
Targeted Destruction of S100A4 Inhibits Metastasis of Triple Negative Breast Cancer Cells
 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Inhibition of Binding of S100A4 to NMIIA
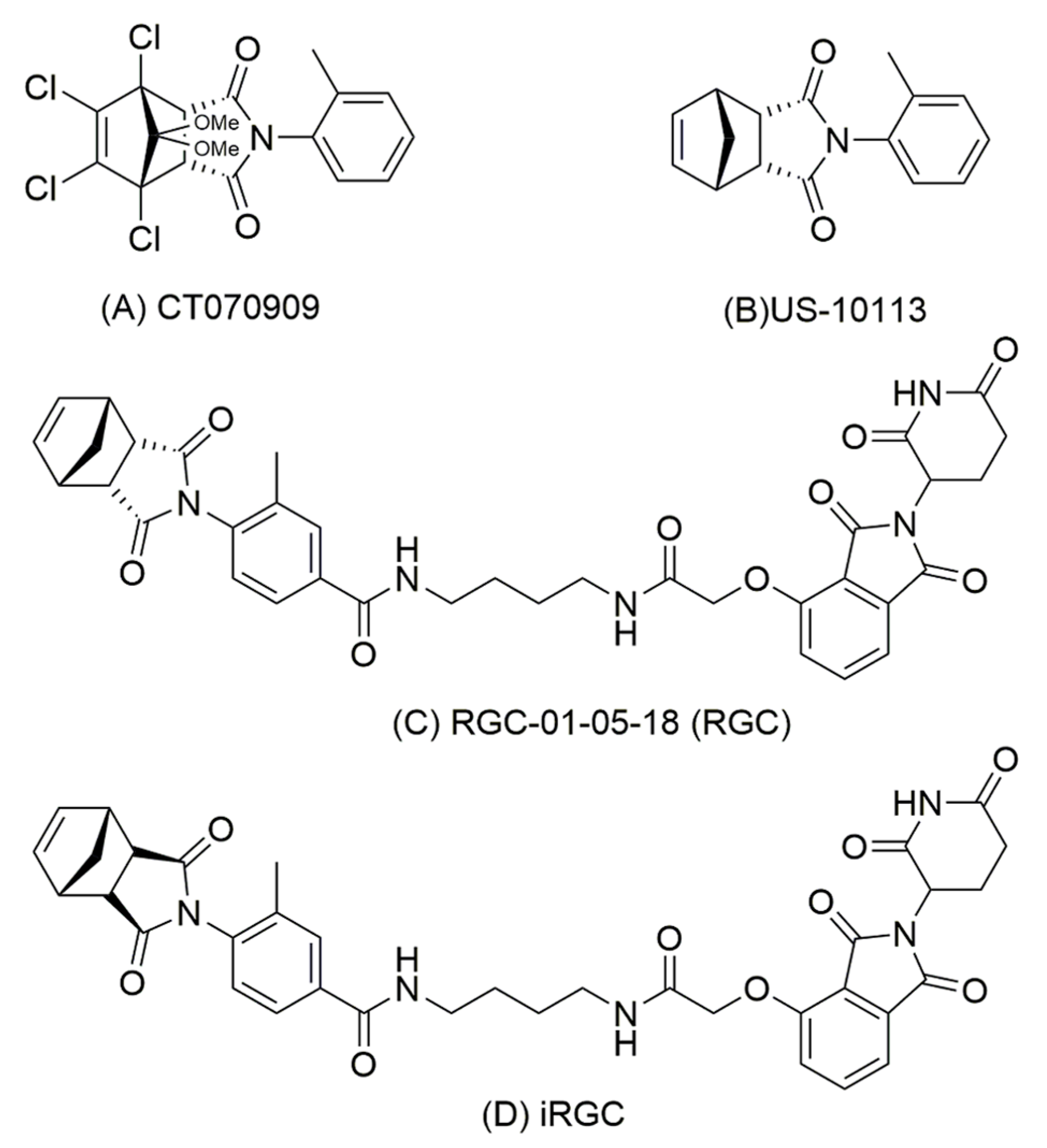
2.2. Chemical Synthesis
2.3. Cell Culture
2.4. Measurement of Cell Migration/Invasion
2.5. Measurement of Cytotoxicity
2.6. Western Blotting and Quantifications
2.7. Depletion of S100A4 in MDA-MB-231 Cells
2.8. In Vivo Metastasis Assay
2.9. Data Analysis
3. Results
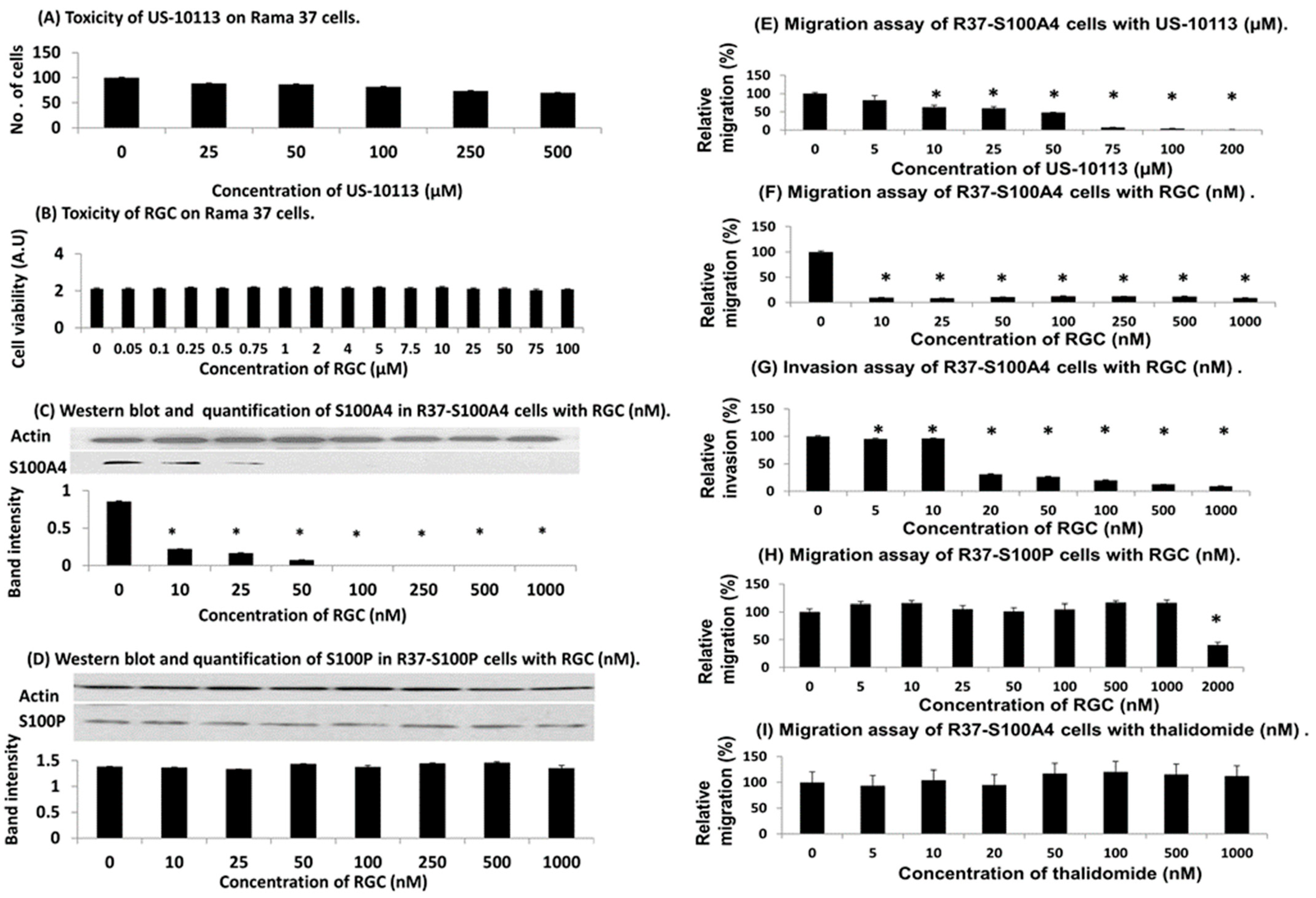
3.1. Identification of Inhibitor of S100A4 Binding to NMIIA and Migration of Rat Mammary Cells
3.2. PROTAC Modification of Inhibitor in Rat Cell Lines
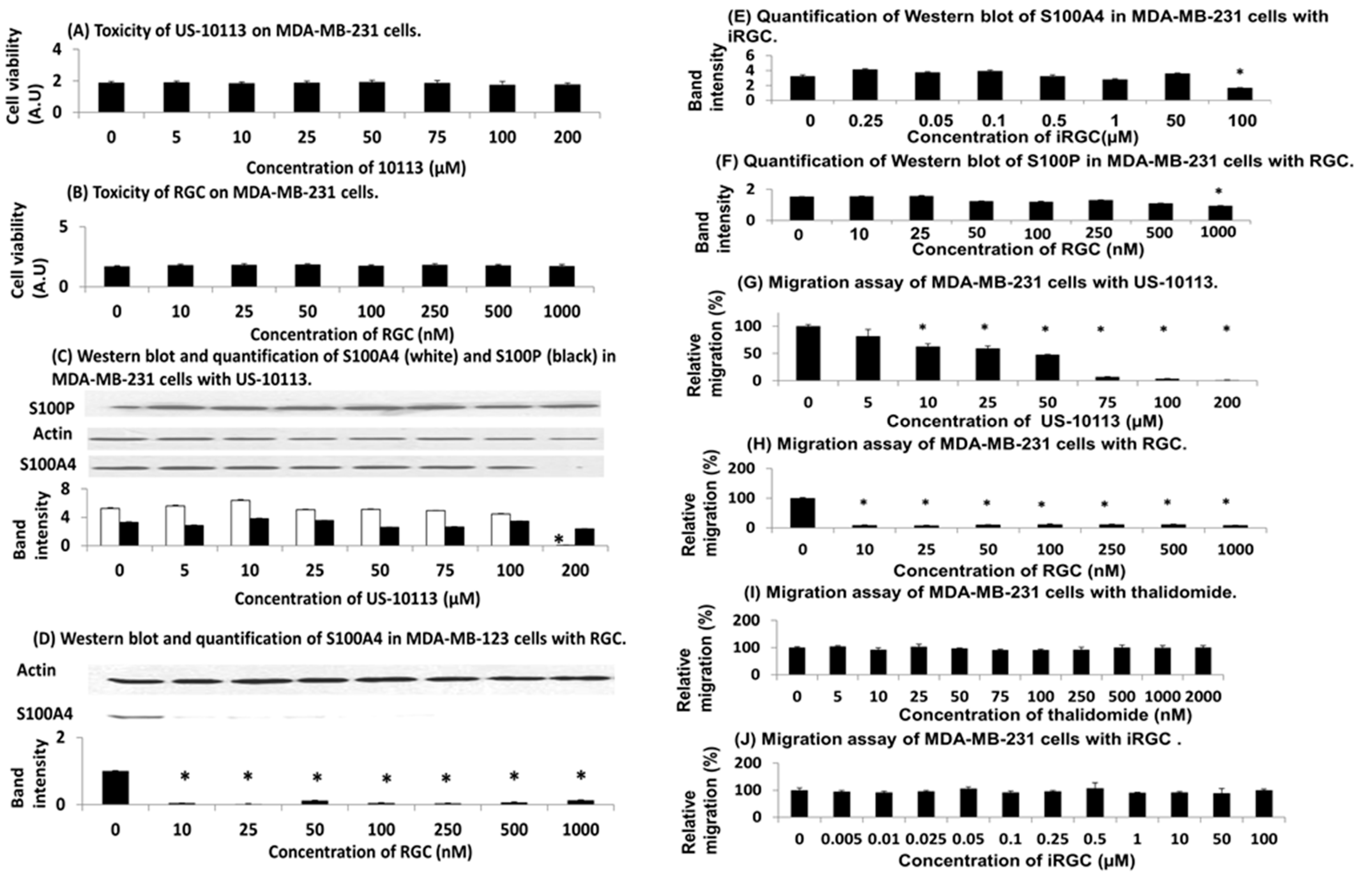
3.3. Effect of Inhibitor and PROTAC on Human TNBC Cell Line
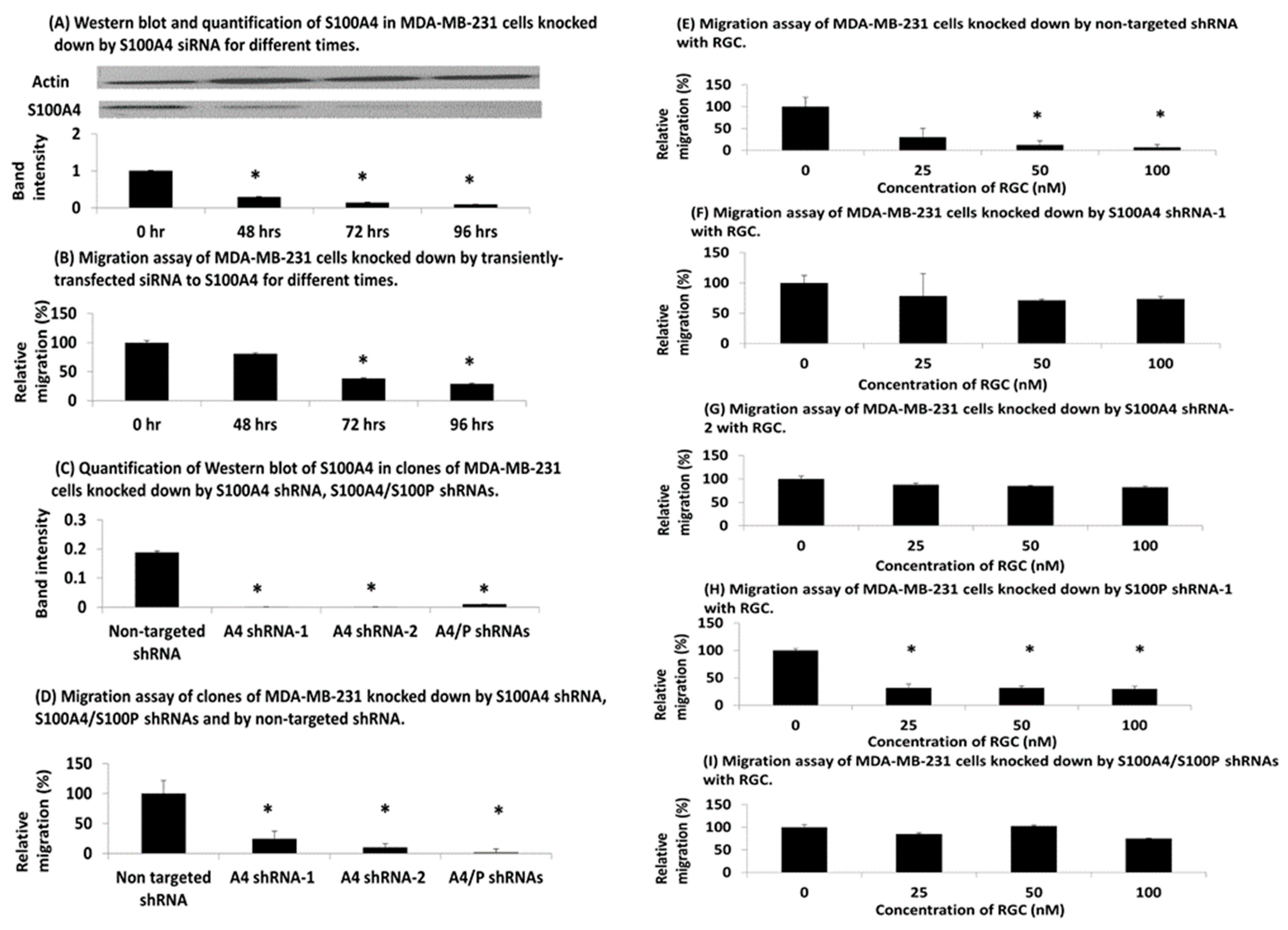
3.4. Knockdown of S100A4 in Human TNBC Cells
3.5. Effect of PROTAC on Metastasis in TNBC Mouse Model
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- International Association of Cancer Research. WHO Globocan. Breast Cancer. 2020. Available online: https://glo.iacr.fr/today/factsheets/cancers/20-fact-sheet.pdf (accessed on 22 November 2022).
- Dawood, S. Triple negative breast cancer. Drugs 2010, 70, 2247–2258. [Google Scholar] [CrossRef]
- Dent, R.; Hanna, M.; Trudeau, M.; Rawlinson, E.; Sun, P.; Norod, S.A. Pattern of meta-static spread in triple negative breast cancer. Breast Cancer Res. Treat. 2009, 115, 423–428. [Google Scholar] [CrossRef] [PubMed]
- Gupta, G.P.; Massagué, J. Cancer metastasis: Building a framework. Cell 2006, 127, 679–695. [Google Scholar] [CrossRef] [Green Version]
- Dunnington, D.J.; Hughes, C.M.; Monaghan, P.; Rudland, P.S. Phenotypic instability of rat mammary tumor epithelial cells. J. Natl. Cancer Inst. 1983, 71, 1227–1240. [Google Scholar] [PubMed]
- Dunnington, D.J.; Kim, U.; Hughes, C.M.; Monaghan, P.; Ormerod, E.J.; Rudland, P.S. Loss of myoepithelial cell characteristics in metastasizing rat mammary tumors relative to their nonmetastasizing counterparts. J. Natl. Cancer Inst. 1984, 72, 455–466. [Google Scholar] [PubMed]
- Steeg, P.S. Tumor metastasis: Mechanistic insights and clinical challenges. Nat. Med. 2006, 12, 895–904. [Google Scholar] [CrossRef]
- Davies, B.R.; Davies, M.P.; Gibbs, F.E.; Barraclough, R.; Rudland, P.S. Induction of the metastatic phenotype by transfection of a benign rat mammary epithelial cell line with the gene for p9ka, a rat calcium-binding protein, but not with the oncogene EJ-ras-1. Oncogene 1993, 82, 999–1008. [Google Scholar]
- Barraclough, R.; Savin, J.; Dube, S.K.; Rudland, P.S. Molecular cloning and sequence of the gene for p9ka a cultured myoepithelial cell protein with strong homology to S-100, a calcium-binding protein. J. Mol. Biol. 1987, 198, 13–20. [Google Scholar] [CrossRef]
- Rudland, P.S.; Platt-Higgins, A.; Renshaw, C.; West, C.R.; Winstanley, J.H.; Robertson, L.; Barraclough, R. Prognostic significance of the metastasis-inducing protein S100A4 (p9ka) in human breast cancer. Cancer Res. 2000, 60, 1595–1603. [Google Scholar]
- Zhao, Y.; Zhang, T.; Wang, Q. S100 calcium binding protein A4 is a novel independent prognostic factor for the poor prognosis of gastric carcinomas. Oncol. Rep. 2013, 30, 111–118. [Google Scholar] [CrossRef]
- Mazzucchelli, L. Protein S100A4: Too long overlooked by pathologists? Am. J. Pathol. 2002, 160, 7–13. [Google Scholar] [CrossRef] [Green Version]
- Salama, I.; Malone, P.S.; Mihaimeed, F.; Jones, J.L. A review of the S100 proteins in cancer. Europ. J. Surg. Oncol. 2008, 34, 357–364. [Google Scholar] [CrossRef]
- Bresnick, A.R.; Weber, D.J.; Zimmer, D.B. S100 proteins in cancer. Nat. Rev. Cancer 2015, 15, 96–109. [Google Scholar] [CrossRef] [Green Version]
- Davies, M.P.; Rudland, P.S.; Robertson, L.; Parry, E.W.; Jolicoeur, P.; Barraclough, R. Expression of the calcium-binding protein S100A4 (p9ka) in MMTV-neu transgenic mice induces metastasis of mammary tumours. Oncogene 1996, 13, 1631–1637. [Google Scholar]
- Ismail, T.M.; Bennett, D.; Platt-Higgins, A.M.; Al-Medhity, M.; Barraclough, R.; Rudland, P.S. Elevation of S100A4 empowers expression of metastasis effector molecules in human breast cancer. Cancer Res. 2017, 77, 780–789. [Google Scholar] [CrossRef] [Green Version]
- Gross, S.R.; Sin, C.G.; Barraclough, R.; Rudland, P.S. Joining S100 proteins and migration: For better or for worse, in sickness and in health. Cell. Mol. Life Sci. 2014, 71, 1551–1579. [Google Scholar] [CrossRef] [Green Version]
- Mathews, C.K.; van Holden, K.E.; Ahern, K.G. Signal transduction, oncogenes and cancer. In Biochemistry, 3rd ed.; Addison, Wesely and Longman Inc.: San Francisco, CA, USA, 2000; pp. 861–868. [Google Scholar]
- Fei, F.; Qu, J.; Zhang, M.; Li, Y.; Zhang, S. S100A4 in cancer progression and metastasis: A systematic review. Oncotarget 2017, 8, 73219–73239. [Google Scholar] [CrossRef] [Green Version]
- Bresnick, A.R. S100 proteins as therapeutic targets. Biophys. Rev. 2018, 10, 1617–1629. [Google Scholar] [CrossRef]
- Pollard, T.D.; Earnshaw, W.C. Membrane pumps. In Cell Biology; Elsevier Science: Philadelphia, PA, USA, 2002; pp. 105–116. [Google Scholar]
- Walter, I.; Wolfesberger, B.; Miller, I.; Mair, G.; Burger, S.; Gallè, B.; Steinborn, R. Human osteosarcoma cells respond to sorafenib chemotherapy by downregulation of the tumor progression factors S100A4, CXCR4 and the oncogene FOS. Oncol. Rep. 2014, 31, 1147–1156. [Google Scholar] [CrossRef] [Green Version]
- Sack, U.; Walther, W.; Scudiero, D.; Selby, M.; Kobelt, D.; Lemm, M.; Fichtner, I.; Schlag, P.M.; Shoemaker, R.H.; Stein, U. Novel effect of antihelminthic Niclosamide on S100A4-mediated metastatic progression in colon cancer. J. Natl. Cancer Inst. 2011, 103, 1018–1036. [Google Scholar] [CrossRef] [Green Version]
- Sack, U.; Walther, W.; Scudiero, D.; Selby, M.; Aumann, J.; Lemos, C.; Fichtner, I.; Schlag, P.M.; Shoemaker, R.H.; Stein, U. S100A4-induced cell motility and metastasis is restricted by the Wnt/β-catenin pathway inhibitor calcimycin in colon cancer cells. Mol. Biol. Cell 2011, 22, 3344–3354. [Google Scholar] [CrossRef] [PubMed]
- Capoccia, E.; Cirillo, C.; Marchetto, A.; Tiberi, S.; Sawikr, Y.; Pesce, M.; D’Alessandro, A.; Scuderi, C.; Sarnelli, G.; Cuomo, R.; et al. S100B-p53 disengagement by pentamidine promotes apoptosis and inhibits cellular migration via aquaporin-4 and metalloproteinase-2 inhibition in C6 glioma cells. Oncol. Lett. 2015, 9, 2864–2870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKnight, L.E.; Raman, E.P.; Bezawada, P.; Kudrimoti, S.; Wilder, P.T.; Hartman, K.G.; Godoy-Ruiz, R.; Toth, E.A.; Coop, A.; MacKerell, A.D.; et al. Structure-based discovery of a novel pentamidine-related inhibitor of the calcium-binding protein S100B. ACS Med. Chem. Lett. 2012, 3, 975–979. [Google Scholar] [CrossRef]
- Markowitz, J.; Chen, I.; Gitti, R.; Baldisseri, D.M.; Pan, Y.; Udan, R.; Carrier, F.; MacKerell, A.D.; Weber, D.J. Identification and characterization of small molecule inhibitors of the calcium-dependent S100B-p53 tumor suppressor interaction. J. Med. Chem. 2004, 47, 5085–5093. [Google Scholar] [CrossRef] [PubMed]
- Kriajevska, M.V.; Cardenas, M.N.; Grigorian, M.S.; Ambartsumian, N.S.; Georgiev, G.P.; Lukanidin, E.M. Nonmuscle myosin heavy chain as a possible target for protein encoded by metastasis-related mts-1 gene. J. Biol. Chem. 1994, 269, 19679–19682. [Google Scholar] [CrossRef]
- Jenkinson, S.R.; Barraclough, R.; West, C.R.; Rudland, P.S. S100A4 regulates cell motility and invasion in an in vitro model for breast cancer metastasis. Br. J. Cancer 2004, 90, 253–262. [Google Scholar] [CrossRef] [Green Version]
- Ito, T.; Ando, H.; Suzuki, T.; Ogura, T.; Hotta, K.; Imamura, Y.; Yamaguchi, Y.; Handa, H. Identification of a primary target of thalidomide teratogenicity. Science 2010, 327, 1345–1350. [Google Scholar] [CrossRef] [Green Version]
- Lu, G.; Middleton, R.E.; Sun, H.; Naniong, M.; Ott, C.J.; Mitsiades, C.S.; Wong, K.-K.; Bradner, J.E.; Kaelin, W.G., Jr. The myeloma drug lenalidomide promotes the cereblon-dependent destruction of Ikaros proteins. Science 2014, 343, 305–309. [Google Scholar] [CrossRef] [Green Version]
- Winter, G.E.; Buckley, D.L.; Paulk, J.; Roberts, J.M.; Souza, A.; Dhe-Paganon, S.; Bradner, J.E. Drug development. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science 2015, 48, 1376–1381. [Google Scholar] [CrossRef] [Green Version]
- Lloyd, B.H.; Platt-Higgins, A.; Rudland, P.; Barraclough, R. Human S100A4 (p9ka) induces the metastatic phenotype upon benign tumour cells. Oncogene 1998, 17, 465–473. [Google Scholar] [CrossRef] [Green Version]
- Rudland, P.S.; Platt-Higgins, A.M.; Davies, L.M.; de Silva Rudland, S.; Wilson, J.B.; Aladwani, A.; Winstanley, J.H.; Barraclough, D.L.; Barraclough, R.; West, C.R.; et al. Significance of the Fanconi Anemia FANCD2 protein in sporadic and metastatic human breast cancer. Am. J. Pathol. 2010, 176, 2935–2947. [Google Scholar] [CrossRef]
- DuPres, S.A.; Redelman, D.; Hunter, K.W. The mouse mammary carcinoma 4T1: Characterisation of the cellular landscape of primary tumours and metastatic tumour foci. Exptl Pathol 2007, 88, 351–360. [Google Scholar] [CrossRef]
- Liu, Y.; Geng, Y.-H.; Yang, H.; Yang, H.; Zhou, Y.-T.; Zhang, H.-Q.; Tian, X.-X.; Fang, W.-G. Extracellular ATP drives breast cancer cell migration and metastasis via S100A4 production by cancer cells and fibroblasts. Cancer Lett. 2018, 430, 1–10. [Google Scholar] [CrossRef]
- Fernig, D.G. Optical biosensor techniques to analyze protein-polysaccharide interactions. Methods Mol. Biol. 2001, 171, 505–518. [Google Scholar]
- Ismail, T.M.; Fernig, D.G.; Rudland, P.S.; Terry, C.J.; Wang, G.; Barraclough, R. The basic C-terminal amino acids of calcium-binding protein S100A4 promote metastasis. Carcinogenesis 2008, 29, 2259–2266. [Google Scholar] [CrossRef] [Green Version]
- Li, P.; Hwang, J.; Maier, J.M.; Zhao, C.; Kaborda, D.V.; Smith, M.D.; Pellechia, P.J.; Shimizu, K.D. Correlation between solid-state and solution conformational ratios in a series of n-(o-tolyl) succinimide molecular rotors. Cryst. Growth Des. 2015, 15, 3561–3564. [Google Scholar] [CrossRef]
- Fischer, E.S.; Böhm, K.; Lydeard, J.R.; Yang, H.; Stadler, M.B.; Cavadini, S.; Nagel, J.; Serluca, F.; Acker, V.; Lingaraju, G.M.; et al. Structure of the DDB1-CRBN E3 ubiquitin ligase in complex with thalidomide. Nature 2014, 512, 49–53. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.; Platt-Higgins, A.; Carroll, J.; de Silva Rudland, S.; Winstanley, J.; Barraclough, R.; Rudland, P.S. Induction of metastasis by S100P in a rat mammary model and its association with poor survival of breast cancer patients. Cancer Res. 2006, 66, 1199–1207. [Google Scholar] [CrossRef] [Green Version]
- Ismail, T.M.; Zhang, S.; Fernig, D.G.; Gross, S.; Martin-Fernandez, M.L.; See, V.; Tozawa, K.; Tynan, C.J.; Wang, G.; Wilkinson, M.C.; et al. Self-association of calcium binding protein S100A4 and metastasis. J. Biol. Chem. 2010, 285, 914–922. [Google Scholar] [CrossRef] [Green Version]
- Ismail, T.M.; Gross, S.R.; Lancaster, T.; Rudland, P.S.; Barraclough, R. The role of C-terminal lysine of S100P in S100P-induced cell migration and metastasis. Biomolecules 2021, 11, 1471. [Google Scholar] [CrossRef]
- Neve, R.M.; Chin, K.; Fridlyand, J.; Yeh, J.; Beahner, F.L.; Fevr, T.; Clark, I.; Bayani, N.; Coppe, J.-P.; Tong, F.; et al. A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell 2006, 10, 515–527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dukhanina, E.A.; Portseva, T.M.; Kotnova, A.P.; Pankratova, E.V.; Georgieva, S.G. The expression level of S100A4 protein affects the migration activity of breast cancer cells. Dokl. Biochem. Biophys. 2019, 485, 104–106. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Zhang, S.; Fernig, D.G.; Martin-Fernandez, M.; Rudland, P.S.; Barraclough, R. Mutually antagonistic actions of S100A4 and S100A1 on normal and metastatic phenotypes. Oncogene 2005, 24, 1445–1454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.; Wang, G.; Liu, D.; Bao, Z.; Fernig, D.; Rudland, P.; Barraclough, R. The C-terminal region of S100A4 is important for its metastasis-inducing properties. Oncogene 2005, 24, 4401–4411. [Google Scholar] [CrossRef] [Green Version]
- Gibbs, F.E.; Barraclough, R.; Platt-Higgins, A.; Rudland, P.S.; Wilkinson, M.C.; Parry, E.W. Immunocytochemical distribution of the calcium-binding protein p9Ka in normal rat tissues: Variation in the cellular location in different tissues. J. Histochem. Cytochem. 1995, 43, 169–180. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Wang, G.; Ding, Y.; Wang, Z.; Barraclough, R.; Rudland, P.S.; Fernig, D.G.; Rao, Z. The crystal structure at 2Å resolution of the Ca+2-binding protein S100P. J. Mol. Biol. 2003, 325, 785–794. [Google Scholar] [CrossRef]
- Du, M.; Wang, G.; Ismail, T.M.; Gross, S.; Fernig, D.G.; Barraclough, R.; Rudland, P.S. S100P dissociates myosin IIA filaments and focal adhesion sites to reduce cell adhesion and enhance cell migration. J. Biol. Chem. 2012, 287, 15330–15344. [Google Scholar] [CrossRef] [Green Version]
- Chan, K.H.; Zengerle, M.; Testa, A.; Ciulli, A. Impact of target warhead and linkage vector on inducing protein degradation. J. Med. Chem. 2018, 61, 504–513. [Google Scholar] [CrossRef] [Green Version]
- Miranda, K.J.; Loeser, R.F.; Yammani, R.R. Sumoylation and nuclear translocation of S100A4 regulate IL-1beta-mediated production of matrix metalloproteinase-13. J. Biol. Chem. 2010, 285, 31517–31524. [Google Scholar] [CrossRef] [Green Version]
- Orre, L.M.; Panizza, E.; Kaminskyy, V.O.; Vernet, E.; Gräslund, T.; Zhivotovsky, B.; Lehtiö, J. S100A4 interacts with p53 in the nucleus and promotes p53 degradation. Oncogene 2013, 32, 5531–5540. [Google Scholar] [CrossRef] [Green Version]
- Sun, B.; Fiskus, W.; Qian, X.; Rajapakshe, K.; Raina, K.; Coleman, K.G.; Crew, A.P.; Shen, A.; Saenz, D.T.; Mill, C.P.; et al. BET protein proteolysis targeting chimera (PROTAC) exerts potential lethal activity against mantle cell lymphoma cells. Leukemia 2018, 32, 343–352. [Google Scholar] [CrossRef]
- Liao, X.; Qian, X.; Zhang, Z.; Tao, Y.; Li, Z.; Zhang, Q. ARV-825 demonstrates antitumor activity in gastric cancer via MYC-targets and G2M-checkpoint signalling pathways. Front. Oncol. 2021, 11, 4205–4219. [Google Scholar] [CrossRef]
- Lu, J.; Qian, Y.; Altieri, M.; Dong, H.; Wang, J.; Raina, K.; Hines, J.; Winkler, J.D.; Crew, A.P.; Coleman, K.; et al. Hijacking the E3 ubiquitin ligase cereblon to efficiently target BRD4. Chem. Biol. 2016, 22, 755–763. [Google Scholar] [CrossRef] [Green Version]
- Toure, M.; Crews, C.M. Small-molecule PROTACs: New approaches to protein degradation. Agnew Chem. Int. Ed. Engl. 2016, 55, 1966–1973. [Google Scholar] [CrossRef]
- Shu, S.; Polyvak, K. BET bromodomain proteins as cancer therapeutic targets. Cold Spr. Harb. Symp.Quant. Biol. 2016, 81, 123–129. [Google Scholar] [CrossRef] [Green Version]
- Noblejas-Lopez, M.; Nieto-Jimenez, C.; Burgos, M.; Gomez-Juarez, M.; Esparis-Ogando, A.; Galan-Moya, E.; Ocaña, A. Activity of BET proteolysis targeting chimeric (PROTAC) compounds in triple negative breast cancer. J. Expt. Clin. Cancer Res. 2019, 38, 383–392. [Google Scholar] [CrossRef] [Green Version]
- Shao, J.; Yan, Y.; Ding, D.; Wang, D.; He, Y.; Pan, Y.; Yan, W.; Kharbanda, A.; Li, H.; Huang, H. Destruction of DNA-binding proteins by programmable oligonucleotide PROTAC (O’PROTAC): Effective targeting of LEF1 and ERG. Adv. Sci. 2021, 8, 2102555. [Google Scholar] [CrossRef]
- He, S.; Gao, F.; Ma, J.; Ma, H.; Dong, G.; Sheng, C. Aptamer-PROTAC conjugates (APCs) for tumor-specific targeting in breast cancer. Angew. Chem. Int. Ed. 2021, 60, 23299–23305. [Google Scholar] [CrossRef]
- Noblejas-Lopez, M.; Nieto-Jimenez, C.; Galen-Moya, E.M.; Tebar-Garcia, D.; Montero, J.C.; Pandiella, A. MZ1 cooperates with trastuzumab in HER2 positive breast cancer. J. Expt. Clinical Res. 2021, 40, 106–118. [Google Scholar]
- Jiang, L.; Lai, Y.K.; Zhang, J.; Wang, H. Targeting S100P inhibits colon cancer growth and metastasis by Lentivirus-mediated RNA interference and proteomic analysis. Mol. Med. 2011, 17, 709–716. [Google Scholar] [CrossRef] [Green Version]
- Padilla, L.; Dakhel, S.; Hernández, J.L. S100 binding to receptor for advanced glycation end-products binding assay: Looking for inhibitors. Biochem. Biophys. Res. Commun. 2014, 446, 404–409. [Google Scholar] [CrossRef] [PubMed]
- Arumugam, T.; Ramachandran, V.; Gomez, S.B.; Schmidt, A.M.; Logsdon, C.D. S100P-derived RAGE antagonistic peptide reduces tumor growth and metastasis. Clin. Cancer Res. 2012, 18, 4356–4364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malashkevich, V.N.; Dulyaninova, N.G.; Ramagopal, U.A.; Liriano, M.A. Phenothiazines inhibit S100A4 function by inducing protein oligomerization. Proc. Natl. Acad. Sci. USA 2010, 107, 8605–8610. [Google Scholar] [CrossRef] [PubMed]
- Garrett, S.C.; Hodgson, L.; Rybin, A.; Toutchkine, A.; Hahn, K.M.; Lawrence, D.S.; Bresnick, A.R. A biosensor of S100A4 metastasis factor activations: Inhibitor screening and cellular activation dynamics. Biochemistry 2008, 47, 986–996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edmonson, S.D.; Yang, B.; Fallan, C. Proteolysis targeting chimeras (PROTACs) in “beyond rule of 5” chemical space: Recent progress and future challenges. Bioorg. Med. Chem. Lett. 2019, 29, 1555–1564. [Google Scholar] [CrossRef]
- Guedeney, M.; Cornu, M.; Schwalen, F.; Kieffer, C. PROTAC technology: A new drug design for chemical biology with many challenges in drug discovery. Drug Discov. Today 2023, 28, 103395. [Google Scholar] [CrossRef]
- Grum-Schwensen, B.; Klingelhofer, J.; Berg, C.H.; El-Naaman, C.; Grigorian, M.; Lukanidin, E.; Ambartsumian, N. Suppression of tumor development and metastasis formation in mice lacking the S100A4 (mts1) gene. Cancer Res. 2005, 65, 3772–3780. [Google Scholar] [CrossRef] [Green Version]
- Grum-Schwensen, B.; Klingelhöfer, J.; Grigorian, M.; Almholt, K.; Nielsen, B.S.; Lukanidin, E.; Ambartsumian, N. Lung metastasis fails in MMTV-PyMT oncomice lacking S100A4 due to a T-cell deficiency in primary tumors. Cancer Res. 2010, 70, 936–947. [Google Scholar] [CrossRef] [Green Version]
- Warburton, M.J.; Mitchell, D.; Ormerod, E.J.; Rudland, P.S. Distribution of myoepithelial cells and basement membrane proteins in the resting, pregnant, lactating and involuting rat mammary gland. J. Histochem. Cytochem. 1982, 30, 667–676. [Google Scholar] [CrossRef] [Green Version]
- Streefkerk, J.G. Inhibition of erythrocyte pseudoperoxidase activity by treatment with hydrogen peroxide following methanol. J. Histochem. Cytochem. 1972, 20, 829–831. [Google Scholar] [CrossRef]
- Hsu, S.-M.; Raine, L.; Fanger, H. Use of avidin-biotin-peroxide complex (ABC) in immunoperoxide techniques. J. Histochem. Cytochem. 1981, 29, 577–580. [Google Scholar] [CrossRef] [Green Version]
- Heras, A.; Roach, C.M.; Key, M.E. Enhanced labelled-polymer system for immunohistochemistry. In Proceedings of the XVth European Congress of Pathology, Copenhagen, Denmark, 3–8 September 1995; pp. 3–8. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Route of Administration a | Inhibitor | Dose (mg/kg) | Invasion b Incidence | p c | Metastasis d Incidence | p c |
|---|---|---|---|---|---|---|
| Intravenous (iv) | None | - | n.a. | 5 of 5 | - | |
| thalidomide | 1.35 | n.a. | 3 of 5 | 0.22 | ||
| US10113 | 13.5 | n.a. | 4 of 5 | 0.50 | ||
| RGC | 0.07 | n.a. | 1 of 5 | 0.024 | ||
| RGC | 0.70 | n.a. | 1 of 5 | 0.024 | ||
| RGC | 3.50 | n.a. | 2 of 5 | 0.083 | ||
| Mammary fat pad (sc) | None | - | 8 of 8 | - | 8 of 8 | - |
| RGC | 0.007 | 0 of 6 e | 0.0003 | 0 of 7 | 0.0002 | |
| RGC | 0.07 | 1 of 7 | 0.0014 | 0 of 7 | 0.0002 | |
| RGC | 0.70 | 3 of 7 e | 0.026 | 0 of 7 | 0.0002 | |
| RGC | 3.50 | 2 of 8 | 0.0035 | 4 of 8 | 0.038 |
| Route of Administration | Inhibitor (mg/kg) | Tissue Staining a | ||
|---|---|---|---|---|
| Spleen | Colon | Tumour | ||
| Percent ±SD (n) (p) b | Percent ±SD (n) (p) b | Percent ±SD (n) (p) b | ||
| Intravenouscells (iv) | Untreated | 32.5 ± 8.7 (4) | 37.5 ± 8.7(4) | 40 ± 18(5) |
| RGC (0.07) | 17.0 ± 4.5(5) (p = 0.01) | 12.5 ± 6.1(5) (p = 0.0014) | 17.5 ± 0(1) (p = 0.32) | |
| RGC (0.7) | 10.5 ± 2.7(5) (p = 0.001) | 10.0 ± 0.0(5) (p = 0.008) | 50 ± 0(1) (p = 0.64) | |
| RGC (3.5) | 13.7 ± 7.5(4) (p = 0.017) | 17.5 ± 10.4(4) (p = 0.026) | 45 ± 0(1) d (p = 0.81) | |
| US10113 (13.5) | 26.2 ± 8.5(4) (p = 0.34) | 40.0 ± 10.8(4) (p = 0.73) | 42 ± 15(4) (p = 0.83) | |
| Thalidomide (1.35) | 26.5 ± 13.2(4) (p = 0.46) | 37.0 ± 4.5(5) (p = 0.91) | 35 ± 21(3) (p = 0.76) | |
| Cells s/c into mammary fat pad | Untreated RGC (0.007) | 23.6 ± 3.8(7) 6.7 ± 1.8(7) c (p < 0.0001) | 39.3 ± 10.2(7) 11.8 ± 3.4(7) (p = 0.0002) | 63.6 ± 18.2(7) e 6.1 ± 2.0(7) (p = 0.0001) |
| RGC (0.07) | 9.9 ± 2.6(7) (p < 0.0001) | 17.1 ± 9.1(7) (p = 0.001) | 13.5 ± 18.6(7) (p = 0.0003) | |
| RGC(0.7) | 11.4 ± 3.1(6)c (p < 0.0001) | 13.3 ± 1.4(7) (p = 0.0005) | 14.0 ± 3.2(7) (p = 0.0003) | |
| RGC(3.5) | 12.5 ± 5.0(8) (p = 0.0004) | 16.9 ± 10.7(8) (p = 0.0012) | 29.6 ± 30.2(7) d (p = 0.026) | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ismail, T.M.; Crick, R.G.; Du, M.; Shivkumar, U.; Carnell, A.; Barraclough, R.; Wang, G.; Cheng, Z.; Yu, W.; Platt-Higgins, A.; et al. Targeted Destruction of S100A4 Inhibits Metastasis of Triple Negative Breast Cancer Cells. Biomolecules 2023, 13, 1099. https://doi.org/10.3390/biom13071099
Ismail TM, Crick RG, Du M, Shivkumar U, Carnell A, Barraclough R, Wang G, Cheng Z, Yu W, Platt-Higgins A, et al. Targeted Destruction of S100A4 Inhibits Metastasis of Triple Negative Breast Cancer Cells. Biomolecules. 2023; 13(7):1099. https://doi.org/10.3390/biom13071099
Chicago/Turabian StyleIsmail, Thamir M., Rachel G. Crick, Min Du, Uma Shivkumar, Andrew Carnell, Roger Barraclough, Guozheng Wang, Zhenxing Cheng, Weiping Yu, Angela Platt-Higgins, and et al. 2023. "Targeted Destruction of S100A4 Inhibits Metastasis of Triple Negative Breast Cancer Cells" Biomolecules 13, no. 7: 1099. https://doi.org/10.3390/biom13071099