
Circulating H3K27 Methylated Nucleosome Plasma Concentration: Synergistic Information with Circulating Tumor DNA Molecular Profiling
 , , , and
, , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Population
2.2. CfDNA Collection
2.3. Library Preparation for DNA Sequencing
2.4. Nu.Q® ImmunoAssays
2.5. Tissue Microarray (TMA)
2.6. Statistics
2.7. Study Limitations
3. Results
3.1. High Levels of Circulating H3K27Me3 Nucleosomes Are Observed in NSCLC Samples at Diagnosis
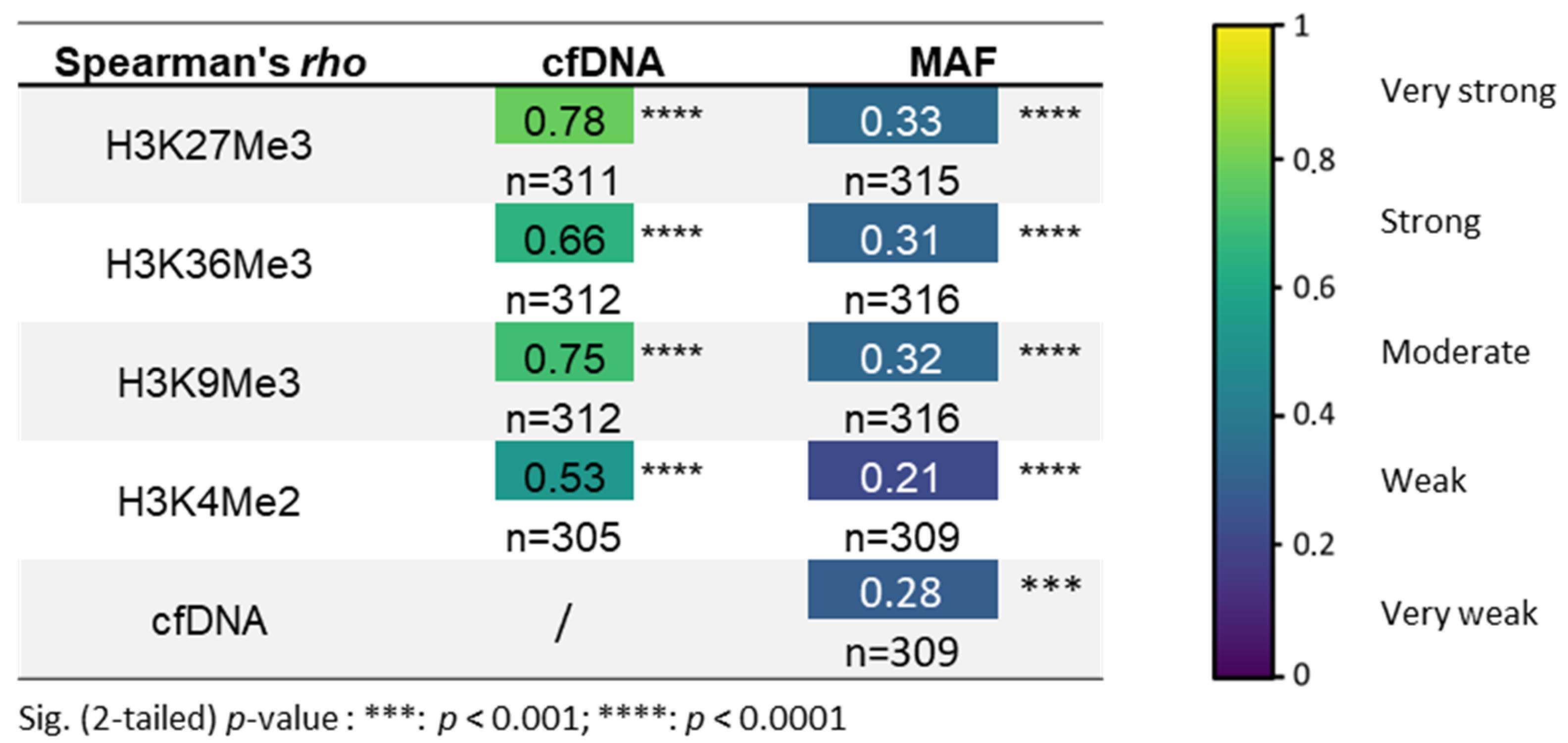
3.2. H3K27Me3-Nucleosome Levels Correlate with cfDNA Quantity and Percentage of Mutational Allele Fraction (MAF) at Diagnosis
3.3. H3K27Me3 Expression Level in Normal and Adenocarcinoma Tissue
3.4. Relevant Combined Information from the Circulating H3K27Me3-Nucleosome Levels and ctDNA Molecular Profile at Diagnosis
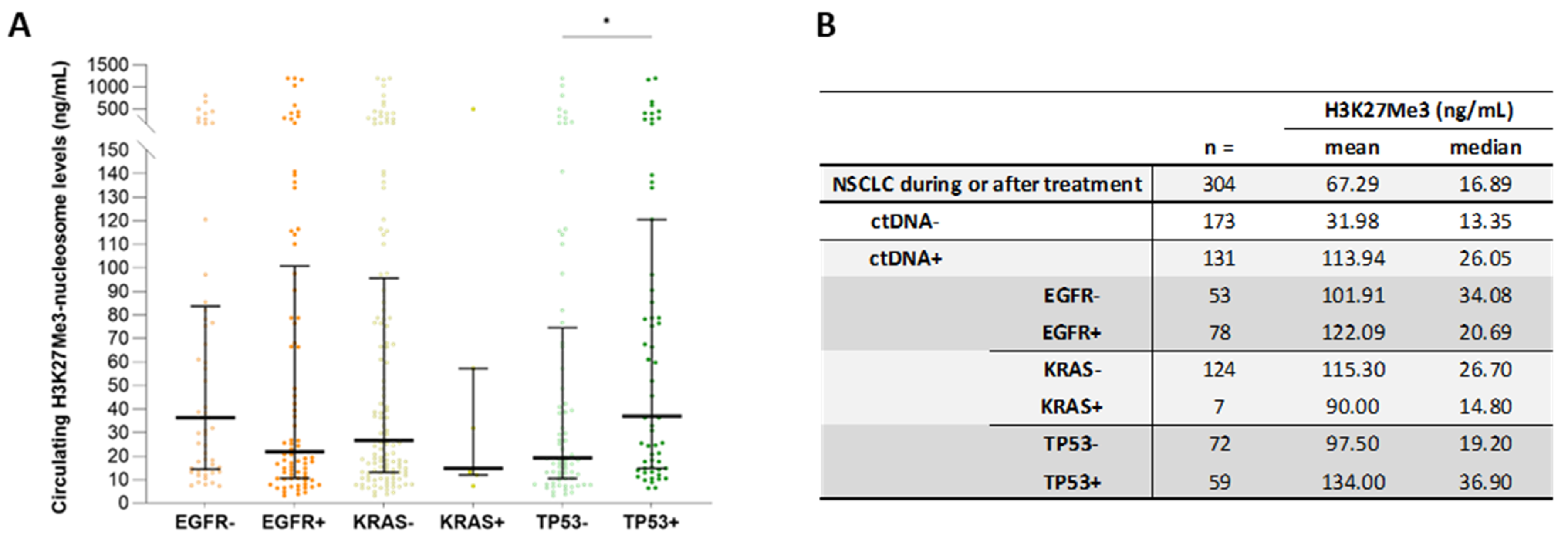
3.5. High Level of Circulating H3K27Me3 Nucleosomes Observed in NSCLC Samples during Treatment Is More Pronounced in the Presence of Mutated ctDNA
3.6. Additive Value of Both Biomarkers: H3K27Me3 Nucleosomes and ctDNA
3.7. High Circulating H3K27Me3-Nucleosome Levels Are Preferentially Associated with TP53 Mutations during Treatment
4. Discussion
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fan, H.C.; Blumenfeld, Y.J.; Chitkara, U.; Hudgins, L.; Quake, S.R. Noninvasive diagnosis of fetal aneuploidy by shotgun sequencing DNA from maternal blood. Proc. Natl. Acad. Sci. USA 2008, 105, 16266–16271. [Google Scholar] [CrossRef] [PubMed]
- Grabuschnig, S.; Bronkhorst, A.J.; Holdenrieder, S.; Rosales Rodriguez, I.; Schliep, K.P.; Schwendenwein, D.; Ungerer, V.; Sensen, C.W. Putative Origins of Cell-Free DNA in Humans: A Review of Active and Passive Nucleic Acid Release Mechanisms. Int. J. Mol. Sci. 2020, 21, 8062. [Google Scholar] [CrossRef] [PubMed]
- Bronkhorst, A.J.; Ungerer, V.; Holdenrieder, S. The emerging role of cell-free DNA as a molecular marker for cancer management. Biomol. Detect. Quantif. 2019, 17, 100087. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, C.; Roch, B.; Mazard, T.; Blache, P.; Dache, Z.A.A.; Pastor, B.; Pisareva, E.; Tanos, R.; Thierry, A.R. Circulating nuclear DNA structural features, origins, and complete size profile revealed by fragmentomics. JCI Insight 2021, 6, e144561. [Google Scholar] [CrossRef]
- Thierry, A.R.; El Messaoudi, S.; Gahan, P.B.; Anker, P.; Stroun, M. Origins, structures, and functions of circulating DNA in oncology. Cancer Metastasis Rev. 2016, 35, 347–376. [Google Scholar] [CrossRef]
- Zhao, Z.; Shilatifard, A. Epigenetic modifications of histones in cancer. Genome Biol. 2019, 20, 245. [Google Scholar] [CrossRef]
- Tamkovich, S.N.; Cherepanova, A.V.; Kolesnikova, E.V.; Rykova, E.Y.; Pyshnyi, D.V.; Vlassov, V.V.; Laktionov, P.P. Circulating DNA and DNase activity in human blood. Ann. N. Y. Acad. Sci. 2006, 1075, 191–196. [Google Scholar] [CrossRef]
- McAnena, P.; Brown, J.A.; Kerin, M.J. Circulating Nucleosomes and Nucleosome Modifications as Biomarkers in Cancer. Cancers 2017, 9, 5. [Google Scholar] [CrossRef]
- Licht, J.D.; Bennett, R.L. Leveraging epigenetics to enhance the efficacy of immunotherapy. Clin. Epigenet. 2021, 13, 115. [Google Scholar] [CrossRef]
- Fenley, A.T.; Anandakrishnan, R.; Kidane, Y.H.; Onufriev, A.V. Modulation of nucleosomal DNA accessibility via charge-altering post-translational modifications in histone core. Epigenet. Chromatin 2018, 11, 11. [Google Scholar] [CrossRef]
- Greer, E.L.; Shi, Y. Histone methylation: A dynamic mark in health, disease and inheritance. Nat. Rev. Genet. 2012, 13, 343–357. [Google Scholar] [CrossRef] [PubMed]
- Varambally, S.; Dhanasekaran, S.M.; Zhou, M.; Barrette, T.R.; Kumar-Sinha, C.; Sanda, M.G.; Ghosh, D.; Pienta, K.J.; Sewalt, R.G.; Otte, A.P.; et al. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature 2002, 419, 624–629. [Google Scholar] [CrossRef] [PubMed]
- Comet, I.; Riising, E.M.; Leblanc, B.; Helin, K. Maintaining cell identity: PRC2-mediated regulation of transcription and cancer. Nat. Rev. Cancer 2016, 16, 803–810. [Google Scholar] [CrossRef] [PubMed]
- Kleer, C.G.; Cao, Q.; Varambally, S.; Shen, R.; Ota, I.; Tomlins, S.A.; Ghosh, D.; Sewalt, R.G.; Otte, A.P.; Hayes, D.F.; et al. EZH2 is a marker of aggressive breast cancer and promotes neoplastic transformation of breast epithelial cells. Proc. Natl. Acad. Sci. USA 2003, 100, 11606–11611. [Google Scholar] [CrossRef]
- Wiles, E.T.; Selker, E.U. H3K27 methylation: A promiscuous repressive chromatin mark. Curr. Opin. Genet. Dev. 2017, 43, 31–37. [Google Scholar] [CrossRef]
- Wan, L.; Li, X.; Shen, H.; Bai, X. Quantitative analysis of EZH2 expression and its correlations with lung cancer patients’ clinical pathological characteristics. Clin. Transl. Oncol. 2013, 15, 132–138. [Google Scholar] [CrossRef]
- Sato, T.; Kaneda, A.; Tsuji, S.; Isagawa, T.; Yamamoto, S.; Fujita, T.; Yamanaka, R.; Tanaka, Y.; Nukiwa, T.; Marquez, V.E.; et al. PRC2 overexpression and PRC2-target gene repression relating to poorer prognosis in small cell lung cancer. Sci. Rep. 2013, 3, 1911. [Google Scholar] [CrossRef]
- Cardenas, H.; Zhao, J.; Vieth, E.; Nephew, K.P.; Matei, D. EZH2 inhibition promotes epithelial-to-mesenchymal transition in ovarian cancer cells. Oncotarget 2016, 7, 84453–84467. [Google Scholar] [CrossRef]
- Ma, J.; Zhang, J.; Weng, Y.C.; Wang, J.C. EZH2-Mediated microRNA-139-5p Regulates Epithelial-Mesenchymal Transition and Lymph Node Metastasis of Pancreatic Cancer. Mol. Cells 2018, 41, 868–880. [Google Scholar] [CrossRef]
- Lachat, C.; Bruyere, D.; Etcheverry, A.; Aubry, M.; Mosser, J.; Warda, W.; Herfs, M.; Hendrick, E.; Ferrand, C.; Borg, C.; et al. EZH2 and KDM6B Expressions Are Associated with Specific Epigenetic Signatures during EMT in Non Small Cell Lung Carcinomas. Cancers 2020, 12, 3649. [Google Scholar] [CrossRef]
- Lu, C.; Han, H.D.; Mangala, L.S.; Ali-Fehmi, R.; Newton, C.S.; Ozbun, L.; Armaiz-Pena, G.N.; Hu, W.; Stone, R.L.; Munkarah, A.; et al. Regulation of tumor angiogenesis by EZH2. Cancer Cell 2010, 18, 185–197. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Lu, X.; Zhen, F.; Jin, S.; Yu, T.; Zhu, Q.; Wang, W.; Xu, K.; Yao, J.; Guo, R. LINC00665 Induces Acquired Resistance to Gefitinib through Recruiting EZH2 and Activating PI3K/AKT Pathway in NSCLC. Mol. Ther. Nucleic Acids 2019, 16, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Passaro, A.; Janne, P.A.; Mok, T.; Peters, S. Overcoming therapy resistance in EGFR-mutant lung cancer. Nat. Cancer 2021, 2, 377–391. [Google Scholar] [CrossRef]
- Gong, H.; Li, Y.; Yuan, Y.; Li, W.; Zhang, H.; Zhang, Z.; Shi, R.; Liu, M.; Liu, C.; Chen, C.; et al. EZH2 inhibitors reverse resistance to gefitinib in primary EGFR wild-type lung cancer cells. BMC Cancer 2020, 20, 1189. [Google Scholar] [CrossRef] [PubMed]
- Duan, R.; Du, W.; Guo, W. EZH2: A novel target for cancer treatment. J. Hematol. Oncol. 2020, 13, 104. [Google Scholar] [CrossRef]
- Garcia, J.; Dusserre, E.; Cheynet, V.; Bringuier, P.P.; Brengle-Pesce, K.; Wozny, A.S.; Rodriguez-Lafrasse, C.; Freyer, G.; Brevet, M.; Payen, L.; et al. Evaluation of pre-analytical conditions and comparison of the performance of several digital PCR assays for the detection of major EGFR mutations in circulating DNA from non-small cell lung cancers: The CIRCAN_0 study. Oncotarget 2017, 8, 87980–87996. [Google Scholar] [CrossRef]
- Bieler, J.; Pozzorini, C.; Garcia, J.; Tuck, A.C.; Macheret, M.; Willig, A.; Couraud, S.; Xing, X.; Menu, P.; Steinmetz, L.M.; et al. High-Throughput Nucleotide Resolution Predictions of Assay Limitations Increase the Reliability and Concordance of Clinical Tests. JCO Clin. Cancer Inform. 2021, 5, 1085–1095. [Google Scholar] [CrossRef]
- Garcia, J.; Gauthier, A.; Lescuyer, G.; Barthelemy, D.; Geiguer, F.; Balandier, J.; Edelstein, D.L.; Jones, F.S.; Holtrup, F.; Duruisseau, M.; et al. Routine Molecular Screening of Patients with Advanced Non-SmallCell Lung Cancer in Circulating Cell-Free DNA at Diagnosis and During Progression Using OncoBEAM(TM) EGFR V2 and NGS Technologies. Mol. Diagn. Ther. 2021, 25, 239–250. [Google Scholar] [CrossRef]
- Garcia, J.; Kamps-Hughes, N.; Geiguer, F.; Couraud, S.; Sarver, B.; Payen, L.; Ionescu-Zanetti, C. Sensitivity, specificity, and accuracy of a liquid biopsy approach utilizing molecular amplification pools. Sci. Rep. 2021, 11, 10761. [Google Scholar] [CrossRef]
- Pennell, N.A.; Arcila, M.E.; Gandara, D.R.; West, H. Biomarker Testing for Patients With Advanced Non-Small Cell Lung Cancer: Real-World Issues and Tough Choices. Am. Soc. Clin. Oncol. Educ. Book 2019, 39, 531–542. [Google Scholar] [CrossRef]
- Leighl, N.B.; Page, R.D.; Raymond, V.M.; Daniel, D.B.; Divers, S.G.; Reckamp, K.L.; Villalona-Calero, M.A.; Dix, D.; Odegaard, J.I.; Lanman, R.B.; et al. Clinical Utility of Comprehensive Cell-free DNA Analysis to Identify Genomic Biomarkers in Patients with Newly Diagnosed Metastatic Non-small Cell Lung Cancer. Clin. Cancer Res. 2019, 25, 4691–4700. [Google Scholar] [CrossRef] [PubMed]
- Mack, P.C.; Banks, K.C.; Espenschied, C.R.; Burich, R.A.; Zill, O.A.; Lee, C.E.; Riess, J.W.; Mortimer, S.A.; Talasaz, A.; Lanman, R.B.; et al. Spectrum of driver mutations and clinical impact of circulating tumor DNA analysis in non-small cell lung cancer: Analysis of over 8000 cases. Cancer 2020, 126, 3219–3228. [Google Scholar] [CrossRef] [PubMed]
- Cui, W.; Milner-Watts, C.; McVeigh, T.P.; Minchom, A.; Bholse, J.; Davidson, M.; Yousaf, N.; MacMahon, S.; Mugalaasi, H.; Gunapala, R.; et al. A pilot of Blood-First diagnostic cell free DNA (cfDNA) next generation sequencing (NGS) in patients with suspected advanced lung cancer. Lung Cancer 2022, 165, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Kuang, P.P.; Li, N.; Liu, Z.; Sun, T.Y.; Wang, S.Q.; Hu, J.; Ou, W.; Wang, S.Y. Circulating Tumor DNA Analyses as a Potential Marker of Recurrence and Effectiveness of Adjuvant Chemotherapy for Resected Non-Small-Cell Lung Cancer. Front. Oncol. 2020, 10, 595650. [Google Scholar] [CrossRef]
- Nagasaka, M.; Uddin, M.H.; Al-Hallak, M.N.; Rahman, S.; Balasubramanian, S.; Sukari, A.; Azmi, A.S. Liquid biopsy for therapy monitoring in early-stage non-small cell lung cancer. Mol. Cancer 2021, 20, 82. [Google Scholar] [CrossRef]
- Markou, A.N.; Londra, D.; Stergiopoulou, D.; Vamvakaris, I.; Potaris, K.; Pateras, I.S.; Kotsakis, A.; Georgoulias, V.; Lianidou, E. Preoperative Mutational Analysis of Circulating Tumor Cells (CTCs) and Plasma-cfDNA Provides Complementary Information for Early Prediction of Relapse: A Pilot Study in Early-Stage Non-Small Cell Lung Cancer. Cancers 2023, 15, 1877. [Google Scholar] [CrossRef]
- Chan, H.T.; Chin, Y.M.; Low, S.K. Circulating Tumor DNA-Based Genomic Profiling Assays in Adult Solid Tumors for Precision Oncology: Recent Advancements and Future Challenges. Cancers 2022, 14, 3275. [Google Scholar] [CrossRef]
- Gale, D.; Heider, K.; Ruiz-Valdepenas, A.; Hackinger, S.; Perry, M.; Marsico, G.; Rundell, V.; Wulff, J.; Sharma, G.; Knock, H.; et al. Residual ctDNA after treatment predicts early relapse in patients with early-stage non-small cell lung cancer. Ann. Oncol. 2022, 33, 500–510. [Google Scholar] [CrossRef]
- Chaudhuri, A.A.; Chabon, J.J.; Lovejoy, A.F.; Newman, A.M.; Stehr, H.; Azad, T.D.; Khodadoust, M.S.; Esfahani, M.S.; Liu, C.L.; Zhou, L.; et al. Early Detection of Molecular Residual Disease in Localized Lung Cancer by Circulating Tumor DNA Profiling. Cancer Discov. 2017, 7, 1394–1403. [Google Scholar] [CrossRef]
- Gong, Y.; Wei, C.; Cheng, L.; Ma, F.; Lu, S.; Peng, Q.; Liu, L.; Wang, Y. Tracking the Dynamic Histone Methylation of H3K27 in Live Cancer Cells. ACS Sens. 2021, 6, 4369–4378. [Google Scholar] [CrossRef]
- Donaldson-Collier, M.C.; Sungalee, S.; Zufferey, M.; Tavernari, D.; Katanayeva, N.; Battistello, E.; Mina, M.; Douglass, K.M.; Rey, T.; Raynaud, F.; et al. EZH2 oncogenic mutations drive epigenetic, transcriptional, and structural changes within chromatin domains. Nat. Genet. 2019, 51, 517–528. [Google Scholar] [CrossRef] [PubMed]
- Cai, M.Y.; Tong, Z.T.; Zhu, W.; Wen, Z.Z.; Rao, H.L.; Kong, L.L.; Guan, X.Y.; Kung, H.F.; Zeng, Y.X.; Xie, D. H3K27me3 protein is a promising predictive biomarker of patients’ survival and chemoradioresistance in human nasopharyngeal carcinoma. Mol. Med. 2011, 17, 1137–1145. [Google Scholar] [CrossRef] [PubMed]
- Haragan, A.; Field, J.K.; Davies, M.P.A.; Escriu, C.; Gruver, A.; Gosney, J.R. Heterogeneity of PD-L1 expression in non-small cell lung cancer: Implications for specimen sampling in predicting treatment response. Lung Cancer 2019, 134, 79–84. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.C.; Chien, M.N.; Chang, Y.C.; Lee, J.J.; Dai, S.H.; Cheng, S.P. Overexpression of Histone H3 Lysine 27 Trimethylation Is Associated with Aggressiveness and Dedifferentiation of Thyroid Cancer. Endocr. Pathol. 2019, 30, 305–311. [Google Scholar] [CrossRef]
- Hoffmann, F.; Niebel, D.; Aymans, P.; Ferring-Schmitt, S.; Dietrich, D.; Landsberg, J. H3K27me3 and EZH2 expression in melanoma: Relevance for melanoma progression and response to immune checkpoint blockade. Clin. Epigenet. 2020, 12, 24. [Google Scholar] [CrossRef] [PubMed]
- Holzel, M.; Bovier, A.; Tuting, T. Plasticity of tumour and immune cells: A source of heterogeneity and a cause for therapy resistance? Nat. Rev. Cancer 2013, 13, 365–376. [Google Scholar] [CrossRef]
- Yuan, S.; Norgard, R.J.; Stanger, B.Z. Cellular Plasticity in Cancer. Cancer Discov. 2019, 9, 837–851. [Google Scholar] [CrossRef]
- Li, J.; Stanger, B.Z. How Tumor Cell Dedifferentiation Drives Immune Evasion and Resistance to Immunotherapy. Cancer Res. 2020, 80, 4037–4041. [Google Scholar] [CrossRef]
- Holdenrieder, S.; Nagel, D.; Schalhorn, A.; Heinemann, V.; Wilkowski, R.; von Pawel, J.; Raith, H.; Feldmann, K.; Kremer, A.E.; Muller, S.; et al. Clinical relevance of circulating nucleosomes in cancer. Ann. N. Y. Acad. Sci. 2008, 1137, 180–189. [Google Scholar] [CrossRef]
- Schwarzenbach, H.; Hoon, D.S.; Pantel, K. Cell-free nucleic acids as biomarkers in cancer patients. Nat. Rev. Cancer 2011, 11, 426–437. [Google Scholar] [CrossRef]
- Holdenrieder, S.; Stieber, P.; von Pawel, J.; Raith, H.; Nagel, D.; Feldmann, K.; Seidel, D. Circulating nucleosomes predict the response to chemotherapy in patients with advanced non-small cell lung cancer. Clin. Cancer Res. 2004, 10, 5981–5987. [Google Scholar] [CrossRef] [PubMed]
- Tsoneva, D.K.; Ivanov, M.N.; Conev, N.V.; Manev, R.; Stoyanov, D.S.; Vinciguerra, M. Circulating Histones to Detect and Monitor the Progression of Cancer. Int. J. Mol. Sci. 2023, 24, 942. [Google Scholar] [CrossRef] [PubMed]
- Barlesi, F.; Mazieres, J.; Merlio, J.P.; Debieuvre, D.; Mosser, J.; Lena, H.; Ouafik, L.; Besse, B.; Rouquette, I.; Westeel, V.; et al. Routine molecular profiling of patients with advanced non-small-cell lung cancer: Results of a 1-year nationwide programme of the French Cooperative Thoracic Intergroup (IFCT). Lancet 2016, 387, 1415–1426. [Google Scholar] [CrossRef]
- Mitsudomi, T.; Hamajima, N.; Ogawa, M.; Takahashi, T. Prognostic significance of p53 alterations in patients with non-small cell lung cancer: A meta-analysis. Clin. Cancer Res. 2000, 6, 4055–4063. [Google Scholar] [PubMed]
- Gu, J.; Zhou, Y.; Huang, L.; Ou, W.; Wu, J.; Li, S.; Xu, J.; Feng, J.; Liu, B. TP53 mutation is associated with a poor clinical outcome for non-small cell lung cancer: Evidence from a meta-analysis. Mol. Clin. Oncol. 2016, 5, 705–713. [Google Scholar] [CrossRef]
- Guo, Y.; Song, J.; Wang, Y.; Huang, L.; Sun, L.; Zhao, J.; Zhang, S.; Jing, W.; Ma, J.; Han, C. Concurrent Genetic Alterations and Other Biomarkers Predict Treatment Efficacy of EGFR-TKIs in EGFR-Mutant Non-Small Cell Lung Cancer: A Review. Front. Oncol. 2020, 10, 610923. [Google Scholar] [CrossRef]
- Assoun, S.; Theou-Anton, N.; Nguenang, M.; Cazes, A.; Danel, C.; Abbar, B.; Pluvy, J.; Gounant, V.; Khalil, A.; Namour, C.; et al. Association of TP53 mutations with response and longer survival under immune checkpoint inhibitors in advanced non-small-cell lung cancer. Lung Cancer 2019, 132, 65–71. [Google Scholar] [CrossRef]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grolleau, E.; Candiracci, J.; Lescuyer, G.; Barthelemy, D.; Benzerdjeb, N.; Haon, C.; Geiguer, F.; Raffin, M.; Hardat, N.; Balandier, J.; et al. Circulating H3K27 Methylated Nucleosome Plasma Concentration: Synergistic Information with Circulating Tumor DNA Molecular Profiling. Biomolecules 2023, 13, 1255. https://doi.org/10.3390/biom13081255
Grolleau E, Candiracci J, Lescuyer G, Barthelemy D, Benzerdjeb N, Haon C, Geiguer F, Raffin M, Hardat N, Balandier J, et al. Circulating H3K27 Methylated Nucleosome Plasma Concentration: Synergistic Information with Circulating Tumor DNA Molecular Profiling. Biomolecules. 2023; 13(8):1255. https://doi.org/10.3390/biom13081255
Chicago/Turabian StyleGrolleau, Emmanuel, Julie Candiracci, Gaelle Lescuyer, David Barthelemy, Nazim Benzerdjeb, Christine Haon, Florence Geiguer, Margaux Raffin, Nathalie Hardat, Julie Balandier, and et al. 2023. "Circulating H3K27 Methylated Nucleosome Plasma Concentration: Synergistic Information with Circulating Tumor DNA Molecular Profiling" Biomolecules 13, no. 8: 1255. https://doi.org/10.3390/biom13081255