
Evolutionary Study of Protein Short Tandem Repeats in Protein Families



{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
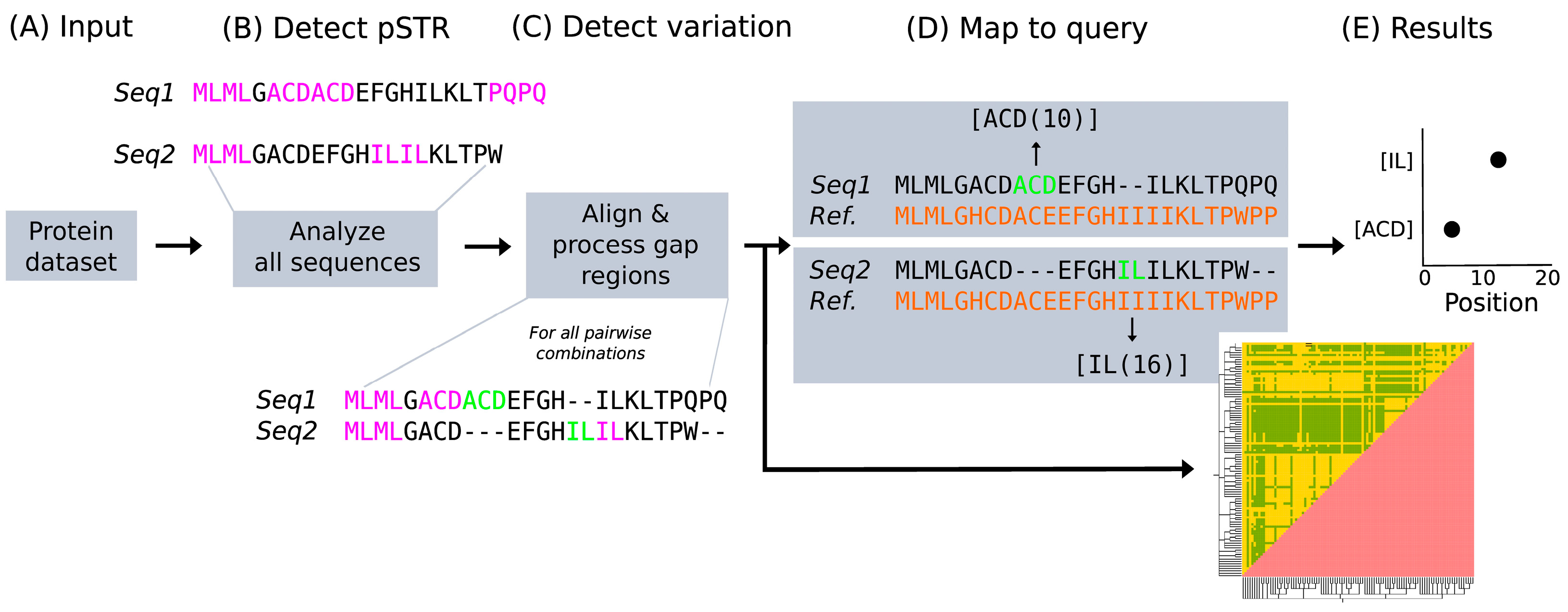
2. Materials and Methods
3. Results
3.1. Protein Short Tandem Repeats Search Strategy
3.2. Comparison of pSTRs in Metazoans
3.3. Analysis of pSTR Unit Variation
3.4. Web Tool to Search for pSTR Unit Variation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mier, P.; Paladin, L.; Tamana, S.; Petrosian, S.; Hajdu-Soltész, B.; Urbanek, A.; Gruca, A.; Plewczynski, D.; Grynberg, M.; Bernadó, P.; et al. Disentangling the complexity of low complexity proteins. Brief. Bioinform. 2020, 21, 458–472. [Google Scholar] [CrossRef] [PubMed]
- Marcotte, E.M.; Pellegrini, M.; Yeates, T.O.; Eisenberg, D. A census of protein repeats. J. Mol. Biol. 1999, 293, 151–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jorda, J.; Kajava, A.V. Protein homorepeats sequences, structures, evolution, and functions. Adv. Protein Chem. Struct. Biol. 2010, 79, 59–88. [Google Scholar]
- Kajava, A.V. Tandem repeats in proteins: From sequence to structure. J. Struct. Biol. 2012, 179, 279–288. [Google Scholar] [CrossRef] [PubMed]
- Andrade, M.A.; Perez-Iratxeta, C.; Ponting, C.P. Protein repeats: Structures, functions, and evolution. J. Struct. Biol. 2001, 134, 117–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrade, M.A.; Ponting, C.P.; Gibson, T.J.; Bork, P. Homology-based method for identification of protein repeats using statistical significance estimates. J. Mol. Biol. 2000, 298, 521–537. [Google Scholar] [CrossRef]
- Jorda, J.; Kajava, A.V. T-REKS: Identification of Tandem REpeats in sequences with a K-meanS based algorithm. Bioinformatics 2009, 25, 2632–2638. [Google Scholar] [CrossRef] [Green Version]
- Biegert, A.; Söding, J. De novo identification of highly diverged protein repeats by probabilistic consistency. Bioinformatics 2008, 24, 807–814. [Google Scholar] [CrossRef] [Green Version]
- Walsh, I.; Sirocco, F.G.; Minervini, G.; Di Domenico, T.; Ferrari, C.; Tosatto, S.C.E. RAPHAEL: Recognition, periodicity and insertion assignment of solenoid protein structures. Bioinformatics 2012, 28, 3257–3264. [Google Scholar] [CrossRef] [Green Version]
- Viet, P.D.; Roche, D.B.; Kajava, A.V. TAPO: A combined method for the identification of tandem repeats in protein structures. FEBS Lett. 2015, 589, 2611–2619. [Google Scholar] [CrossRef] [Green Version]
- Bolognini, D.; Magi, A.; Benes, V.; Korbel, J.O.; Rausch, T. TRiCoLOR: Tandem repeat profiling using whole-genome long-read sequencing data. Gigascience 2020, 9, giaa101. [Google Scholar] [CrossRef] [PubMed]
- Kamel, M.; Kastano, K.; Mier, P.; Andrade-Navarro, M.A. REP2: A Web Server to Detect Common Tandem Repeats in Protein Sequences. J. Mol. Biol. 2021, 433, 166895. [Google Scholar] [CrossRef] [PubMed]
- Paladin, L.; Bevilacqua, M.; Errigo, S.; Piovesan, D.; Mičetić, I.; Necci, M.; Monzon, A.M.; Fabre, M.L.; Lopez, J.L.; Nilsson, J.F.; et al. RepeatsDB in 2021: Improved data and extended classification for protein tandem repeat structures. Nucleic Acids Res. 2020, 49, D452–D457. [Google Scholar] [CrossRef]
- Hirsh, L.; Paladin, L.; Piovesan, D.; Tosatto, S.C.E. RepeatsDB-lite: A web server for unit annotation of tandem repeat proteins. Nucleic Acids Res. 2018, 46, W402–W407. [Google Scholar] [CrossRef] [Green Version]
- Albà, M.M.; Santibáñez-Koref, M.F.; Hancock, J.M. The comparative genomics of polyglutamine repeats: Extreme differences in the codon organization of repeat-encoding regions between mammals and Drosophila. J. Mol. Biol. 2001, 52, 249–259. [Google Scholar] [CrossRef]
- Shoubridge, C.; Gecz, J. Polyalanine tract disorders and neurocogniive phynotypes. In Madame Curie Bioscience Database; Landes Bioscience: Austin, TX, USA, 2011. Available online: https://www.ncbi.nlm.nih.gov/books/NBK51932/ (accessed on 5 July 2023).
- Viguera, E.; Canceill, D.; Ehrlich, S. Replication slippage involves DNA polymerase pausing and dissociation. EMBO J. 2001, 20, 2587–2595. [Google Scholar] [CrossRef] [Green Version]
- Kamel, M.; Mier, P.; Tari, A.; Andrade-Navarro, M.A. Repeatability in protein sequences. J. Struct. Biol. 2019, 208, 86–91. [Google Scholar] [CrossRef]
- Schaefer, M.H.; Wanker, E.E.; Andrade-Navarro, M.A. Evolution and function of CAG/polyglutamine repeats in protein–protein interaction networks. Nucleic Acids Res. 2012, 40, 4273–4287. [Google Scholar] [CrossRef] [Green Version]
- Chong, P.A.; Vernon, R.M.; Forman-Kay, J.D. RGG/RG Motif Regions in RNA Binding and Phase Separation. J. Mol. Biol. 2018, 430, 4650–4665. [Google Scholar] [CrossRef]
- Reddy, A.S.N.; Ali, G.S. Plant serine/arginine-rich proteins: Roles in precursor messenger RNA splicing, plant development, and stress responses. Wiley Interdiscip. Rev. RNA 2011, 2, 875–889. [Google Scholar] [CrossRef]
- Emms, D.M.; Kelly, S. Benchmarking Orthogroup Inference Accuracy: Revisiting Orthobench. Genome Biol. Evol. 2020, 12, 2258–2266. [Google Scholar] [CrossRef] [PubMed]
- UniProt Consortium. UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [Green Version]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mier, P.; Elena-Real, C.; Urbanek, A.; Bernadó, P.; Andrade-Navarro, M.A. The importance of definitions in the study of polyQ regions: A tale of thresholds, impurities and sequence context. Comput. Struct. Biotechnol. J. 2020, 18, 306–313. [Google Scholar] [CrossRef] [PubMed]
- Kastano, K.; Mier, P.; Dosztányi, Z.; Promponas, V.J.; Andrade-Navarro, M.A. Functional Tuning of Intrinsically Disordered Regions in Human Proteins by Composition Bias. Biomolecules 2022, 12, 1486. [Google Scholar] [CrossRef]
- Farahi, N.; Lazar, T.; Wodak, S.J.; Tompa, P.; Pancsa, R. Integration of Data from Liquid–Liquid Phase Separation Databases Highlights Concentration and Dosage Sensitivity of LLPS Drivers. Int. J. Mol. Sci. 2021, 22, 3017. [Google Scholar] [CrossRef]
- Hardenberg, M.; Horvath, A.; Ambrus, V.; Fuxreiter, M.; Vendruscolo, M. Widespread occurrence of the droplet state of proteins in the human proteome. Proc. Natl. Acad. Sci. USA 2020, 117, 33254–33262. [Google Scholar] [CrossRef]
- Piovesan, D.; Del Conte, A.; Clementel, D.; Monzon, A.M.; Bevilacqua, M.; Aspromonte, M.C.; Iserte, J.A.; Orti, F.E.; Marino-Buslje, C.; Tosatto, S.C.E. MobiDB: 10 years of intrinsically disordered proteins. Nucleic Acids Res. 2023, 51, D438–D444. [Google Scholar] [CrossRef]
- Huang, S.; Zhu, S.; Kumar, P.; MacMicking, J.D. A phase-separated nuclear GBPL circuit controls immunity in plants. Nature 2021, 594, 424–429. [Google Scholar] [CrossRef]
- Zhu, S.; Gu, J.; Yao, J.; Li, Y.; Zhang, Z.; Xia, W.; Wang, Z.; Gui, X.; Li, L.; Li, D.; et al. Liquid-liquid phase separation of RBGD2/4 is required for heat stress resistance in Arabidopsis. Dev. Cell 2022, 57, 583–597. [Google Scholar] [CrossRef]
- Chin, L.-S.; Olzmann, J.A.; Li, L. Parkin-mediated ubiquitin signalling in aggresome formation and autophagy. Biochem. Soc. Trans. 2010, 38, 144–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [PubMed] [Green Version]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mier, P.; Andrade-Navarro, M.A. Evolutionary Study of Protein Short Tandem Repeats in Protein Families. Biomolecules 2023, 13, 1116. https://doi.org/10.3390/biom13071116
Mier P, Andrade-Navarro MA. Evolutionary Study of Protein Short Tandem Repeats in Protein Families. Biomolecules. 2023; 13(7):1116. https://doi.org/10.3390/biom13071116
Chicago/Turabian StyleMier, Pablo, and Miguel A. Andrade-Navarro. 2023. "Evolutionary Study of Protein Short Tandem Repeats in Protein Families" Biomolecules 13, no. 7: 1116. https://doi.org/10.3390/biom13071116