
Membrane Transporters Involved in Iron Trafficking: Physiological and Pathological Aspects

 , , and
, , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
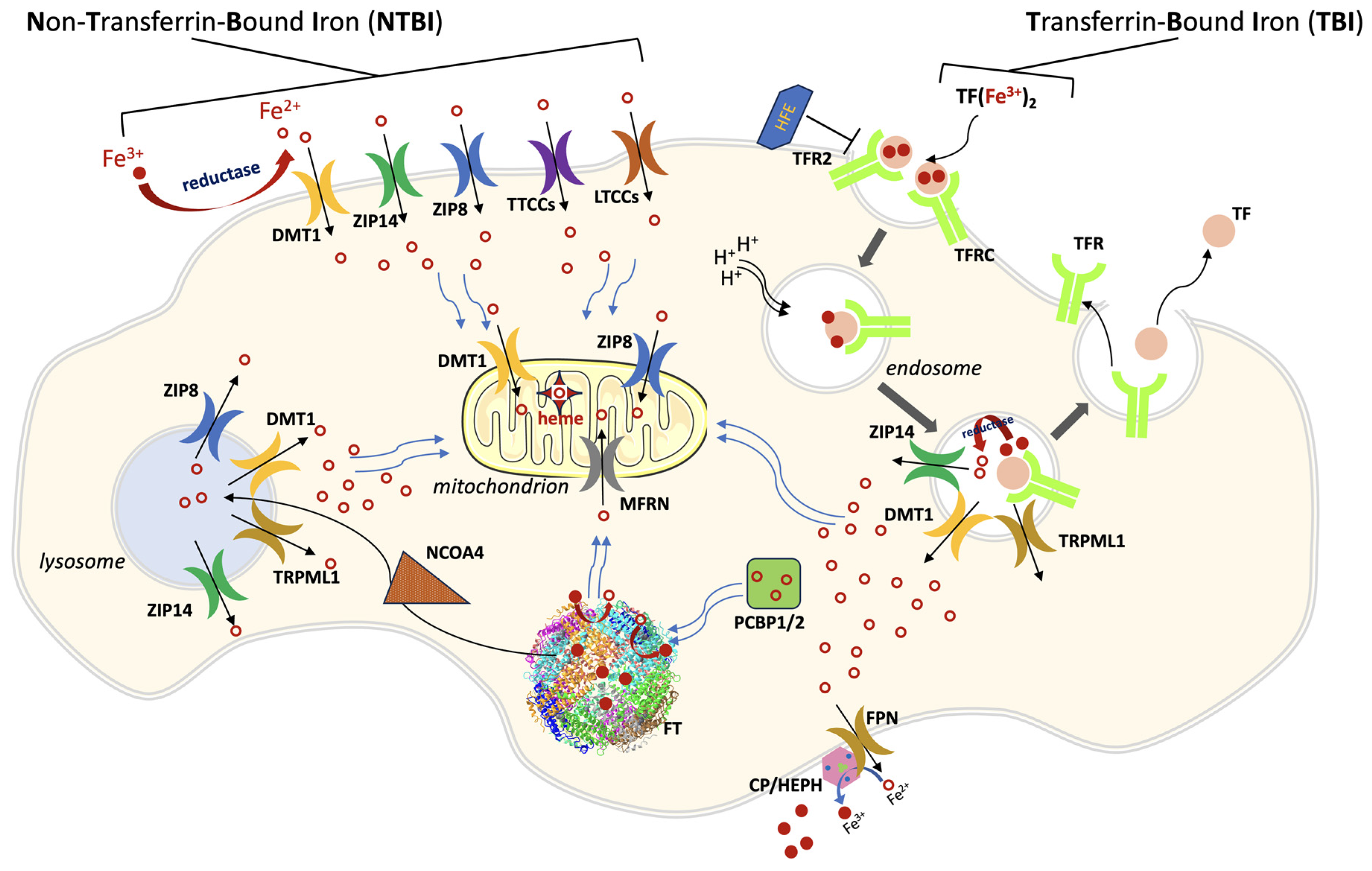
:1. General Iron Metabolism and Trafficking
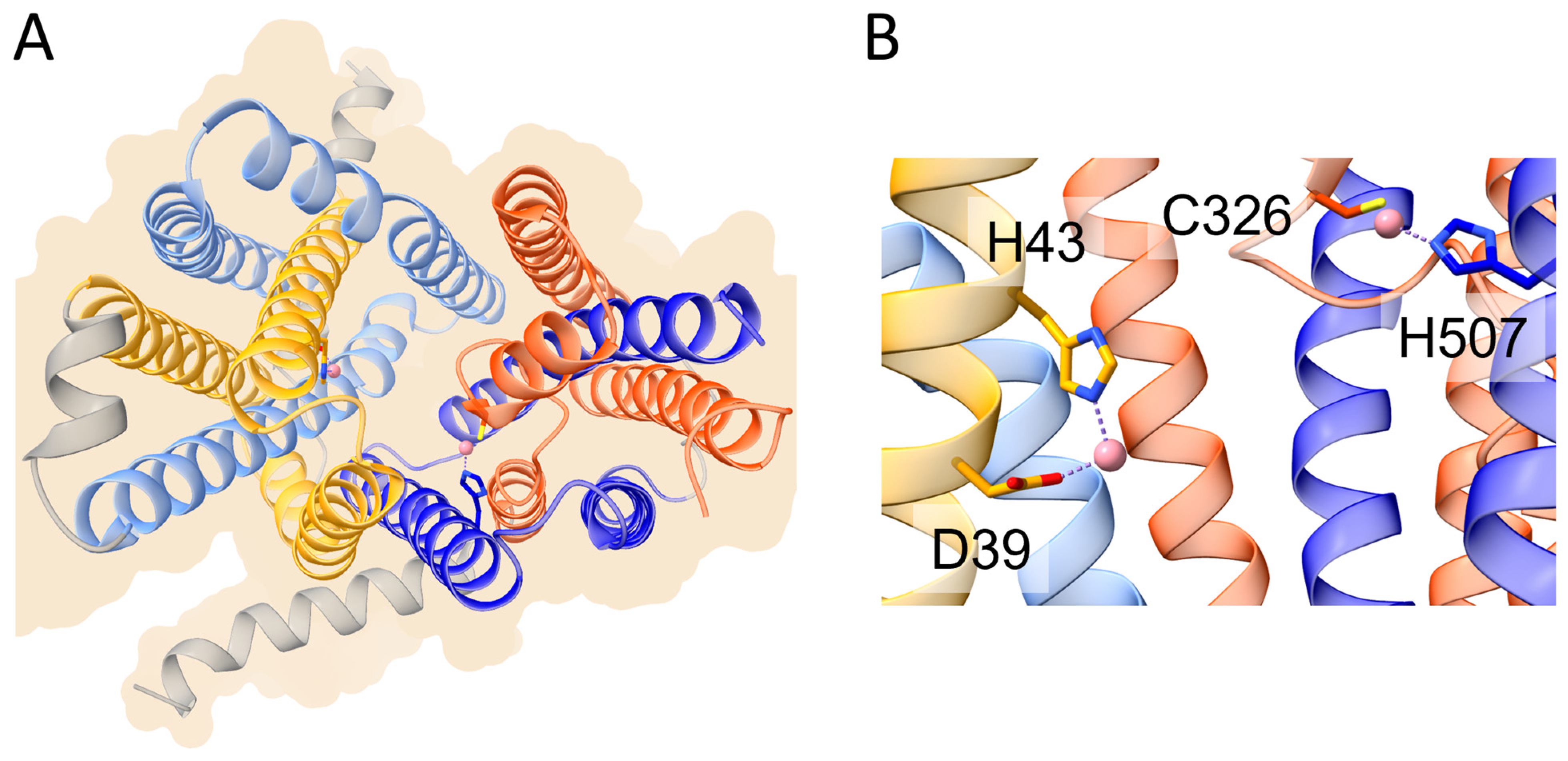
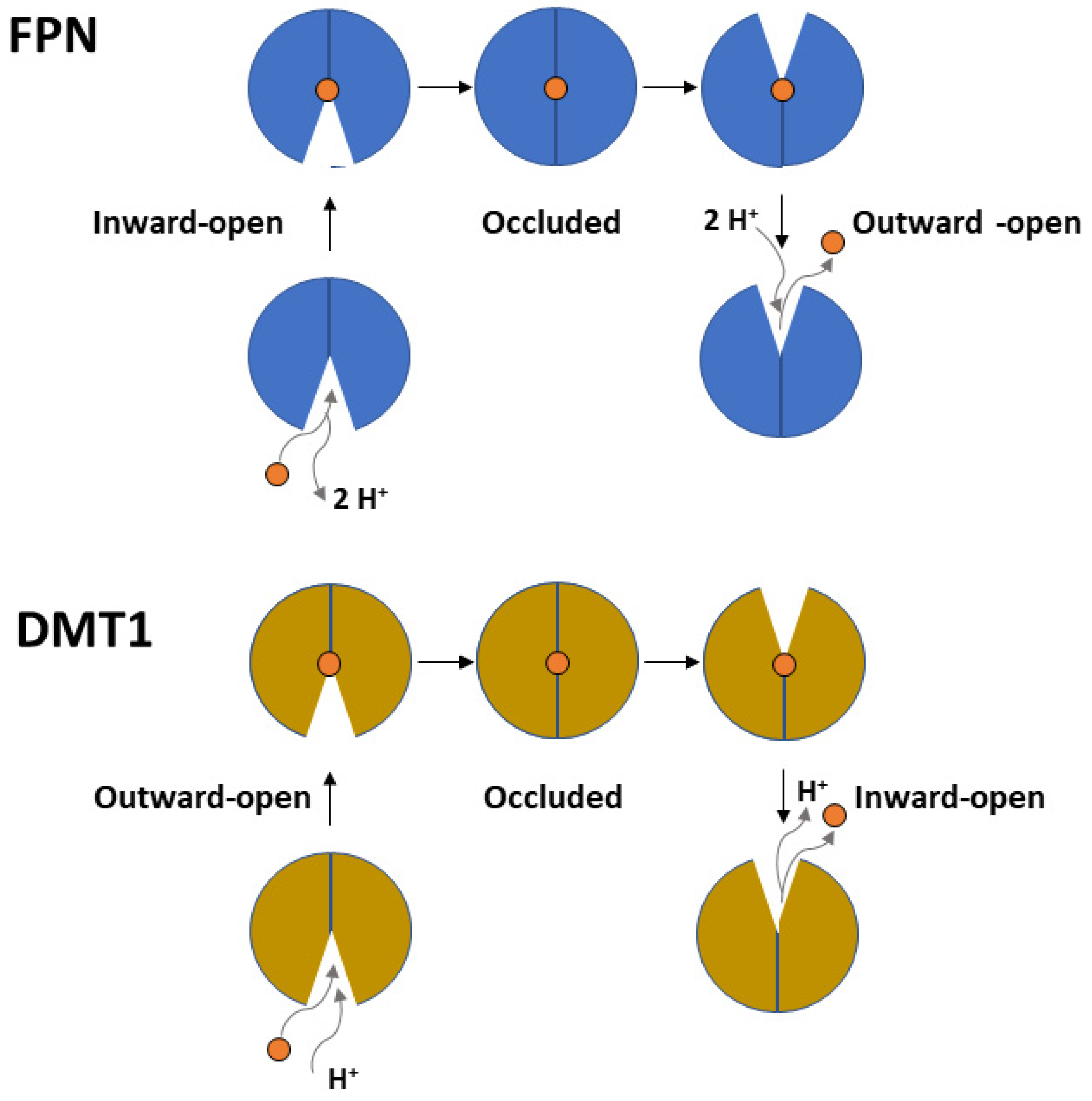
2. Ferroportin


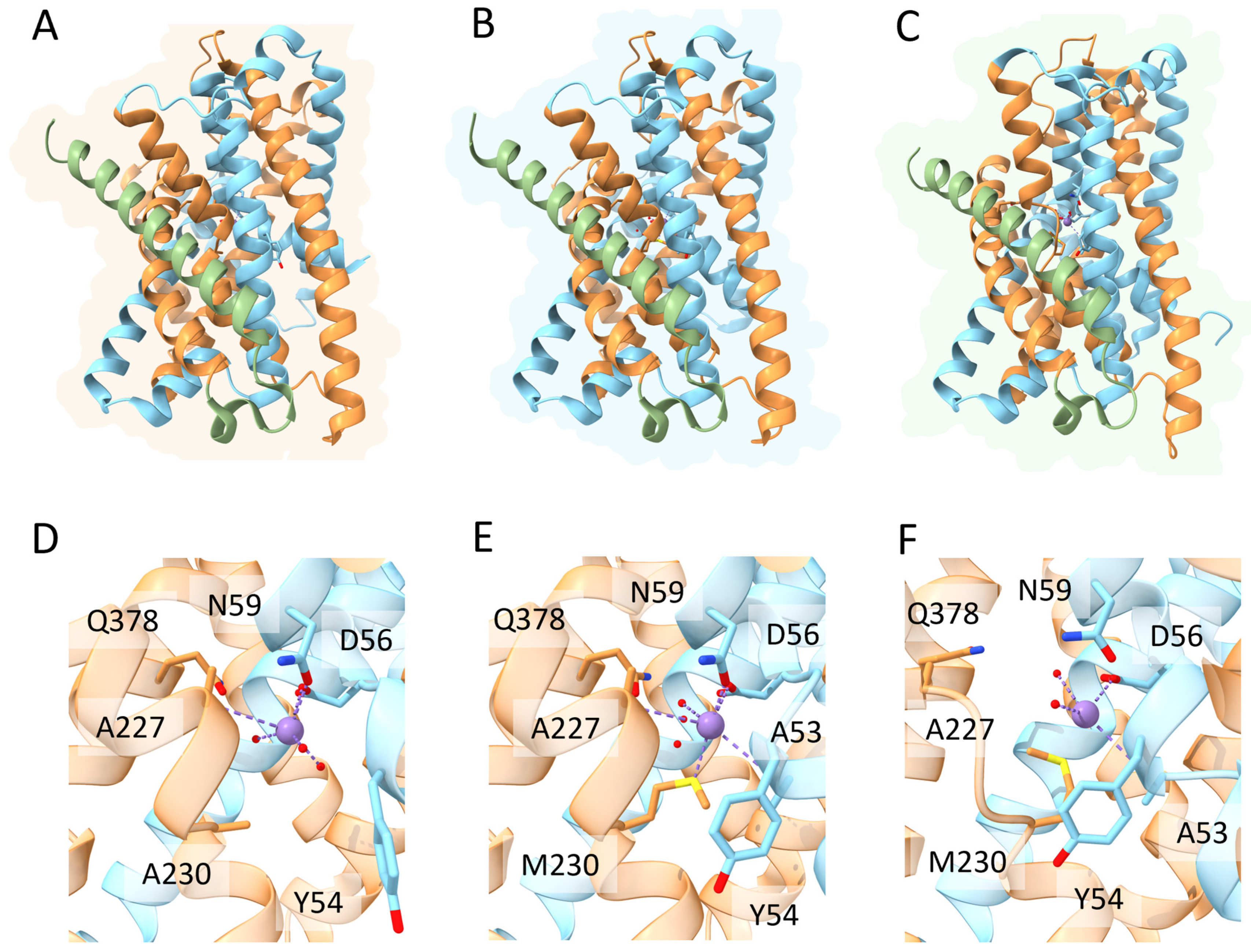
3. DMT1
4. ZIP8 and ZIP14

5. Mitoferrin
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Hentze, M.W.; Muckenthaler, M.U.; Galy, B.; Camaschella, C. Two to Tango: Regulation of Mammalian Iron Metabolism. Cell 2010, 142, 24–38. [Google Scholar] [CrossRef] [Green Version]
- Anderson, G.J.; Frazer, D.M. Current Understanding of Iron Homeostasis. Am. J. Clin. Nutr. 2017, 106, 1559S–1566S. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Liu, C.; Zhao, Y.; Gao, G. Mitochondria Regulation in Ferroptosis. Eur. J. Cell Biol. 2020, 99, 151058. [Google Scholar] [CrossRef]
- Huo, C.; Li, G.; Hu, Y.; Sun, H. The Impacts of Iron Overload and Ferroptosis on Intestinal Mucosal Homeostasis and Inflammation. Int. J. Mol. Sci. 2022, 23, 14195. [Google Scholar] [CrossRef] [PubMed]
- Gunshin, H.; Fujiwara, Y.; Custodio, A.O.; DiRenzo, C.; Robine, S.; Andrews, N.C. Slc11a2 Is Required for Intestinal Iron Absorption and Erythropoiesis but Dispensable in Placenta and Liver. J. Clin. Invest. 2005, 115, 1258–1266. [Google Scholar] [CrossRef]
- McKie, A.T.; Barrow, D.; Latunde-Dada, G.O.; Rolfs, A.; Sager, G.; Mudaly, E.; Mudaly, M.; Richardson, C.; Barlow, D.; Bomford, A.; et al. An Iron-Regulated Ferric Reductase Associated with the Absorption of Dietary Iron. Science 2001, 291, 1755–1759. [Google Scholar] [CrossRef]
- Fuqua, B.K.; Lu, Y.; Darshan, D.; Frazer, D.M.; Wilkins, S.J.; Wolkow, N.; Bell, A.G.; Hsu, J.; Yu, C.C.; Chen, H.; et al. The Multicopper Ferroxidase Hephaestin Enhances Intestinal Iron Absorption in Mice. PLoS ONE 2014, 9, e98792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Musci, G.; Polticelli, F.; Bonaccorsi di Patti, M.C. Ceruloplasmin-Ferroportin System of Iron Traffic in Vertebrates. World J. Biol. Chem. 2014, 5, 204–215. [Google Scholar] [CrossRef]
- Zhang, D.-L.; Wu, J.; Shah, B.N.; Greutélaers, K.C.; Ghosh, M.C.; Ollivierre, H.; Su, X.; Thuma, P.E.; Bedu-Addo, G.; Mockenhaupt, F.P.; et al. Erythrocytic Ferroportin Reduces Intracellular Iron Accumulation, Hemolysis, and Malaria Risk. Science 2018, 359, 1520–1523. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.-L.; Ghosh, M.C.; Ollivierre, H.; Li, Y.; Rouault, T.A. Ferroportin Deficiency in Erythroid Cells Causes Serum Iron Deficiency and Promotes Hemolysis Due to Oxidative Stress. Blood 2018, 132, 2078–2087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aisen, P.; Leibman, A.; Zweier, J. Stoichiometric and Site Characteristics of the Binding of Iron to Human Transferrin. J. Biol. Chem. 1978, 253, 1930–1937. [Google Scholar] [CrossRef] [PubMed]
- Enns, C.A.; Rutledge, E.A.; Williams, A.M. The Transferrin Receptor. In Biomembranes: A Multi-Volume Treatise; Lee, A.G., Ed.; Endoctosis and Exocytosis: Amsterdam, The Netherlands, 1996; Volume 4, pp. 255–287. [Google Scholar]
- Ohgami, R.S.; Campagna, D.R.; Greer, E.L.; Antiochos, B.; McDonald, A.; Chen, J.; Sharp, J.J.; Fujiwara, Y.; Barker, J.E.; Fleming, M.D. Identification of a Ferrireductase Required for Efficient Transferrin-Dependent Iron Uptake in Erythroid Cells. Nat. Genet. 2005, 37, 1264–1269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fleming, M.D.; Romano, M.A.; Su, M.A.; Garrick, L.M.; Garrick, M.D.; Andrews, N.C. Nramp2 Is Mutated in the Anemic Belgrade (b) Rat: Evidence of a Role for Nramp2 in Endosomal Iron Transport. Proc. Natl. Acad. Sci. USA 1998, 95, 1148–1153. [Google Scholar] [CrossRef]
- Zhao, N.; Gao, J.; Enns, C.A.; Knutson, M.D. ZRT/IRT-like Protein 14 (ZIP14) Promotes the Cellular Assimilation of Iron from Transferrin*. J. Biol. Chem. 2010, 285, 32141–32150. [Google Scholar] [CrossRef] [Green Version]
- Dong, X.-P.; Cheng, X.; Mills, E.; Delling, M.; Wang, F.; Kurz, T.; Xu, H. The Type IV Mucolipidosis-Associated Protein TRPML1 Is an Endolysosomal Iron Release Channel. Nature 2008, 455, 992–996. [Google Scholar] [CrossRef] [Green Version]
- Kawabata, H. Transferrin and Transferrin Receptors Update. Free Radic. Biol. Med. 2019, 133, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Liuzzi, J.P.; Aydemir, F.; Nam, H.; Knutson, M.D.; Cousins, R.J. Zip14 (Slc39a14) Mediates Non-Transferrin-Bound Iron Uptake into Cells. Proc. Natl. Acad. Sci. USA 2006, 103, 13612–13617. [Google Scholar] [CrossRef]
- Wang, C.-Y.; Jenkitkasemwong, S.; Duarte, S.; Sparkman, B.K.; Shawki, A.; Mackenzie, B.; Knutson, M.D. ZIP8 Is an Iron and Zinc Transporter Whose Cell-Surface Expression Is Up-Regulated by Cellular Iron Loading*. J. Biol. Chem. 2012, 287, 34032–34043. [Google Scholar] [CrossRef] [Green Version]
- Garrick, L.M.; Dolan, K.G.; Romano, M.A.; Garrick, M.D. Non-Transferrin-Bound Iron Uptake in Belgrade and Normal Rat Erythroid Cells. J. Cell. Physiol. 1999, 178, 349–358. [Google Scholar] [CrossRef]
- Tsushima, R.G.; Wickenden, A.D.; Bouchard, R.A.; Oudit, G.Y.; Liu, P.P.; Backx, P.H. Modulation of Iron Uptake in Heart by L-Type Ca2+ Channel Modifiers. Circ. Res. 1999, 84, 1302–1309. [Google Scholar] [CrossRef]
- Kumfu, S.; Chattipakorn, S.; Srichairatanakool, S.; Settakorn, J.; Fucharoen, S.; Chattipakorn, N. T-Type Calcium Channel as a Portal of Iron Uptake into Cardiomyocytes of Beta-Thalassemic Mice. Eur. J. Haematol. 2011, 86, 156–166. [Google Scholar] [CrossRef] [PubMed]
- Lopin, K.V.; Gray, I.P.; Obejero-Paz, C.A.; Thévenod, F.; Jones, S.W. Fe2+ Block and Permeation of CaV3.1 (A1G) T-Type Calcium Channels: Candidate Mechanism for Non–Transferrin-Mediated Fe2+ Influx. Mol. Pharmacol. 2012, 82, 1194–1204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knutson, M.D. Non-Transferrin-Bound Iron Transporters. Free Radic. Biol. Med. 2019, 133, 101–111. [Google Scholar] [CrossRef]
- Liu, Q.; Barker, S.; Knutson, M.D. Iron and Manganese Transport in Mammalian Systems. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2021, 1868, 118890. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.Y.; Oliva, C.R.; Flor, S.; Griguer, C.E. Mitoferrin, Cellular and Mitochondrial Iron Homeostasis. Cells 2022, 11, 3464. [Google Scholar] [CrossRef] [PubMed]
- Besecker, B.; Bao, S.; Bohacova, B.; Papp, A.; Sadee, W.; Knoell, D.L. The Human Zinc Transporter SLC39A8 (Zip8) Is Critical in Zinc-Mediated Cytoprotection in Lung Epithelia. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2008, 294, L1127–L1136. [Google Scholar] [CrossRef] [Green Version]
- Wolff, N.A.; Garrick, L.M.; Zhao, L.; Garrick, M.D.; Thévenod, F. Mitochondria Represent Another Locale for the Divalent Metal Transporter 1 (DMT1). Channels 2014, 8, 458–466. [Google Scholar] [CrossRef] [Green Version]
- Wolff, N.A.; Ghio, A.J.; Garrick, L.M.; Garrick, M.D.; Zhao, L.; Fenton, R.A.; Thévenod, F. Evidence for Mitochondrial Localization of Divalent Metal Transporter 1 (DMT1). FASEB J. 2014, 28, 2134–2145. [Google Scholar] [CrossRef]
- Wolff, N.A.; Garrick, M.D.; Zhao, L.; Garrick, L.M.; Ghio, A.J.; Thévenod, F. A Role for Divalent Metal Transporter (DMT1) in Mitochondrial Uptake of Iron and Manganese. Sci. Rep. 2018, 8, 211. [Google Scholar] [CrossRef] [Green Version]
- Melino, G.; Stefanini, S.; Chiancone, E.; Antonini, E. Stoichiometry of Iron Oxidation by Apoferritin. FEBS Lett. 1978, 86, 136–138. [Google Scholar] [CrossRef] [Green Version]
- Yanatori, I.; Richardson, D.R.; Toyokuni, S.; Kishi, F. The New Role of Poly (RC)-Binding Proteins as Iron Transport Chaperones: Proteins That Could Couple with Inter-Organelle Interactions to Safely Traffic Iron. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2020, 1864, 129685. [Google Scholar] [CrossRef]
- Leidgens, S.; Bullough, K.Z.; Shi, H.; Li, F.; Shakoury-Elizeh, M.; Yabe, T.; Subramanian, P.; Hsu, E.; Natarajan, N.; Nandal, A.; et al. Each Member of the Poly-r(C)-Binding Protein 1 (PCBP) Family Exhibits Iron Chaperone Activity toward Ferritin*. J. Biol. Chem. 2013, 288, 17791–17802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Pantopoulos, K. Regulation of Cellular Iron Metabolism. Biochem. J. 2011, 434, 365–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baringer, S.L.; Palsa, K.; Spiegelman, V.S.; Simpson, I.A.; Connor, J.R. Apo- and Holo-Transferrin Differentially Interact with Hephaestin and Ferroportin in a Novel Mechanism of Cellular Iron Release Regulation. J. Biomed. Sci. 2023, 30, 36. [Google Scholar] [CrossRef] [PubMed]
- Pietrangelo, A. Iron and the Liver. Liver Int. 2016, 36 (Suppl. S1), 116–123. [Google Scholar] [CrossRef]
- Camaschella, C. Iron-Deficiency Anemia. N. Engl. J. Med. 2015, 372, 1832–1843. [Google Scholar] [CrossRef] [Green Version]
- Zumerle, S.; Mathieu, J.R.R.; Delga, S.; Heinis, M.; Viatte, L.; Vaulont, S.; Peyssonnaux, C. Targeted Disruption of Hepcidin in the Liver Recapitulates the Hemochromatotic Phenotype. Blood 2014, 123, 3646–3650. [Google Scholar] [CrossRef] [Green Version]
- Muckenthaler, M.U.; Rivella, S.; Hentze, M.W.; Galy, B. A Red Carpet for Iron Metabolism. Cell 2017, 168, 344–361. [Google Scholar] [CrossRef] [Green Version]
- Aschemeyer, S.; Qiao, B.; Stefanova, D.; Valore, E.V.; Sek, A.C.; Ruwe, T.A.; Vieth, K.R.; Jung, G.; Casu, C.; Rivella, S.; et al. Structure-Function Analysis of Ferroportin Defines the Binding Site and an Alternative Mechanism of Action of Hepcidin. Blood 2018, 131, 899–910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nemeth, E.; Tuttle, M.S.; Powelson, J.; Vaughn, M.B.; Donovan, A.; Ward, D.M.; Ganz, T.; Kaplan, J. Hepcidin Regulates Cellular Iron Efflux by Binding to Ferroportin and Inducing Its Internalization. Science 2004, 306, 2090–2093. [Google Scholar] [CrossRef] [Green Version]
- Drakesmith, H.; Nemeth, E.; Ganz, T. Ironing out Ferroportin. Cell Metab. 2015, 22, 777–787. [Google Scholar] [CrossRef] [Green Version]
- Ward, D.M.; Kaplan, J. Ferroportin-Mediated Iron Transport: Expression and Regulation. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2012, 1823, 1426–1433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallace, D.F.; Harris, J.M.; Subramaniam, V.N. Functional Analysis and Theoretical Modeling of Ferroportin Reveals Clustering of Mutations According to Phenotype. Am. J. Physiol.-Cell Physiol. 2010, 298, C75–C84. [Google Scholar] [CrossRef] [PubMed]
- Le Gac, G.; Ka, C.; Joubrel, R.; Gourlaouen, I.; Lehn, P.; Mornon, J.-P.; Férec, C.; Callebaut, I. Structure-Function Analysis of the Human Ferroportin Iron Exporter (SLC40A1): Effect of Hemochromatosis Type 4 Disease Mutations and Identification of Critical Residues. Hum. Mutat. 2013, 34, 1371–1380. [Google Scholar] [CrossRef] [PubMed]
- Bonaccorsi Di Patti, M.C.; Polticelli, F.; Cece, G.; Cutone, A.; Felici, F.; Persichini, T.; Musci, G. A Structural Model of Human Ferroportin and of Its Iron Binding Site. FEBS J. 2014, 281, 2851–2860. [Google Scholar] [CrossRef]
- Pao, S.S.; Paulsen, I.T.; Saier, M.H. Major Facilitator Superfamily. Microbiol. Mol. Biol. Rev. 1998, 62, 1–34. [Google Scholar] [CrossRef] [Green Version]
- Quistgaard, E.M.; Löw, C.; Guettou, F.; Nordlund, P. Understanding Transport by the Major Facilitator Superfamily (MFS): Structures Pave the Way. Nat. Rev. Mol. Cell Biol. 2016, 17, 123–132. [Google Scholar] [CrossRef]
- Yan, N. Structural Advances for the Major Facilitator Superfamily (MFS) Transporters. Trends Biochem. Sci. 2013, 38, 151–159. [Google Scholar] [CrossRef]
- Law, C.J.; Maloney, P.C.; Wang, D.-N. Ins and Outs of Major Facilitator Superfamily Antiporters. Annu. Rev. Microbiol. 2008, 62, 289–305. [Google Scholar] [CrossRef] [Green Version]
- Pan, Y.; Ren, Z.; Gao, S.; Shen, J.; Wang, L.; Xu, Z.; Yu, Y.; Bachina, P.; Zhang, H.; Fan, X.; et al. Structural Basis of Ion Transport and Inhibition in Ferroportin. Nat. Commun. 2020, 11, 5686. [Google Scholar] [CrossRef]
- Li, S.; Yang, Y.; Li, W. Human Ferroportin Mediates Proton-Coupled Active Transport of Iron. Blood Adv. 2020, 4, 4758–4768. [Google Scholar] [CrossRef]
- Billesbølle, C.B.; Azumaya, C.M.; Kretsch, R.C.; Powers, A.S.; Gonen, S.; Schneider, S.; Arvedson, T.; Dror, R.O.; Cheng, Y.; Manglik, A. Structure of Hepcidin-Bound Ferroportin Reveals Iron Homeostatic Mechanisms. Nature 2020, 586, 807–811. [Google Scholar] [CrossRef]
- Shen, J.; Wilbon, A.S.; Zhou, M.; Pan, Y. Mechanism of Ca2+ Transport by Ferroportin. eLife 2023, 12, e82947. [Google Scholar] [CrossRef]
- Pietrangelo, A. Ferroportin Disease: Pathogenesis, Diagnosis and Treatment. Haematologica 2017, 102, 1972–1984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vlasveld, L.T.; Janssen, R.; Bardou-Jacquet, E.; Venselaar, H.; Hamdi-Roze, H.; Drakesmith, H.; Swinkels, D.W. Twenty Years of Ferroportin Disease: A Review or An Update of Published Clinical, Biochemical, Molecular, and Functional Features. Pharmaceuticals 2019, 12, 132. [Google Scholar] [CrossRef] [Green Version]
- Majore, S.; Bonaccorsi di Patti, M.C.; Valiante, M.; Polticelli, F.; Cortese, A.; Di Bartolomeo, S.; De Bernardo, C.; De Muro, M.; Faienza, F.; Radio, F.C.; et al. Characterization of Three Novel Pathogenic SLC40A1 Mutations and Genotype/Phenotype Correlations in 7 Italian Families with Type 4 Hereditary Hemochromatosis. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2018, 1864, 464–470. [Google Scholar] [CrossRef] [PubMed]
- Ka, C.; Guellec, J.; Pepermans, X.; Kannengiesser, C.; Ged, C.; Wuyts, W.; Cassiman, D.; de Ledinghen, V.; Varet, B.; de Kerguenec, C.; et al. The SLC40A1 R178Q Mutation Is a Recurrent Cause of Hemochromatosis and Is Associated with a Novel Pathogenic Mechanism. Haematologica 2018, 103, 1796–1805. [Google Scholar] [CrossRef] [Green Version]
- Tortosa, V.; di Patti, M.C.B.; Brandi, V.; Musci, G.; Polticelli, F. An Improved Structural Model of the Human Iron Exporter Ferroportin. Insight into the Role of Pathogenic Mutations in Hereditary Hemochromatosis Type 4. Bio-Algorithms Med-Syst. 2017, 13, 215–222. [Google Scholar] [CrossRef]
- Honma, Y.; Karasuyama, T.; Kumamoto, K.; Shimajiri, S.; Toki, Y.; Tatsumi, Y.; Sumida, K.; Koikawa, K.; Morino, K.; Oe, S.; et al. Type 4B Hereditary Hemochromatosis Due to Heterozygous p.D157A Mutation in SLC40A1 Complicated with Hypopituitarism. Med. Mol. Morphol. 2021, 54, 60–67. [Google Scholar] [CrossRef]
- Jiang, L.; Wang, J.; Wang, K.; Wang, H.; Wu, Q.; Yang, C.; Yu, Y.; Ni, P.; Zhong, Y.; Song, Z.; et al. RNF217 Regulates Iron Homeostasis through Its E3 Ubiquitin Ligase Activity by Modulating Ferroportin Degradation. Blood 2021, 138, 689–705. [Google Scholar] [CrossRef] [PubMed]
- Traeger, L.; Wiegand, S.B.; Sauer, A.J.; Corman, B.H.P.; Peneyra, K.M.; Wunderer, F.; Fischbach, A.; Bagchi, A.; Malhotra, R.; Zapol, W.M.; et al. UBA6 and NDFIP1 Regulate the Degradation of Ferroportin. Hematology 2021, 107, 478–488. [Google Scholar] [CrossRef] [PubMed]
- Bayele, H.K.; Srai, S.K.S. A Disease-Causing Mutation K240E Disrupts Ferroportin Trafficking by SUMO (Ferroportin SUMOylation). Biochem. Biophys. Rep. 2021, 25, 100873. [Google Scholar] [CrossRef]
- Yanatori, I.; Richardson, D.R.; Imada, K.; Kishi, F. Iron Export through the Transporter Ferroportin 1 Is Modulated by the Iron Chaperone PCBP2. J. Biol. Chem. 2016, 291, 17303–17318. [Google Scholar] [CrossRef] [Green Version]
- Manatschal, C.; Dutzler, R. The Structural Basis for Metal Ion Transport in the SLC11/NRAMP Family. Chimia 2022, 76, 1005. [Google Scholar] [CrossRef]
- Montalbetti, N.; Simonin, A.; Kovacs, G.; Hediger, M.A. Mammalian Iron Transporters: Families SLC11 and SLC40. Mol. Asp. Med. 2013, 34, 270–287. [Google Scholar] [CrossRef] [PubMed]
- Illing, A.C.; Shawki, A.; Cunningham, C.L.; Mackenzie, B. Substrate Profile and Metal-Ion Selectivity of Human Divalent Metal-Ion Transporter-1. J. Biol. Chem. 2012, 287, 30485–30496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ehrnstorfer, I.A.; Geertsma, E.R.; Pardon, E.; Steyaert, J.; Dutzler, R. Crystal Structure of a SLC11 (NRAMP) Transporter Reveals the Basis for Transition-Metal Ion Transport. Nat. Struct. Mol. Biol. 2014, 21, 990–996. [Google Scholar] [CrossRef] [PubMed]
- Bozzi, A.T.; Bane, L.B.; Weihofen, W.A.; Singharoy, A.; Guillen, E.R.; Ploegh, H.L.; Schulten, K.; Gaudet, R. Crystal Structure and Conformational Change Mechanism of a Bacterial Nramp-Family Divalent Metal Transporter. Structure 2016, 24, 2102–2114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bozzi, A.T.; Bane, L.B.; Zimanyi, C.M.; Gaudet, R. Unique Structural Features in an Nramp Metal Transporter Impart Substrate-Specific Proton Cotransport and a Kinetic Bias to Favor Import. J. Gen. Physiol. 2019, 151, 1413–1429. [Google Scholar] [CrossRef]
- Ehrnstorfer, I.A.; Manatschal, C.; Arnold, F.M.; Laederach, J.; Dutzler, R. Structural and Mechanistic Basis of Proton-Coupled Metal Ion Transport in the SLC11/NRAMP Family. Nat. Commun. 2017, 8, 14033. [Google Scholar] [CrossRef] [Green Version]
- Yamashita, A.; Singh, S.K.; Kawate, T.; Jin, Y.; Gouaux, E. Crystal Structure of a Bacterial Homologue of Na+/Cl--Dependent Neurotransmitter Transporters. Nature 2005, 437, 215–223. [Google Scholar] [CrossRef] [PubMed]
- Ray, S.; Berry, S.P.; Wilson, E.A.; Zhang, C.H.; Shekhar, M.; Singharoy, A.; Gaudet, R. High-Resolution Structures with Bound Mn2+ and Cd2+ Map the Metal Import Pathway in an Nramp Transporter. eLife 2023, 12, e84006. [Google Scholar] [CrossRef] [PubMed]
- Iolascon, A.; De Falco, L. Mutations in the Gene Encoding DMT1: Clinical Presentation and Treatment. Semin. Hematol. 2009, 46, 358–370. [Google Scholar] [CrossRef] [PubMed]
- Romero-Cortadellas, L.; Hernández, G.; Ferrer-Cortès, X.; Zalba-Jadraque, L.; Fuster, J.L.; Bermúdez-Cortés, M.; Galera-Miñarro, A.M.; Pérez-Montero, S.; Tornador, C.; Sánchez, M. New Cases of Hypochromic Microcytic Anemia Due to Mutations in the SLC11A2 Gene and Functional Characterization of the G75R Mutation. IJMS 2022, 23, 4406. [Google Scholar] [CrossRef] [PubMed]
- Mims, M.P.; Guan, Y.; Pospisilova, D.; Priwitzerova, M.; Indrak, K.; Ponka, P.; Divoky, V.; Prchal, J.T. Identification of a Human Mutation of DMT1 in a Patient with Microcytic Anemia and Iron Overload. Blood 2005, 105, 1337–1342. [Google Scholar] [CrossRef] [PubMed]
- Iolascon, A.; d’Apolito, M.; Servedio, V.; Cimmino, F.; Piga, A.; Camaschella, C. Microcytic Anemia and Hepatic Iron Overload in a Child with Compound Heterozygous Mutations in DMT1 (SCL11A2). Blood 2006, 107, 349–354. [Google Scholar] [CrossRef] [Green Version]
- Casale, M.; Borriello, A.; Scianguetta, S.; Roberti, D.; Caiazza, M.; Bencivenga, D.; Tartaglione, I.; Ladogana, S.; Maruzzi, M.; Della Ragione, F.; et al. Hereditary Hypochromic Microcytic Anemia Associated with Loss-of-Function DMT1 Gene Mutations and Absence of Liver Iron Overload. Am. J. Hematol. 2018, 93, E58–E60. [Google Scholar] [CrossRef] [Green Version]
- Beaumont, C. Two New Human DMT1 Gene Mutations in a Patient with Microcytic Anemia, Low Ferritinemia, and Liver Iron Overload. Blood 2006, 107, 4168–4170. [Google Scholar] [CrossRef]
- Bardou-Jacquet, E.; Island, M.-L.; Jouanolle, A.-M.; Détivaud, L.; Fatih, N.; Ropert, M.; Brissot, E.; Mosser, A.; Maisonneuve, H.; Brissot, P.; et al. A Novel N491S Mutation in the Human SLC11A2 Gene Impairs Protein Trafficking and in Association with the G212V Mutation Leads to Microcytic Anemia and Liver Iron Overload. Blood Cells Mol. Dis. 2011, 47, 243–248. [Google Scholar] [CrossRef]
- De Falco, L.; Bruno, M.; Andolfo, I.; David, B.P.; Girelli, D.; Noce, F.D.; Camaschella, C.; Iolascon, A. Identification and Characterization of the First SLC11A2 Isoform 1a Mutation Causing a Defect in Splicing Process and an Hypomorphic Allele Expression of the SLC11A2 Gene. Br. J. Haematol. 2012, 159, 492–495. [Google Scholar] [CrossRef]
- Zhang, D.-L.; Hughes, R.M.; Ollivierre-Wilson, H.; Ghosh, M.C.; Rouault, T.A. A Ferroportin Transcript That Lacks an Iron-Responsive Element Enables Duodenal and Erythroid Precursor Cells to Evade Translational Repression. Cell Metab. 2009, 9, 461–473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yanatori, I.; Kishi, F. DMT1 and Iron Transport. Free Radic. Biol. Med. 2019, 133, 55–63. [Google Scholar] [CrossRef]
- Foot, N.J.; Dalton, H.E.; Shearwin-Whyatt, L.M.; Dorstyn, L.; Tan, S.-S.; Yang, B.; Kumar, S. Regulation of the Divalent Metal Ion Transporter DMT1 and Iron Homeostasis by a Ubiquitin-Dependent Mechanism Involving Ndfips and WWP2. Blood 2008, 112, 4268–4275. [Google Scholar] [CrossRef] [PubMed]
- Yanatori, I.; Yasui, Y.; Tabuchi, M.; Kishi, F. Chaperone Protein Involved in Transmembrane Transport of Iron. Biochem. J. 2014, 462, 25–37. [Google Scholar] [CrossRef] [PubMed]
- Hu, J. Toward Unzipping the ZIP Metal Transporters: Structure, Evolution, and Implications on Drug Discovery against Cancer. FEBS J. 2021, 288, 5805–5825. [Google Scholar] [CrossRef] [PubMed]
- Wiuf, A.; Steffen, J.H.; Becares, E.R.; Grønberg, C.; Mahato, D.R.; Rasmussen, S.G.F.; Andersson, M.; Croll, T.; Gotfryd, K.; Gourdon, P. The Two-Domain Elevator-Type Mechanism of Zinc-Transporting ZIP Proteins. Sci. Adv. 2022, 8, eabn4331. [Google Scholar] [CrossRef]
- Pasquadibisceglie, A.; Leccese, A.; Polticelli, F. A Computational Study of the Structure and Function of Human Zrt and Irt-like Proteins Metal Transporters: An Elevator-Type Transport Mechanism Predicted by AlphaFold2. Front. Chem. 2022, 10, 1004815. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Sui, D.; Hu, J. Structural Insights of ZIP4 Extracellular Domain Critical for Optimal Zinc Transport. Nat. Commun. 2016, 7, 11979. [Google Scholar] [CrossRef] [Green Version]
- Fujishiro, H.; Miyamoto, S.; Sumi, D.; Kambe, T.; Himeno, S. Effects of Individual Amino Acid Mutations of Zinc Transporter ZIP8 on Manganese- and Cadmium-Transporting Activity. Biochem. Biophys. Res. Commun. 2022, 616, 26–32. [Google Scholar] [CrossRef]
- Jiang, Y.; Li, Z.; Sui, D.; Sharma, G.; Wang, T.; MacRenaris, K.; Takahashi, H.; Merz, K.; Hu, J. Rational Engineering of an Elevator-Type Metal Transporter ZIP8 Reveals a Conditional Selectivity Filter Critically Involved in Determining Substrate Specificity. bioRxiv 2023, 545588. [Google Scholar] [CrossRef]
- Jenkitkasemwong, S.; Wang, C.-Y.; Mackenzie, B.; Knutson, M.D. Physiologic Implications of Metal-Ion Transport by ZIP14 and ZIP8. Biometals 2012, 25, 643–655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Girijashanker, K.; He, L.; Soleimani, M.; Reed, J.M.; Li, H.; Liu, Z.; Wang, B.; Dalton, T.P.; Nebert, D.W. Slc39a14 Gene Encodes ZIP14, A Metal/Bicarbonate Symporter: Similarities to the ZIP8 Transporter. Mol. Pharmacol. 2008, 73, 1413–1423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aydemir, T.B.; Liuzzi, J.P.; McClellan, S.; Cousins, R.J. Zinc Transporter ZIP8 (SLC39A8) and Zinc Influence IFN-γ Expression in Activated Human T Cells. J. Leukoc. Biol. 2009, 86, 337–348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, J.; Zhao, N.; Knutson, M.D.; Enns, C.A. The Hereditary Hemochromatosis Protein, HFE, Inhibits Iron Uptake via Down-Regulation of Zip14 in HepG2 Cells*. J. Biol. Chem. 2008, 283, 21462–21468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Núñez, M.T.; Gaete, V.; Watkins, J.A.; Glass, J. Mobilization of Iron from Endocytic Vesicles. The Effects of Acidification and Reduction. J. Biol. Chem. 1990, 265, 6688–6692. [Google Scholar] [CrossRef]
- Zhang, V.; Jenkitkasemwong, S.; Liu, Q.; Ganz, T.; Nemeth, E.; Knutson, M.D.; Kim, A. A Mouse Model Characterizes the Roles of ZIP8 in Systemic Iron Recycling and Lung Inflammation and Infection. Blood Adv. 2023, 7, 1336–1349. [Google Scholar] [CrossRef]
- Gálvez-Peralta, M.; He, L.; Jorge-Nebert, L.F.; Wang, B.; Miller, M.L.; Eppert, B.L.; Afton, S.; Nebert, D.W. ZIP8 Zinc Transporter: Indispensable Role for Both Multiple-Organ Organogenesis and Hematopoiesis In Utero. PLoS ONE 2012, 7, e36055. [Google Scholar] [CrossRef] [Green Version]
- Boycott, K.M.; Beaulieu, C.L.; Kernohan, K.D.; Gebril, O.H.; Mhanni, A.; Chudley, A.E.; Redl, D.; Qin, W.; Hampson, S.; Küry, S.; et al. Autosomal-Recessive Intellectual Disability with Cerebellar Atrophy Syndrome Caused by Mutation of the Manganese and Zinc Transporter Gene SLC39A8. Am. J. Hum. Genet. 2015, 97, 886–893. [Google Scholar] [CrossRef] [Green Version]
- Park, J.H.; Hogrebe, M.; Grüneberg, M.; DuChesne, I.; von der Heiden, A.L.; Reunert, J.; Schlingmann, K.P.; Boycott, K.M.; Beaulieu, C.L.; Mhanni, A.A.; et al. SLC39A8 Deficiency: A Disorder of Manganese Transport and Glycosylation. Am. J. Hum. Genet. 2015, 97, 894–903. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Achkar, J.-P.; Haritunians, T.; Jacobs, J.P.; Hui, K.Y.; D’Amato, M.; Brand, S.; Radford-Smith, G.; Halfvarson, J.; Niess, J.-H.; et al. A Pleiotropic Missense Variant in SLC39A8 Is Associated With Crohn’s Disease and Human Gut Microbiome Composition. Gastroenterology 2016, 151, 724–732. [Google Scholar] [CrossRef] [Green Version]
- Zhang, R.; Witkowska, K.; Afonso Guerra-Assunção, J.; Ren, M.; Ng, F.L.; Mauro, C.; Tucker, A.T.; Caulfield, M.J.; Ye, S. A Blood Pressure-Associated Variant of the SLC39A8 Gene Influences Cellular Cadmium Accumulation and Toxicity. Hum. Mol. Genet. 2016, 25, 4117–4126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riley, L.G.; Cowley, M.J.; Gayevskiy, V.; Roscioli, T.; Thorburn, D.R.; Prelog, K.; Bahlo, M.; Sue, C.M.; Balasubramaniam, S.; Christodoulou, J. A SLC39A8 Variant Causes Manganese Deficiency, and Glycosylation and Mitochondrial Disorders. J. Inherit. Metab. Dis. 2017, 40, 261–269. [Google Scholar] [CrossRef] [PubMed]
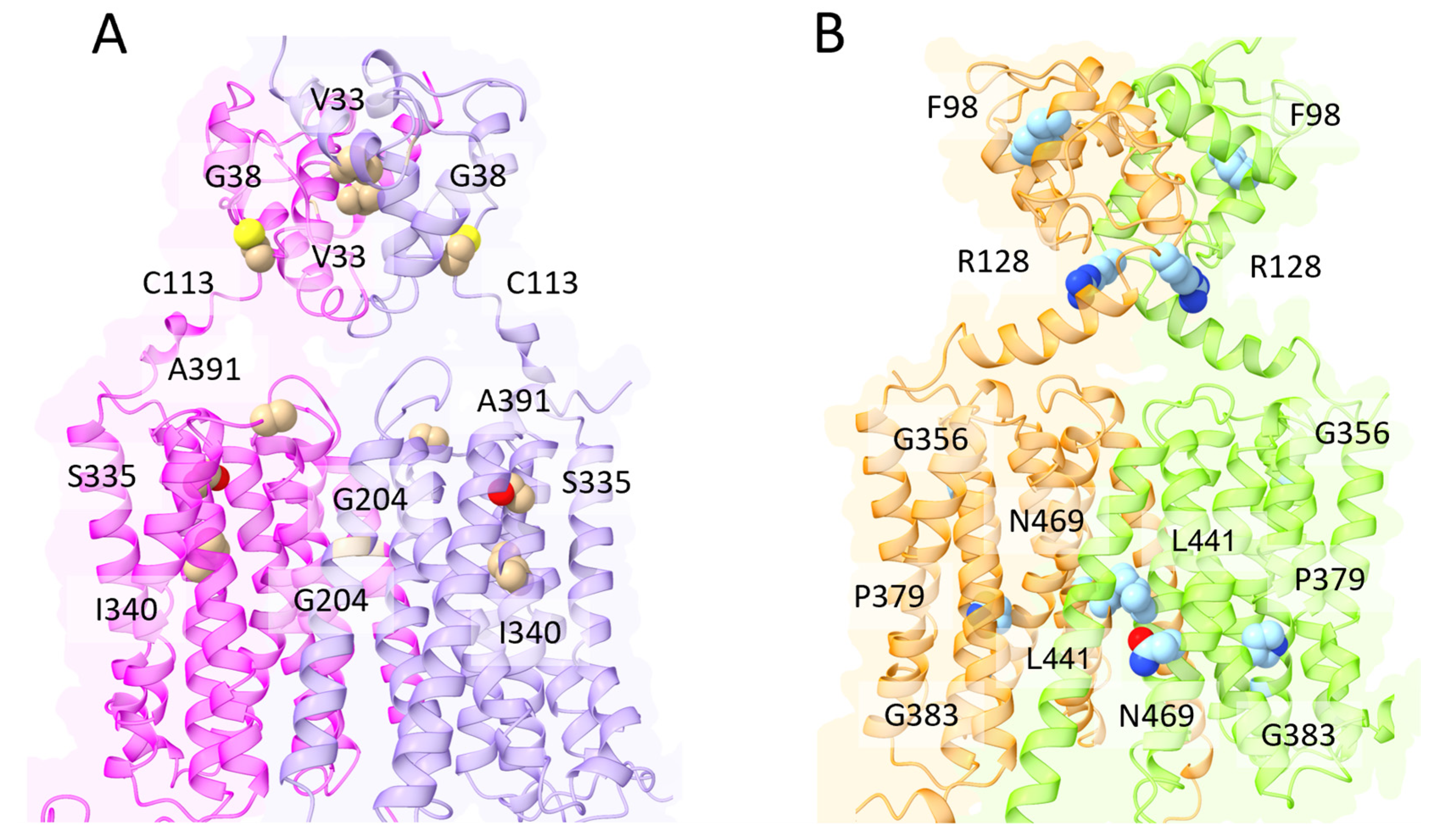
- Liang, Z.-L.; Tan, H.W.; Wu, J.-Y.; Chen, X.-L.; Wang, X.-Y.; Xu, Y.-M.; Lau, A.T.Y. The Impact of ZIP8 Disease-Associated Variants G38R, C113S, G204C, and S335T on Selenium and Cadmium Accumulations: The First Characterization. Int. J. Mol. Sci. 2021, 22, 11399. [Google Scholar] [CrossRef] [PubMed]
- Choi, E.-K.; Nguyen, T.-T.; Gupta, N.; Iwase, S.; Seo, Y.A. Functional Analysis of SLC39A8 Mutations and Their Implications for Manganese Deficiency and Mitochondrial Disorders. Sci. Rep. 2018, 8, 3163. [Google Scholar] [CrossRef] [Green Version]
- Jenkitkasemwong, S.; Wang, C.-Y.; Coffey, R.; Zhang, W.; Chan, A.; Biel, T.; Kim, J.-S.; Hojyo, S.; Fukada, T.; Knutson, M.D. SLC39A14 Is Required for the Development of Hepatocellular Iron Overload in Murine Models of Hereditary Hemochromatosis. Cell Metab. 2015, 22, 138–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheiber, I.F.; Wu, Y.; Morgan, S.E.; Zhao, N. The Intestinal Metal Transporter ZIP14 Maintains Systemic Manganese Homeostasis. J. Biol. Chem. 2019, 294, 9147–9160. [Google Scholar] [CrossRef] [PubMed]
- Jenkitkasemwong, S.; Akinyode, A.; Paulus, E.; Weiskirchen, R.; Hojyo, S.; Fukada, T.; Giraldo, G.; Schrier, J.; Garcia, A.; Janus, C.; et al. SLC39A14 Deficiency Alters Manganese Homeostasis and Excretion Resulting in Brain Manganese Accumulation and Motor Deficits in Mice. Proc. Natl. Acad. Sci. USA 2018, 115, E1769–E1778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tuschl, K.; Meyer, E.; Valdivia, L.E.; Zhao, N.; Dadswell, C.; Abdul-Sada, A.; Hung, C.Y.; Simpson, M.A.; Chong, W.K.; Jacques, T.S.; et al. Mutations in SLC39A14 Disrupt Manganese Homeostasis and Cause Childhood-Onset Parkinsonism–Dystonia. Nat. Commun. 2016, 7, 11601. [Google Scholar] [CrossRef] [Green Version]
- Hendrickx, G.; Borra, V.M.; Steenackers, E.; Yorgan, T.A.; Hermans, C.; Boudin, E.; Waterval, J.J.; Jansen, I.D.C.; Aydemir, T.B.; Kamerling, N.; et al. Conditional Mouse Models Support the Role of SLC39A14 (ZIP14) in Hyperostosis Cranialis Interna and in Bone Homeostasis. PLOS Genet. 2018, 14, e1007321. [Google Scholar] [CrossRef] [Green Version]
- Juneja, M.; Shamim, U.; Joshi, A.; Mathur, A.; Uppili, B.; Sairam, S.; Ambawat, S.; Dixit, R.; Faruq, M. A Novel Mutation in SLC39A14 Causing Hypermanganesemia Associated with Infantile Onset Dystonia. J. Gene Med. 2018, 20, e3012. [Google Scholar] [CrossRef]
- Zeglam, A.; Abugrara, A.; Kabuka, M. Autosomal-Recessive Iron Deficiency Anemia, Dystonia and Hypermanganesemia Caused by New Variant Mutation of the Manganese Transporter Gene SLC39A14. Acta Neurol. Belg. 2019, 119, 379–384. [Google Scholar] [CrossRef] [PubMed]
- Namnah, M.; Bauer, M.; Mor-Shaked, H.; Bressman, S.B.; Raymond, D.; Ozelius, L.J.; Arkadir, D. Benign SLC39A14 Course of Dystonia-Parkinsonism Secondary to Inherited Manganese Accumulation. Mov. Disord. Clin. Pract. 2020, 7, 569–570. [Google Scholar] [CrossRef] [PubMed]
- Waterval, J.J.; Stokroos, R.J.; Bauer, N.J.C.; De Bondt, R.B.J.; Manni, J.J. Phenotypic Manifestations and Management of Hyperostosis Cranialis Interna, a Hereditary Bone Dysplasia Affecting the Calvaria and the Skull Base. Am. J. Med. Genet. Part A 2010, 152A, 547–555. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly Accurate Protein Structure Prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Shaw, G.C.; Cope, J.J.; Li, L.; Corson, K.; Hersey, C.; Ackermann, G.E.; Gwynn, B.; Lambert, A.J.; Wingert, R.A.; Traver, D.; et al. Mitoferrin Is Essential for Erythroid Iron Assimilation. Nature 2006, 440, 96–100. [Google Scholar] [CrossRef]
- Ruprecht, J.J.; Kunji, E.R.S. The SLC25 Mitochondrial Carrier Family: Structure and Mechanism. Trends Biochem. Sci. 2020, 45, 244–258. [Google Scholar] [CrossRef]
- Pierri, C.L.; Palmieri, F.; De Grassi, A. Single-Nucleotide Evolution Quantifies the Importance of Each Site along the Structure of Mitochondrial Carriers. Cell. Mol. Life Sci. 2014, 71, 349–364. [Google Scholar] [CrossRef]
- Robinson, A.J.; Kunji, E.R.S. Mitochondrial Carriers in the Cytoplasmic State Have a Common Substrate Binding Site. Proc. Natl. Acad. Sci. USA 2006, 103, 2617–2622. [Google Scholar] [CrossRef] [PubMed]
- Wiesenberger, G.; Link, T.A.; von Ahsen, U.; Waldherr, M.; Schweyen, R.J. MRS3 and MRS4, Two Suppressors of MtRNA Splicing Defects in Yeast, Are New Members of the Mitochondrial Carrier Family. J. Mol. Biol. 1991, 217, 23–37. [Google Scholar] [CrossRef]
- Christenson, E.T.; Gallegos, A.S.; Banerjee, A. In Vitro Reconstitution, Functional Dissection, and Mutational Analysis of Metal Ion Transport by Mitoferrin-1. J. Biol. Chem. 2018, 293, 3819–3828. [Google Scholar] [CrossRef] [Green Version]
- Hyde, B.B.; Liesa, M.; Elorza, A.A.; Qiu, W.; Haigh, S.E.; Richey, L.; Mikkola, H.K.; Schlaeger, T.M.; Shirihai, O.S. The Mitochondrial Transporter ABC-Me (ABCB10), a Downstream Target of GATA-1, Is Essential for Erythropoiesis in Vivo. Cell Death Differ. 2012, 19, 1117–1126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
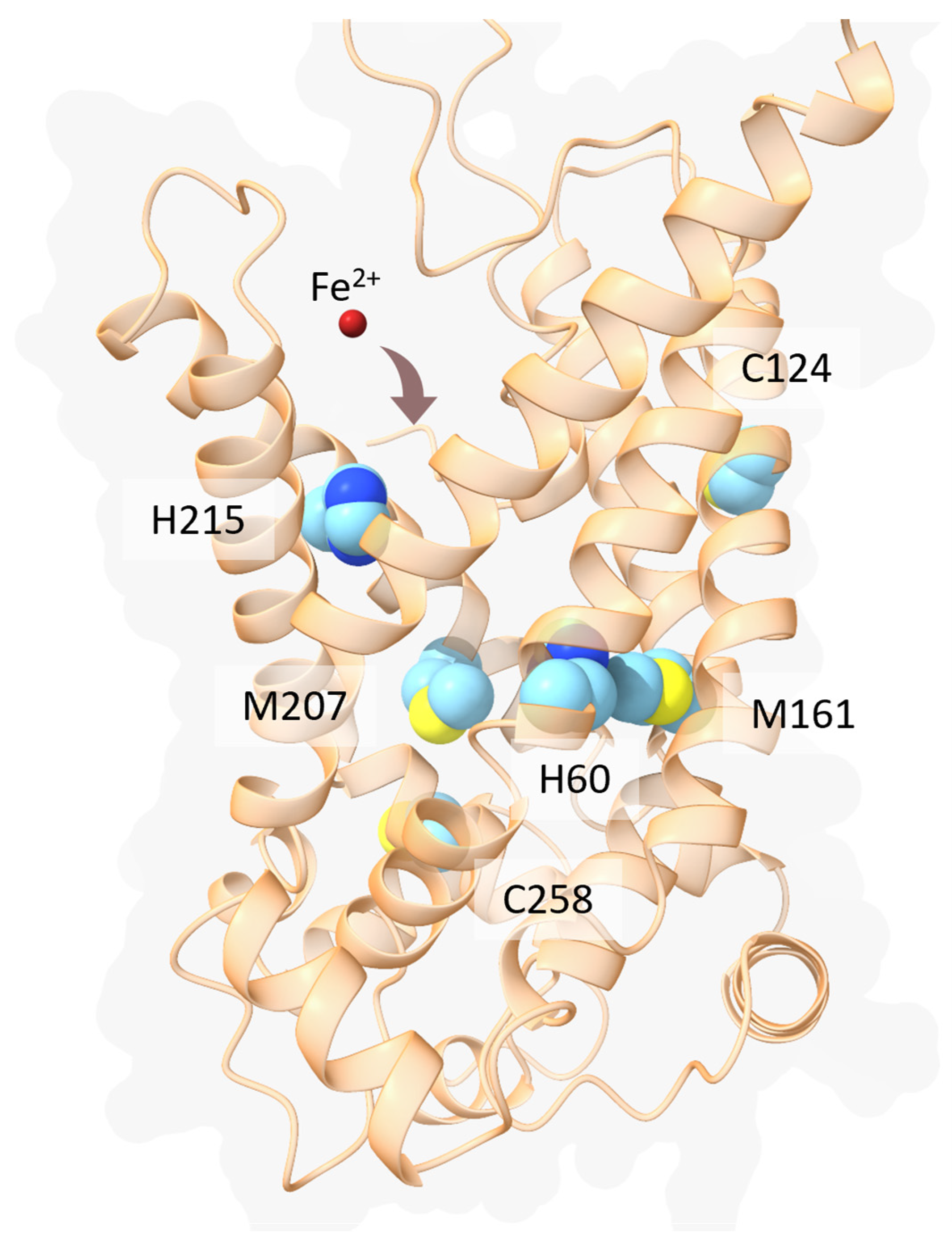
- Brazzolotto, X.; Pierrel, F.; Pelosi, L. Three Conserved Histidine Residues Contribute to Mitochondrial Iron Transport through Mitoferrins. Biochem. J. 2014, 460, 79–92. [Google Scholar] [CrossRef]
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A.; et al. AlphaFold Protein Structure Database: Massively Expanding the Structural Coverage of Protein-Sequence Space with High-Accuracy Models. Nucleic Acids Res. 2022, 50, D439–D444. [Google Scholar] [CrossRef]
- Pinilla-Tenas, J.J.; Sparkman, B.K.; Shawki, A.; Illing, A.C.; Mitchell, C.J.; Zhao, N.; Liuzzi, J.P.; Cousins, R.J.; Knutson, M.D.; Mackenzie, B. Zip14 Is a Complex Broad-Scope Metal-Ion Transporter Whose Functional Properties Support Roles in the Cellular Uptake of Zinc and Nontransferrin-Bound Iron. Am. J. Physiol. -Cell Physiol. 2011, 301, C862–C871. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, C.J.; Shawki, A.; Ganz, T.; Nemeth, E.; Mackenzie, B. Functional Properties of Human Ferroportin, a Cellular Iron Exporter Reactive Also with Cobalt and Zinc. Am. J. Physiol. -Cell Physiol. 2014, 306, C450–C459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Troadec, M.-B.; Warner, D.; Wallace, J.; Thomas, K.; Spangrude, G.J.; Phillips, J.; Khalimonchuk, O.; Paw, B.H.; Ward, D.M.; Kaplan, J. Targeted Deletion of the Mouse Mitoferrin1 Gene: From Anemia to Protoporphyria. Blood 2011, 117, 5494–5502. [Google Scholar] [CrossRef] [PubMed]
- Visconte, V.; Avishai, N.; Mahfouz, R.; Tabarroki, A.; Cowen, J.; Sharghi-Moshtaghin, R.; Hitomi, M.; Rogers, H.J.; Hasrouni, E.; Phillips, J.; et al. Distinct Iron Architecture in SF3B1-Mutant Myelodysplastic Syndrome Patients Is Linked to an SLC25A37 Splice Variant with a Retained Intron. Leukemia 2015, 29, 188–195. [Google Scholar] [CrossRef] [PubMed]
- Huo, Y.-X.; Huang, L.; Zhang, D.-F.; Yao, Y.-G.; Fang, Y.-R.; Zhang, C.; Luo, X.-J. Identification of SLC25A37 as a Major Depressive Disorder Risk Gene. J. Psychiatr. Res. 2016, 83, 168–175. [Google Scholar] [CrossRef]
- Baldauf, L.; Endres, T.; Scholz, J.; Kirches, E.; Ward, D.M.; Lessmann, V.; Borucki, K.; Mawrin, C. Mitoferrin-1 Is Required for Brain Energy Metabolism and Hippocampus-Dependent Memory. Neurosci. Lett. 2019, 713, 134521. [Google Scholar] [CrossRef] [PubMed]
- Navarro, J.A.; Botella, J.A.; Metzendorf, C.; Lind, M.I.; Schneuwly, S. Mitoferrin Modulates Iron Toxicity in a Drosophila Model of Friedreich׳s Ataxia. Free Radic. Biol. Med. 2015, 85, 71–82. [Google Scholar] [CrossRef]
- Huang, J.; Chen, S.; Hu, L.; Niu, H.; Sun, Q.; Li, W.; Tan, G.; Li, J.; Jin, L.; Lyu, J.; et al. Mitoferrin-1 Is Involved in the Progression of Alzheimer’s Disease Through Targeting Mitochondrial Iron Metabolism in a Caenorhabditis Elegans Model of Alzheimer’s Disease. Neuroscience 2018, 385, 90–101. [Google Scholar] [CrossRef] [PubMed]
- Paradkar, P.N.; Zumbrennen, K.B.; Paw, B.H.; Ward, D.M.; Kaplan, J. Regulation of Mitochondrial Iron Import through Differential Turnover of Mitoferrin 1 and Mitoferrin 2. Mol. Cell. Biol. 2009, 29, 1007–1016. [Google Scholar] [CrossRef] [Green Version]
- Seguin, A.; Jia, X.; Earl, A.M.; Li, L.; Wallace, J.; Qiu, A.; Bradley, T.; Shrestha, R.; Troadec, M.-B.; Hockin, M.; et al. The Mitochondrial Metal Transporters Mitoferrin1 and Mitoferrin2 Are Required for Liver Regeneration and Cell Proliferation in Mice. J. Biol. Chem. 2020, 295, 11002–11020. [Google Scholar] [CrossRef] [PubMed]
- Metzendorf, C.; Lind, M.I. Drosophila Mitoferrinis Essential for Male Fertility: Evidence for a Role of Mitochondrial Iron Metabolism during Spermatogenesis. BMC Dev. Biol. 2010, 10, 68. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Ye, P.; Kong, C.; Chao, Y.; Yu, W.; Jiang, X.; Luo, J.; Gu, Y.; Chen, S.-L. Mitoferrin 2 Deficiency Prevents Mitochondrial Iron Overload-Induced Endothelial Injury and Alleviates Atherosclerosis. Exp. Cell Res. 2021, 402, 112552. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, S.; Fox, J.; Thyagarajan, B.; Fox, J.H. Brain Mitochondrial Iron Accumulates in Huntington’s Disease, Mediates Mitochondrial Dysfunction, and Can Be Removed Pharmacologically. Free Radic. Biol. Med. 2018, 120, 317–329. [Google Scholar] [CrossRef]
- Hung, H.-I.; Schwartz, J.M.; Maldonado, E.N.; Lemasters, J.J.; Nieminen, A.-L. Mitoferrin-2-Dependent Mitochondrial Iron Uptake Sensitizes Human Head and Neck Squamous Carcinoma Cells to Photodynamic Therapy*. J. Biol. Chem. 2013, 288, 677–686. [Google Scholar] [CrossRef] [Green Version]
- Tomita, K.; Fukumoto, M.; Itoh, K.; Kuwahara, Y.; Igarashi, K.; Nagasawa, T.; Suzuki, M.; Kurimasa, A.; Sato, T. MiR-7-5p Is a Key Factor That Controls Radioresistance via Intracellular Fe2+ Content in Clinically Relevant Radioresistant Cells. Biochem. Biophys. Res. Commun. 2019, 518, 712–718. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pasquadibisceglie, A.; Bonaccorsi di Patti, M.C.; Musci, G.; Polticelli, F. Membrane Transporters Involved in Iron Trafficking: Physiological and Pathological Aspects. Biomolecules 2023, 13, 1172. https://doi.org/10.3390/biom13081172
Pasquadibisceglie A, Bonaccorsi di Patti MC, Musci G, Polticelli F. Membrane Transporters Involved in Iron Trafficking: Physiological and Pathological Aspects. Biomolecules. 2023; 13(8):1172. https://doi.org/10.3390/biom13081172
Chicago/Turabian StylePasquadibisceglie, Andrea, Maria Carmela Bonaccorsi di Patti, Giovanni Musci, and Fabio Polticelli. 2023. "Membrane Transporters Involved in Iron Trafficking: Physiological and Pathological Aspects" Biomolecules 13, no. 8: 1172. https://doi.org/10.3390/biom13081172