, 719–727. [Google Scholar] [CrossRef]
- Stocco, A.; Smolina, N.; Sabatelli, P.; Šileikytė, J.; Artusi, E.; Mouly, V.; Cohen, M.; Forte, M.; Schiavone, M.; Bernardi, P. Treatment with a Triazole Inhibitor of the Mitochondrial Permeability Transition Pore Fully Corrects the Pathology of Sapje Zebrafish Lacking Dystrophin. Pharmacol. Res. 2021, 165, 105421. [Google Scholar] [CrossRef]
- Polimeni, G.; Esposito, E.; Bevelacqua, V.; Guarneri, C.; Cuzzocrea, S. Role of Melatonin Supplementation in Neurodegenerative Disorders. Front. Biosci. 2014, 19, 429–446. [Google Scholar] [CrossRef]
- Jou, M.-J. Melatonin Preserves the Transient Mitochondrial Permeability Transition for Protection during Mitochondrial Ca2+ Stress in Astrocyte. J. Pineal Res. 2011, 50, 427–435. [Google Scholar] [CrossRef]
- Andrabi, S.A.; Sayeed, I.; Siemen, D.; Wolf, G.; Horn, T.F.W. Direct Inhibition of the Mitochondrial Permeability Transition Pore: A Possible Mechanism Responsible for Anti-Apoptotic Effects of Melatonin. FASEB J. 2004, 18, 869–871. [Google Scholar] [CrossRef]
- Reiter, R.J.; Tan, D.X.; Rosales-Corral, S.; Galano, A.; Jou, M.-J.; Acuna-Castroviejo, D. Melatonin Mitigates Mitochondrial Meltdown: Interactions with SIRT3. Int. J. Mol. Sci. 2018, 19, 2439. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Herrmann, C.J.; Simonovic, M.; Szklarczyk, D.; von Mering, C. Version 4.0 of PaxDb: Protein Abundance Data, Integrated across Model Organisms, Tissues, and Cell-Lines. Proteomics 2015, 15, 3163–3168. [Google Scholar] [CrossRef] [PubMed]
- Rosenwirth, B.; Billich, A.; Datema, R.; Donatsch, P.; Hammerschmid, F.; Harrison, R.; Hiestand, P.; Jaksche, H.; Mayer, P.; Peichl, P. Inhibition of Human Immunodeficiency Virus Type 1 Replication by SDZ NIM 811, a Nonimmunosuppressive Cyclosporine Analog. Antimicrob. Agents Chemother. 1994, 38, 1763–1772. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Boerner, J.E.; TiongYip, C.; Weidmann, B.; Ryder, N.S.; Cooreman, M.P.; Lin, K. NIM811, a Cyclophilin Inhibitor, Exhibits Potent In Vitro Activity against Hepatitis C Virus Alone or in Combination with Alpha Interferon. Antimicrob. Agents Chemother. 2006, 50, 2976–2982. [Google Scholar] [CrossRef]
- Ptak, R.G.; Gallay, P.A.; Jochmans, D.; Halestrap, A.P.; Ruegg, U.T.; Pallansch, L.A.; Bobardt, M.D.; de Béthune, M.-P.; Neyts, J.; De Clercq, E.; et al. Inhibition of Human Immunodeficiency Virus Type 1 Replication in Human Cells by Debio-025, a Novel Cyclophilin Binding Agent. Antimicrob. Agents Chemother. 2008, 52, 1302–1317. [Google Scholar] [CrossRef]
- Flisiak, R.; Horban, A.; Gallay, P.; Bobardt, M.; Selvarajah, S.; Wiercinska-Drapalo, A.; Siwak, E.; Cielniak, I.; Higersberger, J.; Kierkus, J.; et al. The Cyclophilin Inhibitor Debio-025 Shows Potent Anti-Hepatitis C Effect in Patients Coinfected with Hepatitis C and Human Immunodeficiency Virus. Hepatology 2008, 47, 817–826. [Google Scholar] [CrossRef]
- Cung, T.-T.; Morel, O.; Cayla, G.; Rioufol, G.; Garcia-Dorado, D.; Angoulvant, D.; Bonnefoy-Cudraz, E.; Guérin, P.; Elbaz, M.; Delarche, N.; et al. Cyclosporine before PCI in Patients with Acute Myocardial Infarction. N. Engl. J. Med. 2015, 373, 1021–1031. [Google Scholar] [CrossRef]
- Javadov, S.; Jang, S.; Parodi-Rullán, R.; Khuchua, Z.; Kuznetsov, A.V. Mitochondrial Permeability Transition in Cardiac Ischemia-Reperfusion: Whether Cyclophilin D Is a Viable Target for Cardioprotection? Cell Mol. Life Sci. 2017, 74, 2795–2813. [Google Scholar] [CrossRef]
- Haleckova, A.; Benek, O.; Zemanová, L.; Dolezal, R.; Musilek, K. Small-Molecule Inhibitors of Cyclophilin D as Potential Therapeutics in Mitochondria-Related Diseases. Med. Res. Rev. 2022, 42, 1822–1855. [Google Scholar] [CrossRef]
- Panel, M.; Ruiz, I.; Brillet, R.; Lafdil, F.; Teixeira-Clerc, F.; Nguyen, C.T.; Calderaro, J.; Gelin, M.; Allemand, F.; Guichou, J.-F.; et al. Small-Molecule Inhibitors of Cyclophilins Block Opening of the Mitochondrial Permeability Transition Pore and Protect Mice from Hepatic Ischemia/Reperfusion Injury. Gastroenterology 2019, 157, 1368–1382. [Google Scholar] [CrossRef]
- Panel, M.; Ahmed-Belkacem, A.; Ruiz, I.; Guichou, J.-F.; Pawlotsky, J.-M.; Ghaleh, B.; Morin, D. A Phenyl-Pyrrolidine Derivative Reveals a Dual Inhibition Mechanism of Myocardial Mitochondrial Permeability Transition Pore, Which Is Limited by Its Myocardial Distribution. J. Pharmacol. Exp. Ther. 2021, 376, 348–357. [Google Scholar] [CrossRef] [PubMed]
- Waldmeier, P.C.; Zimmermann, K.; Qian, T.; Tintelnot-Blomley, M.; Lemasters, J.J. Cyclophilin D as a Drug Target. Curr. Med. Chem. 2003, 10, 1485–1506. [Google Scholar] [CrossRef] [PubMed]
- Valasani, K.R.; Sun, Q.; Fang, D.; Zhang, Z.; Yu, Q.; Guo, Y.; Li, J.; Roy, A.; ShiDu Yan, S. Identification of a Small Molecule Cyclophilin D Inhibitor for Rescuing Aβ-Mediated Mitochondrial Dysfunction. ACS Med. Chem. Lett. 2016, 7, 294–299. [Google Scholar] [CrossRef]
- Agrawal, M.; Ajazuddin; Tripathi, D.K.; Saraf, S.; Saraf, S.; Antimisiaris, S.G.; Mourtas, S.; Hammarlund-Udenaes, M.; Alexander, A. Recent Advancements in Liposomes Targeting Strategies to Cross Blood-Brain Barrier (BBB) for the Treatment of Alzheimer’s Disease. J. Control. Release 2017, 260, 61–77. [Google Scholar] [CrossRef] [PubMed]
- Rinaldi, C.; Wood, M.J.A. Antisense Oligonucleotides: The next Frontier for Treatment of Neurological Disorders. Nat. Rev. Neurol. 2018, 14, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Min, H.S.; Kim, H.J.; Naito, M.; Ogura, S.; Toh, K.; Hayashi, K.; Kim, B.S.; Fukushima, S.; Anraku, Y.; Miyata, K.; et al. Systemic Brain Delivery of Antisense Oligonucleotides across the Blood-Brain Barrier with a Glucose-Coated Polymeric Nanocarrier. Angew. Chem. Int. Ed. 2020, 59, 8173–8180. [Google Scholar] [CrossRef]



| Mature CyPD | Immature CyPD (with MTS) | Crystal/CyPA |
|---|---|---|
| i | i + 29 | i − 13 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Coluccino, G.; Muraca, V.P.; Corazza, A.; Lippe, G. Cyclophilin D in Mitochondrial Dysfunction: A Key Player in Neurodegeneration? Biomolecules 2023, 13, 1265. https://doi.org/10.3390/biom13081265
Coluccino G, Muraca VP, Corazza A, Lippe G. Cyclophilin D in Mitochondrial Dysfunction: A Key Player in Neurodegeneration? Biomolecules. 2023; 13(8):1265. https://doi.org/10.3390/biom13081265
Chicago/Turabian StyleColuccino, Gabriele, Valentina Pia Muraca, Alessandra Corazza, and Giovanna Lippe. 2023. "Cyclophilin D in Mitochondrial Dysfunction: A Key Player in Neurodegeneration?" Biomolecules 13, no. 8: 1265. https://doi.org/10.3390/biom13081265