

Unconventional Source of Neurotoxic Protein Aggregation from Organelle Off-Target Bax∆2 in Alzheimer’s Disease

 ,
,
Abstract
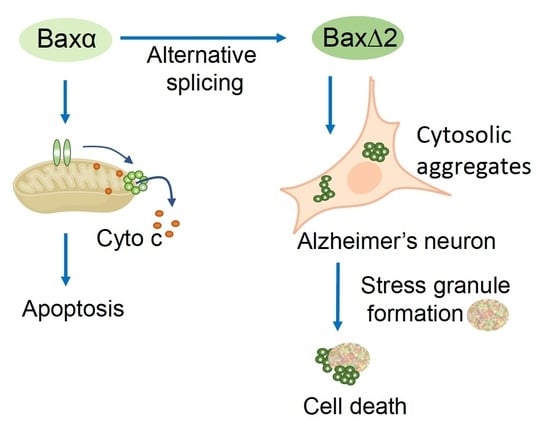
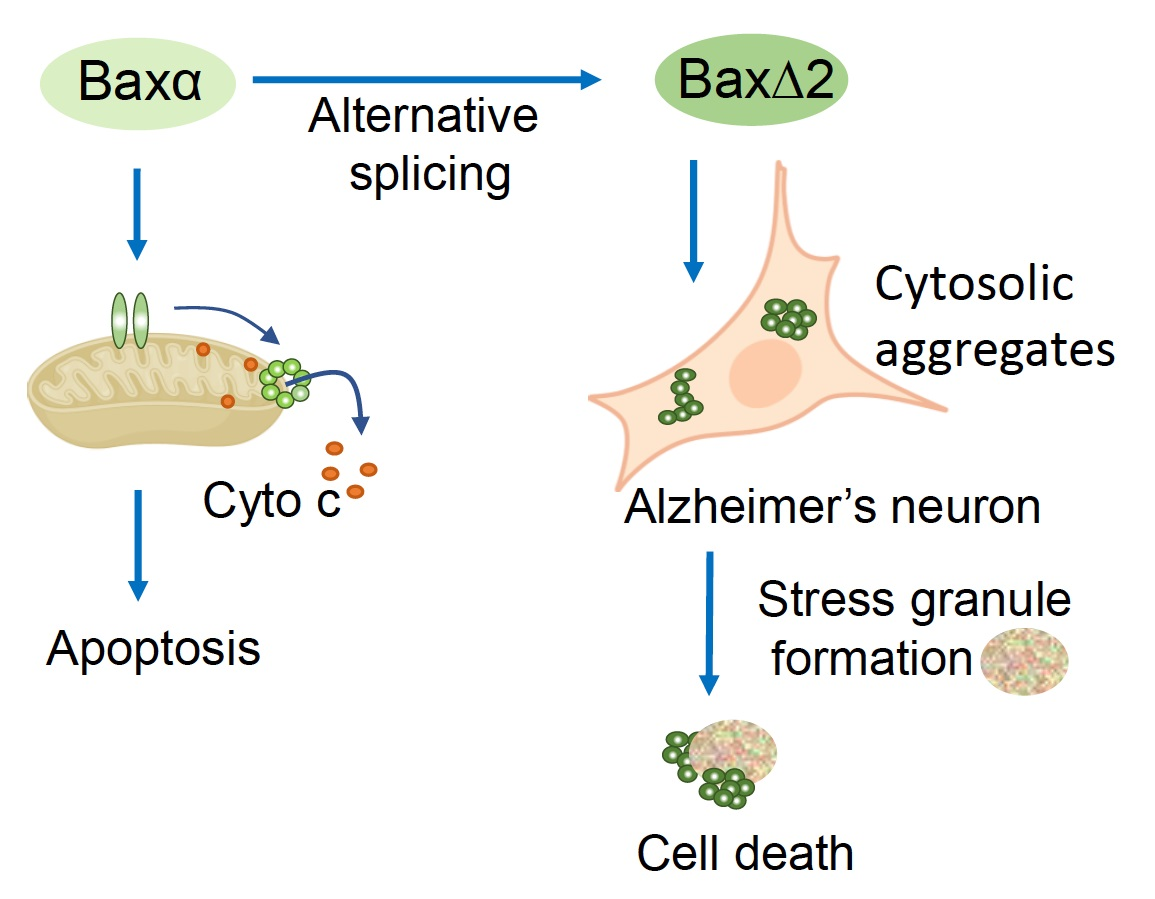
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. RNA-Seq Mapping and Analysis
2.3. Cell Culture and Transfection
2.4. Cell Death Assay
2.5. Immunoblotting
2.6. Immunofluorescence Staining
2.7. Immunohistochemical Staining
2.8. Image Acquisition, Process, and Quantitation
2.9. Statistics
3. Results
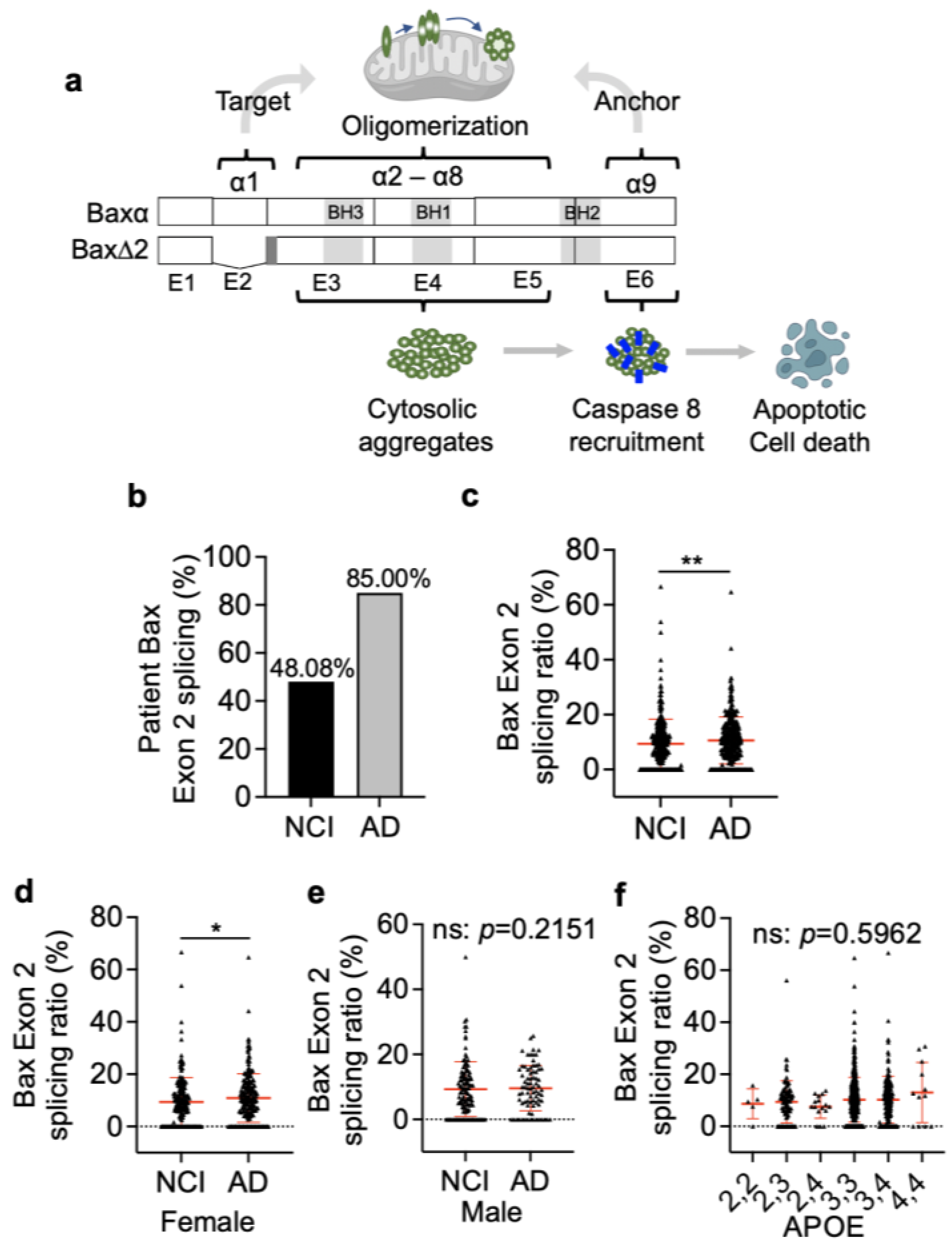
3.1. Bax Exon 2 Alternative Splicing Is Significantly Higher in AD Brains
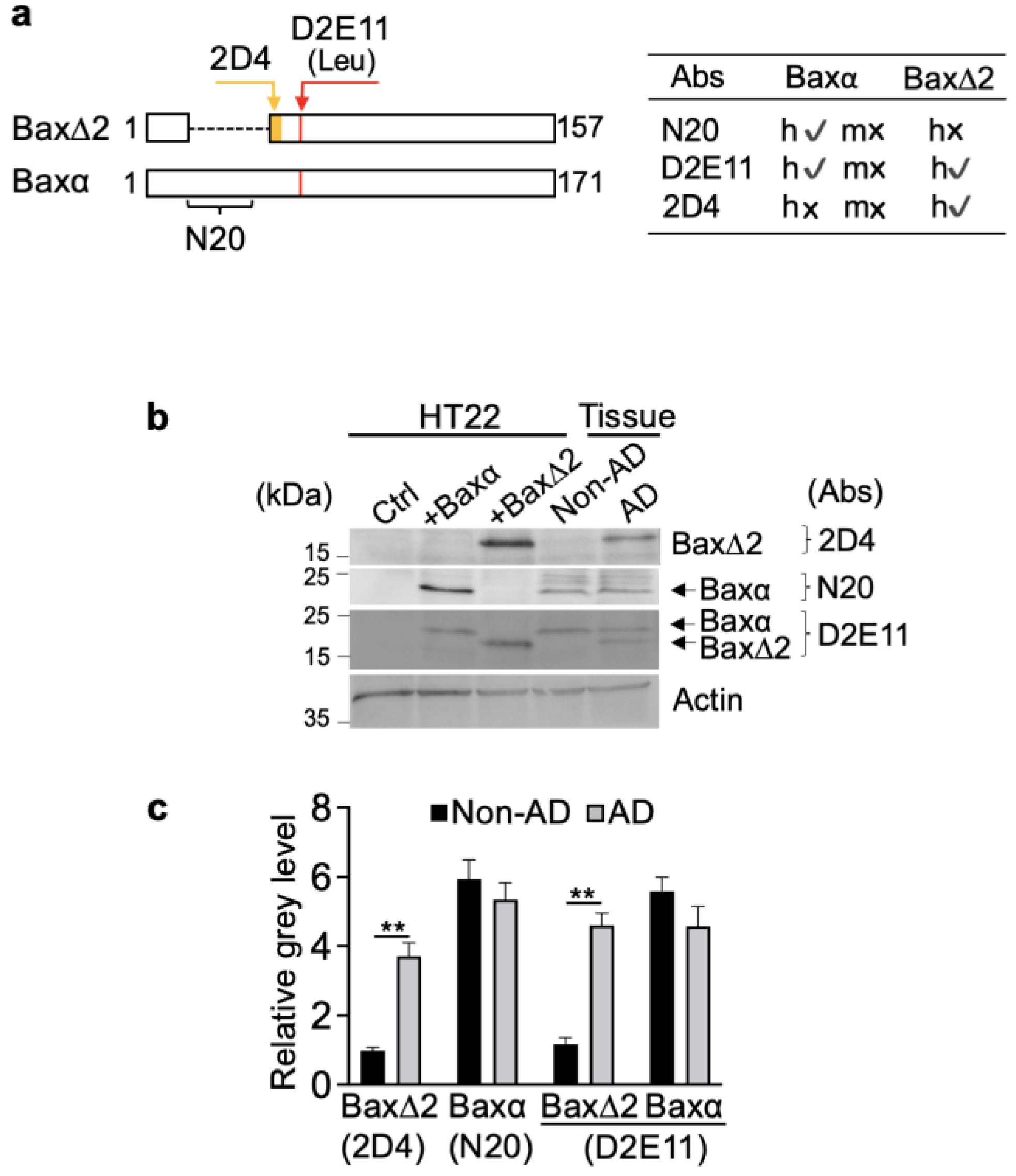
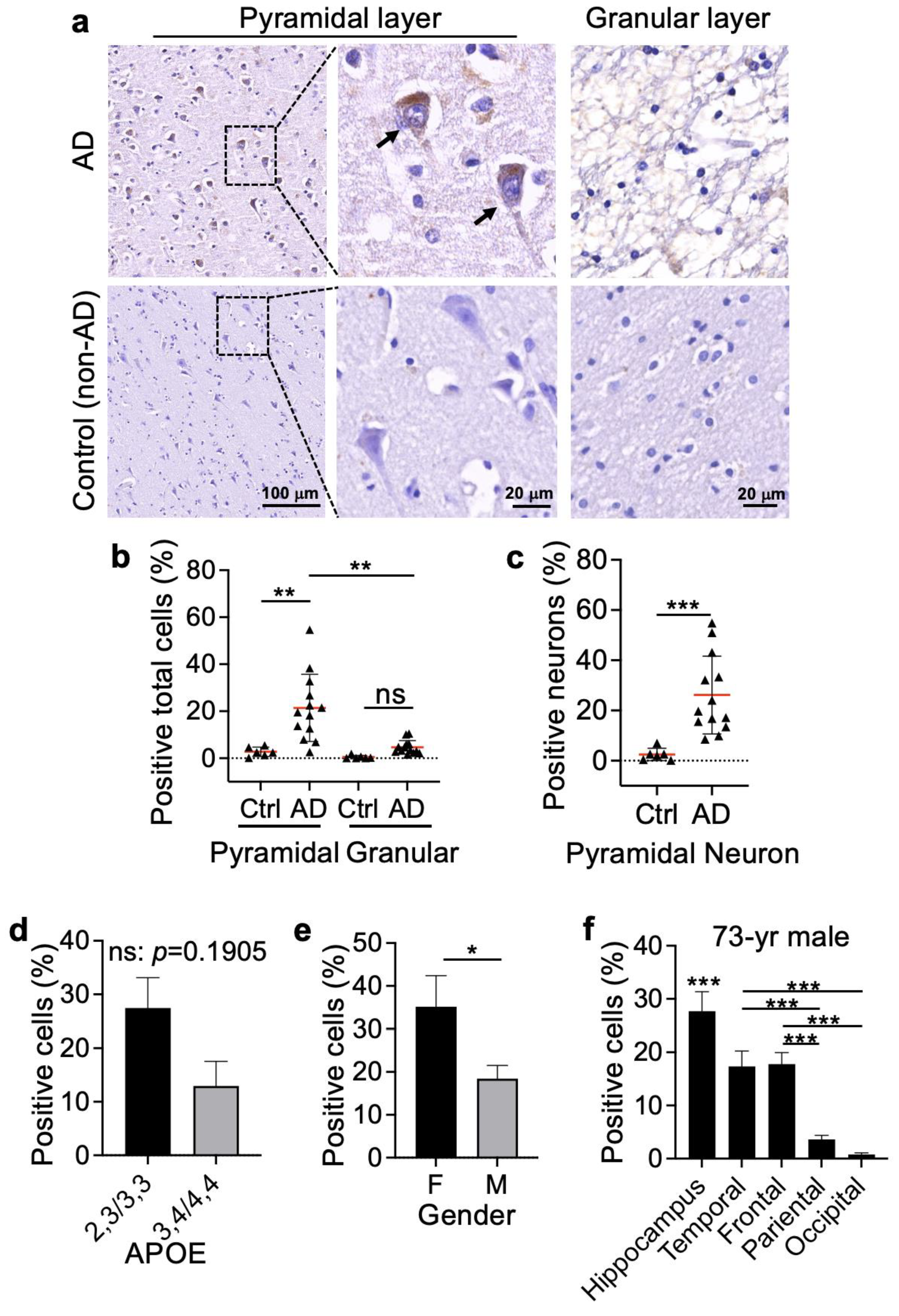
3.2. Bax∆2 Protein Accumulated in Neuronal Cells from AD-Affected Brain Regions
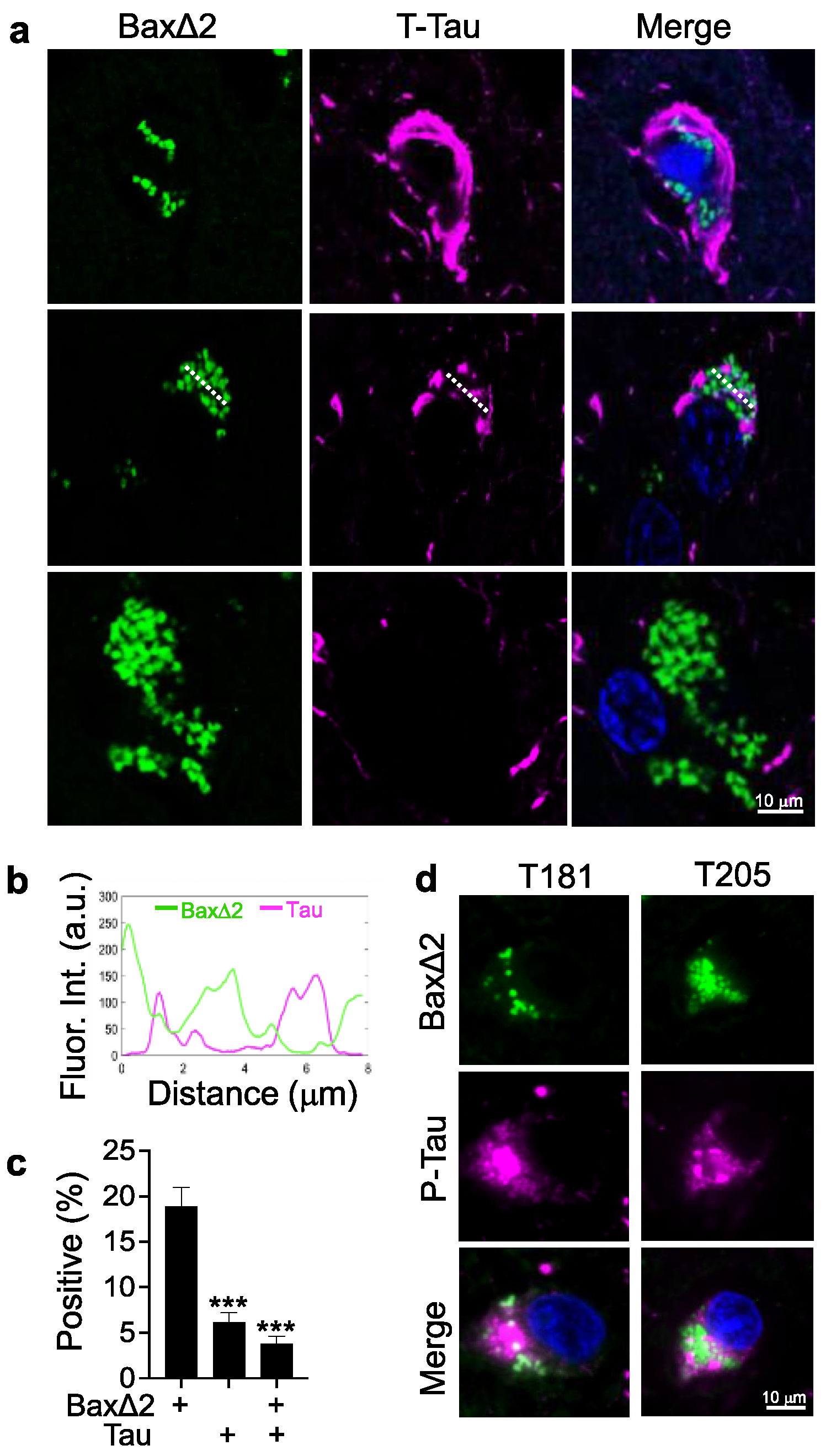
3.3. Bax∆2 Aggregate Distribution Is Independent of Tau Tangles in AD Neurons
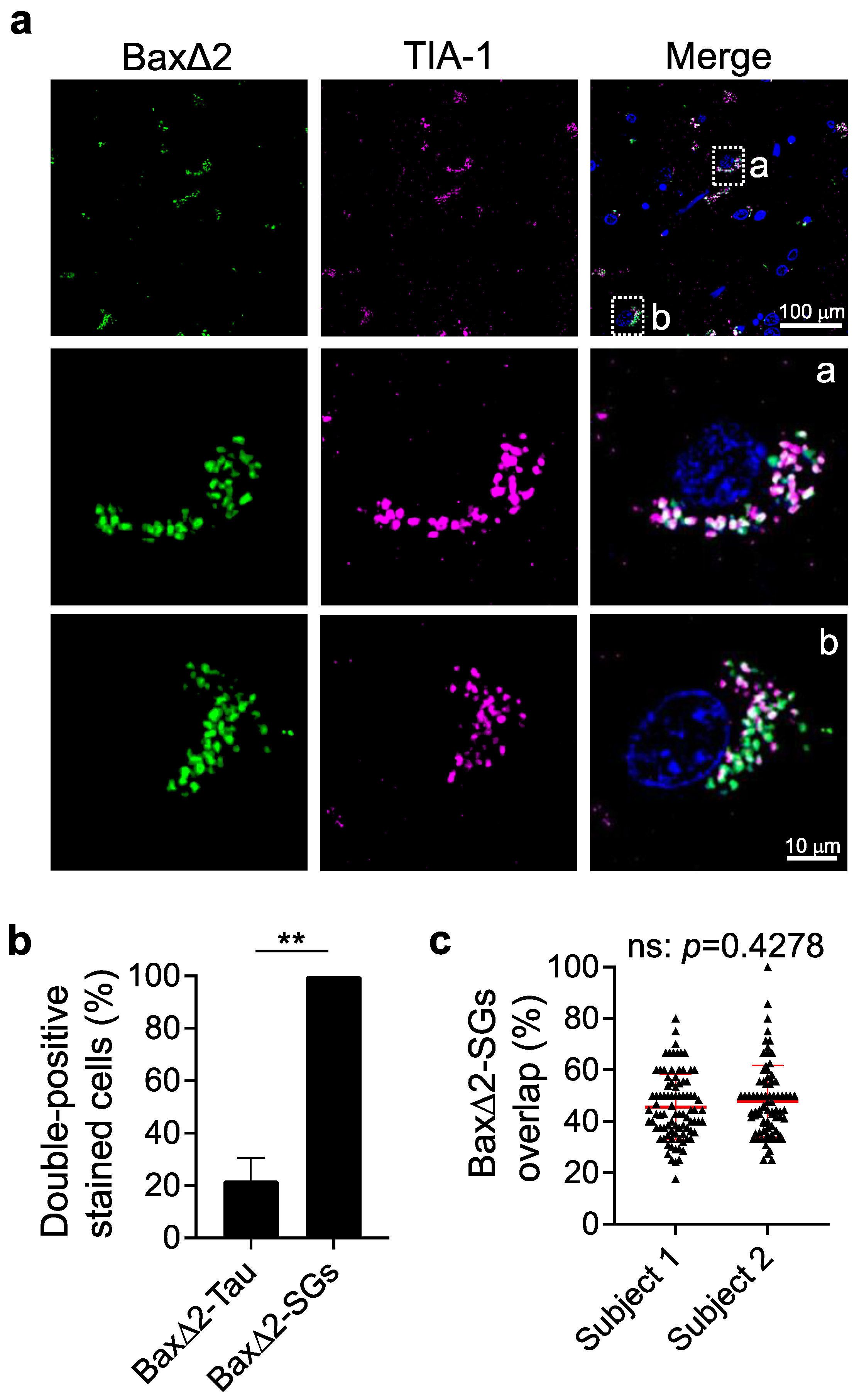
3.4. Bax∆2 Aggregates Coexist and Colocalize with Stress Granules in AD Neurons
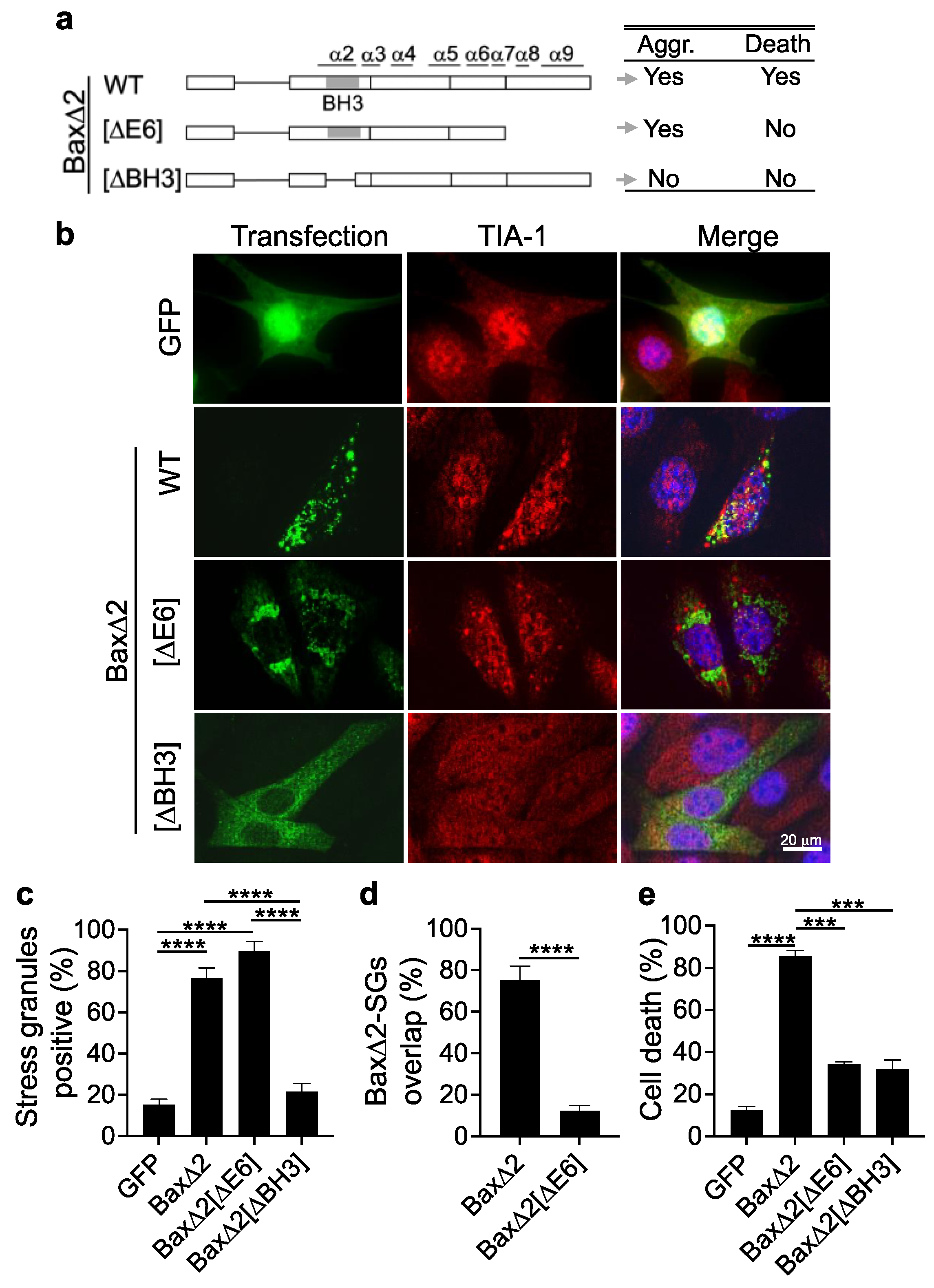
3.5. Aggregation of Bax∆2 Is Critical for SG Formation, and the Bax∆2 C-Terminal Tail Is Required for Bax∆2-SG Colocalization and Cell Death
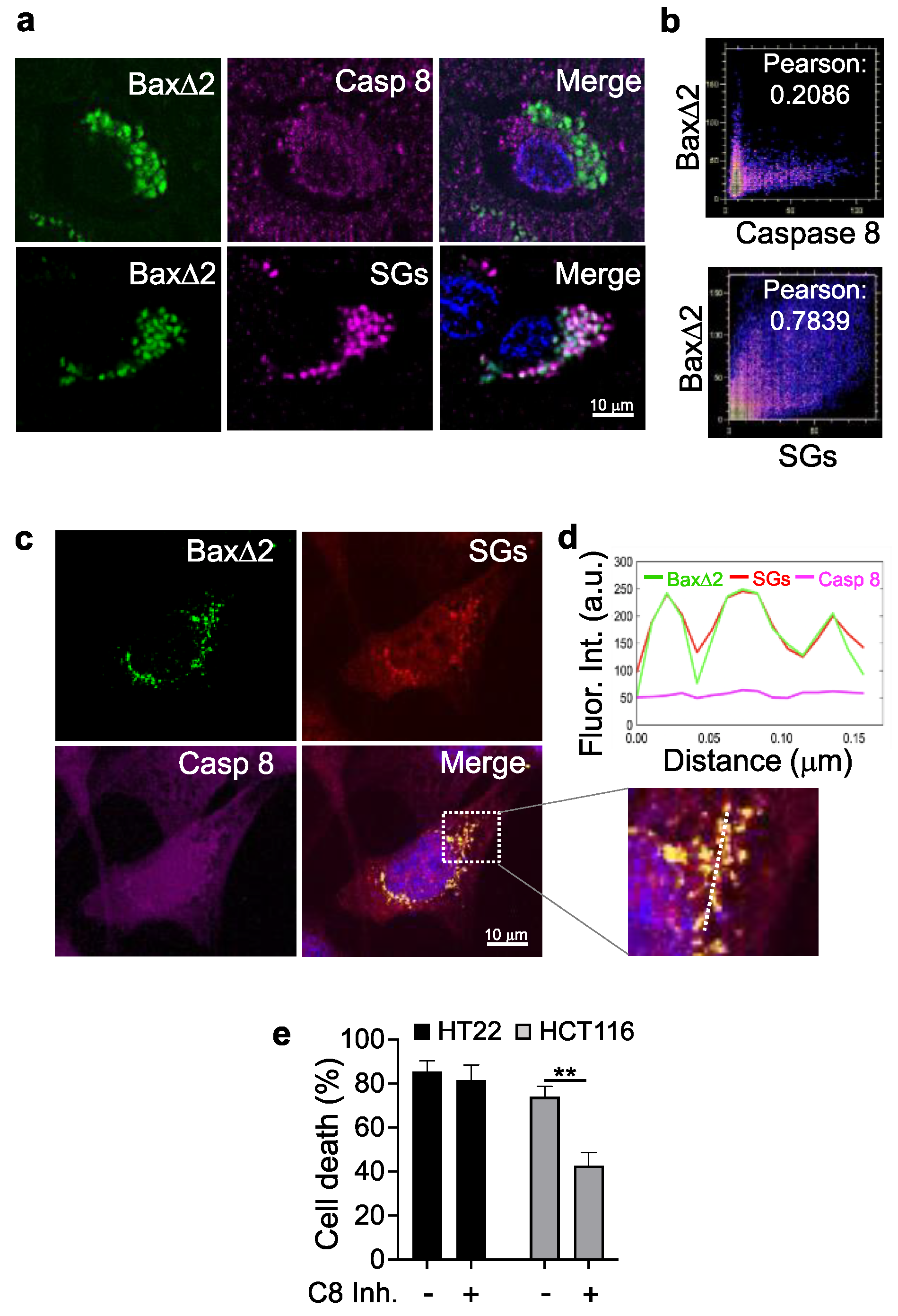
3.6. Bax∆2 Aggregates Prefer SGs and Not Caspase 8 to Induce Cell Death in Neurons
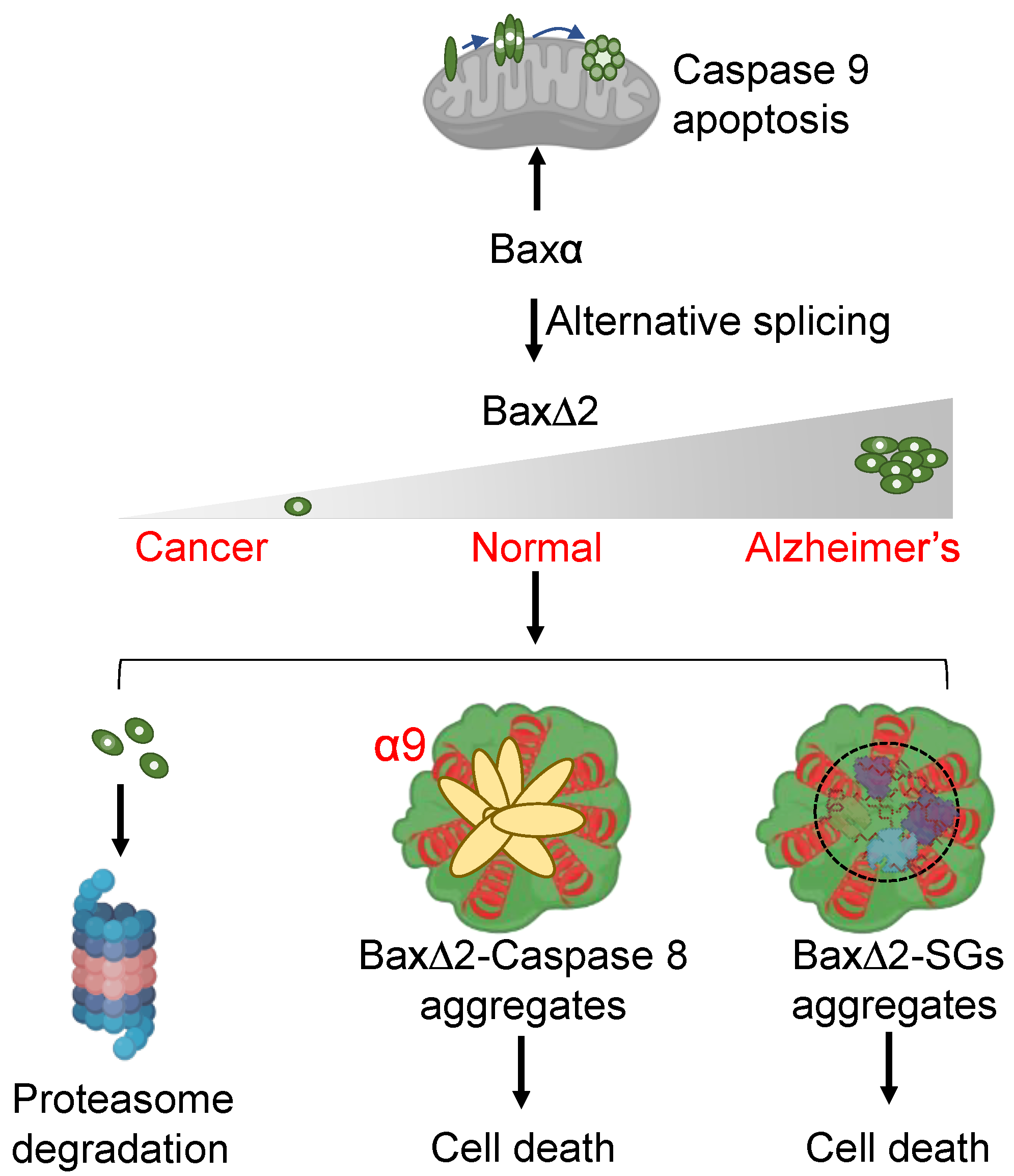
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chao, D.T.; Korsmeyer, S.J. BCL-2 FAMILY: Regulators Of Cell Death. Annu. Rev. Immunol. 1998, 16, 395–419. [Google Scholar] [CrossRef] [PubMed]
- Oltval, Z.N.; Milliman, C.L.; Korsmeyer, S.J. Bcl-2 Heterodimerizes in Vivo with a Conserved Homolog, Bax, That Accelerates Programed Cell Death. Cell 1993, 74, 609–619. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.M.; Oltvai, Z.N.; Veis-Novack, D.J.; Linette, G.P.; Korsmeyer, S.J. Bcl-2 Gene Family and the Regulation of Programmed Cell Death. Cold Spring Harb. Symp. Quant. Biol. 1994, 59, 387–393. [Google Scholar] [CrossRef]
- Chen, M.; Wang, J. Initiator Caspases in Apoptosis Signaling Pathways. Apoptosis 2002, 7, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Upton, J.P.; Valentijn, A.J.; Zhang, L.; Gilmore, A.P. The N-Terminal Conformation of Bax Regulates Cell Commitment to Apoptosis. Cell Death Differ. 2007, 14, 932–942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, C.; Zhang, Z.; Kale, J.; Andrews, D.W.; Lin, J.; Li, J. Conformational Heterogeneity of Bax Helix 9 Dimer for Apoptotic Pore Formation. Sci. Rep. 2016, 6, 29502. [Google Scholar] [CrossRef] [Green Version]
- George, N.M.; Targy, N.; Evans, J.J.D.; Zhang, L.; Luo, X. Bax Contains Two Functional Mitochondrial Targeting Sequences and Translocates to Mitochondria in a Conformational Change- and Homo-Oligomerization-Driven Process. J. Biol. Chem. 2010, 285, 1384–1392. [Google Scholar] [CrossRef] [Green Version]
- Zha, H.; Aimé-Sempé, C.; Sato, T.; Reed, J.C. Proapoptotic Protein Bax Heterodimerizes with Bcl-2 and Homodimerizes with Bax via a Novel Domain (BH3) Distinct from BH1 and BH2. J. Biol. Chem. 1996, 271, 7440–7444. [Google Scholar] [CrossRef] [Green Version]
- Yin, X.M.; Oltvai, Z.N.; Korsmeyer, S.J. BH1 and BH2 Domains of Bcl-2 Are Required for Inhibition of Apoptosis and Heterodimerization with Bax. Nature 1994, 369, 321–323. [Google Scholar] [CrossRef]
- Sedlak, T.W.; Oltvai, Z.N.; Yang, E.; Wang, K.; Boise, L.H.; Thompson, C.B.; Korsmeyer, S.J. Multiple Bcl-2 Family Members Demonstrate Selective Dimerizations with Bax. Proc. Natl. Acad. Sci. USA 1995, 92, 7834–7838. [Google Scholar] [CrossRef] [Green Version]
- Haferkamp, B.; Zhang, H.; Kissinger, S.; Wang, X.; Lin, Y.; Schultz, M.; Xiang, J. BaxΔ2 Family Alternative Splicing Salvages Bax Microsatellite-Frameshift Mutations. Genes Cancer 2013, 4, 501–512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haferkamp, B.; Zhang, H.; Lin, Y.; Yeap, X.; Bunce, A.; Sharpe, J.; Xiang, J. BaxΔ2 Is a Novel Bax Isoform Unique to Microsatellite Unstable Tumors. J. Biol. Chem. 2012, 287, 34722–34729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mañas, A.; Yao, Q.; Davis, A.; Basheer, S.; Beatty, E.; Zhang, H.; Li, J.; Nelson, A.; Zhang, H.; Xiang, J. Immunohistochemical Detection of the Pro-Apoptotic Bax∆2 Protein in Human Tissues. Histochem. Cell Biol. 2020, 154, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Sarosiek, K.A.; Fraser, C.; Muthalagu, N.; Bhola, P.D.; Chang, W.; McBrayer, S.K.; Cantlon, A.; Fisch, S.; Golomb-Mello, G.; Ryan, J.A.; et al. Developmental Regulation of Mitochondrial Apoptosis by C-Myc Governs Age- and Tissue-Specific Sensitivity to Cancer Therapeutics. Cancer Cell 2017, 31, 142–156. [Google Scholar] [CrossRef] [Green Version]
- Shimohama, S.; Fujimoto, S.; Sumida, Y.; Tanino, H. Differential Expression of Rat Brain Bcl-2 Family Proteins in Development and Aging. Biochem. Biophys. Res. Commun. 1998, 252, 92–96. [Google Scholar] [CrossRef] [PubMed]
- Krajewska, M.; Mai, J.K.; Zapata, J.M.; Ashwell, K.W.; Schendel, S.L.; Reed, J.C.; Krajewski, S. Dynamics of Expression of Apoptosis-Regulatory Proteins Bid, Bcl-2, Bcl-X, Bax and Bak during Development of Murine Nervous System. Cell Death Differ. 2002, 9, 145–157. [Google Scholar] [CrossRef]
- Yao, Q.; Zhang, H.; Standish, C.; Grube, J.; Mañas, A.; Xiang, J. Expression Profile of the Proapoptotic Protein Bax in the Human Brain. Histochem. Cell Biol. 2022, 159, 209–220. [Google Scholar] [CrossRef]
- Mañas, A.; Chen, W.; Nelson, A.; Yao, Q.; Xiang, J. BaxΔ2 Sensitizes Colorectal Cancer Cells to Proteasome Inhibitor-Induced Cell Death. Biochem. Biophys. Res. Commun. 2018, 496, 18–24. [Google Scholar] [CrossRef]
- Mañas, A.; Wang, S.; Nelson, A.; Li, J.; Zhao, Y.; Zhang, H.; Davis, A.; Xie, B.; Maltsev, N.; Xiang, J. The Functional Domains for Bax∆2 Aggregate-Mediated Caspase 8-Dependent Cell Death. Exp. Cell Res. 2017, 359, 342–355. [Google Scholar] [CrossRef]
- Zhang, H.; Lin, Y.; Mañas, A.; Zhao, Y.; Denning, M.F.; Ma, L.; Xiang, J. Bax∆2 Promotes Apoptosis through Caspase-8 Activation in Microsatellite-Unstable Colon Cancer. Mol. Cancer Res. 2014, 12, 1225–1232. [Google Scholar] [CrossRef] [Green Version]
- Xie, B.; Yao, Q.; Xiang, J.; Minh, D.D.L. A Structural Model for Bax∆2-Mediated Activation of Caspase 8-Dependent Apoptosis. Int. J. Mol. Sci. 2020, 21, 5476. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J. Alzheimer ’ s Disease: Genes, Proteins, and Therapy. Phys. Rev. 2018, 81, 4110. [Google Scholar]
- Gandhi, J.; Antonelli, A.C.; Afridi, A.; Vatsia, S.; Joshi, G.; Romanov, V.; Murray, I.V.J.; Khan, S.A. Protein Misfolding and Aggregation in Neurodegenerative Diseases: A Review of Pathogeneses, Novel Detection Strategies, and Potential Therapeutics. Rev. Neurosci. 2019, 30, 339–358. [Google Scholar] [CrossRef]
- Whitehouse, P.J.; Price, D.L.; Struble, R.G.; Clark, A.W.; Coyle, J.T.; Delon, M.R. Alzheimer’s Disease and Senile Dementia: Loss of Neurons in the Basal Forebrain. Science 1981, 251, 1038–1040. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Wang, L.M.; Geng, M.Y. Tau Protein and Neurodegeneration. Chin. J. Clin. Rehabil. 2006, 10, 124–126. [Google Scholar]
- Hardy, J.; Selkoe, D.J. The Amyloid Hypothesis of Alzheimer’s Disease: Progress and Problems on the Road to Therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef] [Green Version]
- Bloom, G.S. Amyloid-β and Tau: The Trigger and Bullet in Alzheimer Disease Pathogenesis. JAMA Neurol. 2014, 71, 505–508. [Google Scholar] [CrossRef] [Green Version]
- Su, J.H. Bax Protein Expression Is Increased in Alzheimer’s Brain: Correlations with DNA Damage, Bcl-2 Expression, and Brain Pathology. J. Neuropathol. Exp. Neurol. 1997, 56, 86–93. [Google Scholar] [CrossRef]
- Tortosa, A.; Löpez, E.; Ferrer, I. Bcl-2 and Bax Protein Expression in Alzheimer’s Disease. Acta Neuropathol. 1998, 95, 407–412. [Google Scholar] [CrossRef]
- Toral-Rios, D.; Pichardo-Rojas, P.S.; Alonso-Vanegas, M.; Campos-Peña, V. GSK3β and Tau Protein in Alzheimer’s Disease and Epilepsy. Front. Cell. Neurosci. 2020, 14, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.Q.; Wei, J.S.; Lei, Z.N.; Zhang, L.M.; Liu, Y.; Sun, F.Y. Induction of Bcl-2 and Bax Was Related to Hyperphosphorylation of Tau and Neuronal Death Induced by Okadaic Acid in Rat Brain. Anat. Rec. - Part A Discov. Mol. Cell. Evol. Biol. 2005, 287, 1236–1245. [Google Scholar] [CrossRef] [PubMed]
- Mañas, A.; Davis, A.; Lamerand, S.; Xiang, J. Detection of Pro-Apoptotic Bax∆2 Proteins in the Human Cerebellum. Histochem. Cell Biol. 2018, 150, 77–82. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.J.; Ma, Y.; Yu, L.; Dawe, R.J.; Arfanakis, K.; Mayeux, R.; Bennett, D.A.; Klein, H.U.; De Jager, P.L. Multi-Region Brain Transcriptomes Uncover Two Subtypes of Aging Individuals with Differences in the Impact of APOEe4. Alzheimer’s Dement. 2021, 17, e057240. [Google Scholar] [CrossRef]
- Bennett, D.A.; Buchman, A.S.; Boyle, P.A.; Barnes, L.L.; Wilson, R.S.; Schneider, J.A. Religious Orders Study and Rush Memory and Aging Project. J. Alzheimer’s Dis. 2018, 64, S161–S189. [Google Scholar] [CrossRef]
- Yu, L.; Tasaki, S.; Schneider, J.A.; Arfanakis, K.; Duong, D.M.; Wingo, A.P.; Wingo, T.S.; Kearns, N.; Thatcher, G.R.J.; Seyfried, N.T.; et al. Cortical Proteins Associated with Cognitive Resilience in Community-Dwelling Older Persons. JAMA Psychiatry 2020, 77, 1172–1180. [Google Scholar] [CrossRef]
- Greenwood, A.K.; Montgomery, K.S.; Kauer, N.; Woo, K.H.; Leanza, Z.J.; Poehlman, W.L.; Gockley, J.; Sieberts, S.K.; Bradic, L.; Logsdon, B.A.; et al. The AD Knowledge Portal: A Repository for Multi-Omic Data on Alzheimer’s Disease and Aging. Curr. Protoc. Hum. Genet. 2020, 108, e105. [Google Scholar] [CrossRef]
- Li, H. Minimap2: Pairwise Alignment for Nucleotide Sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef] [Green Version]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M. Twelve Years of SAMtools and BCFtools. Gigascience 2021, 10, giab008. [Google Scholar] [CrossRef]
- Kwon, M.; Lee, S.; Berselli, M.; Chu, C.; Park, P.J. BamSnap: A Lightweight Viewer for Sequencing Reads in BAM Files. Bioinformatics 2021, 37, 263–264. [Google Scholar] [CrossRef]
- Carpenter, A.E.; Jones, T.R.; Lamprecht, M.R.; Clarke, C.; Kang, I.H.; Friman, O.; Guertin, D.A.; Chang, J.H.; Lindquist, R.A.; Moffat, J.; et al. CellProfiler: Image Analysis Software for Identifying and Quantifying Cell Phenotypes. Genome Biol. 2006, 7, R100. [Google Scholar] [CrossRef] [Green Version]
- Saunders, A.M.; Strittmatter, W.J.; Schmechel, D.; St. George-Hyslop, P.H.; Pericak-Vance, M.A.; Joo, S.H.; Rosi, B.L.; Gusella, J.F.; Crapper-Mac Lachlan, D.R.; Alberts, M.J.; et al. Association of Apolipoprotein E Allele Ε4 with Late-Onset Familial and Sporadic Alzheimer’s Disease. Neurology 1993, 43, 1467–1472. [Google Scholar] [CrossRef] [Green Version]
- Vanderweyde, T.; Youmans, K.; Liu-Yesucevitz, L.; Wolozin, B. Role of Stress Granules and RNA-Binding Proteins in Neurodegeneration: A Mini-Review. Gerontology 2013, 59, 524–533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ash, P.E.A.; Vanderweyde, T.E.; Youmans, K.L.; Apicco, D.J.; Wolozin, B. Pathological Stress Granules in Alzheimer’s Disease. Brain Res. 2014, 1584, 52–58. [Google Scholar] [CrossRef] [Green Version]
- Biamonti, G.; Amato, A.; Belloni, E.; Di Matteo, A.; Infantino, L.; Pradella, D.; Ghigna, C. Alternative Splicing in Alzheimer’s Disease. Aging Clin. Exp. Res. 2021, 33, 747–758. [Google Scholar] [CrossRef]
- Tseng, B.P.; Green, K.N.; Chan, J.L.; Blurton-Jones, M.; LaFerla, F.M. Aβ Inhibits the Proteasome and Enhances Amyloid and Tau Accumulation. Neurobiol. Aging 2008, 29, 1607–1618. [Google Scholar]
- Protter, D.S.W.; Parker, R. Principles and Properties of Stress Granules. Trends Cell Biol. 2016, 26, 668–679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monahan, Z.; Shewmaker, F.; Pandey, U.B. Stress Granules at the Intersection of Autophagy and ALS. Brain Res. 2016, 1649, 189–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buchan, J.R.; Kolaitis, R.M.; Taylor, J.P.; Parker, R. Eukaryotic Stress Granules Are Cleared by Autophagy and Cdc48/VCP Function. Cell 2013, 153, 1461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Omer, A.; Patel, D.; Moran, J.L.; Lian, X.J.; Di Marco, S.; Gallouzi, I.E. Autophagy and Heat-Shock Response Impair Stress Granule Assembly during Cellular Senescence. Mech. Ageing Dev. 2020, 192, 111382. [Google Scholar] [CrossRef]
- Das, G.; Shravage, B.V.; Baehrecke, E.H. Regulation and Function of Autophagy during Cell Survival and Cell Death. Cold Spring Harb. Perspect. Biol. 2012, 4, a008813. [Google Scholar] [CrossRef] [Green Version]
- Arimoto, K.; Fukuda, H.; Imajoh-Ohmi, S.; Saito, H.; Takekawa, M. Formation of Stress Granules Inhibits Apoptosis by Suppressing Stress-Responsive MAPK Pathways. Nat. Cell Biol. 2008, 10, 1324–1332. [Google Scholar] [CrossRef] [PubMed]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yao, Q.; Mascarenhas dos Santos, A.C.; Zhang, H.; Mañas, A.; Hussaini, A.; Kim, U.; Xu, C.; Basheer, S.; Tasaki, S.; Xiang, J. Unconventional Source of Neurotoxic Protein Aggregation from Organelle Off-Target Bax∆2 in Alzheimer’s Disease. Biomolecules 2023, 13, 970. https://doi.org/10.3390/biom13060970
Yao Q, Mascarenhas dos Santos AC, Zhang H, Mañas A, Hussaini A, Kim U, Xu C, Basheer S, Tasaki S, Xiang J. Unconventional Source of Neurotoxic Protein Aggregation from Organelle Off-Target Bax∆2 in Alzheimer’s Disease. Biomolecules. 2023; 13(6):970. https://doi.org/10.3390/biom13060970
Chicago/Turabian StyleYao, Qi, Anne Caroline Mascarenhas dos Santos, Huaiyuan Zhang, Adriana Mañas, Ammarah Hussaini, Ujin Kim, Congtai Xu, Sana Basheer, Shinya Tasaki, and Jialing Xiang. 2023. "Unconventional Source of Neurotoxic Protein Aggregation from Organelle Off-Target Bax∆2 in Alzheimer’s Disease" Biomolecules 13, no. 6: 970. https://doi.org/10.3390/biom13060970