
Microglia NLRP3 Inflammasome and Neuroimmune Signaling in Substance Use Disorders

 ,
,
Abstract
:1. Introduction
2. Microglia, Inflammasomes, and SUDs
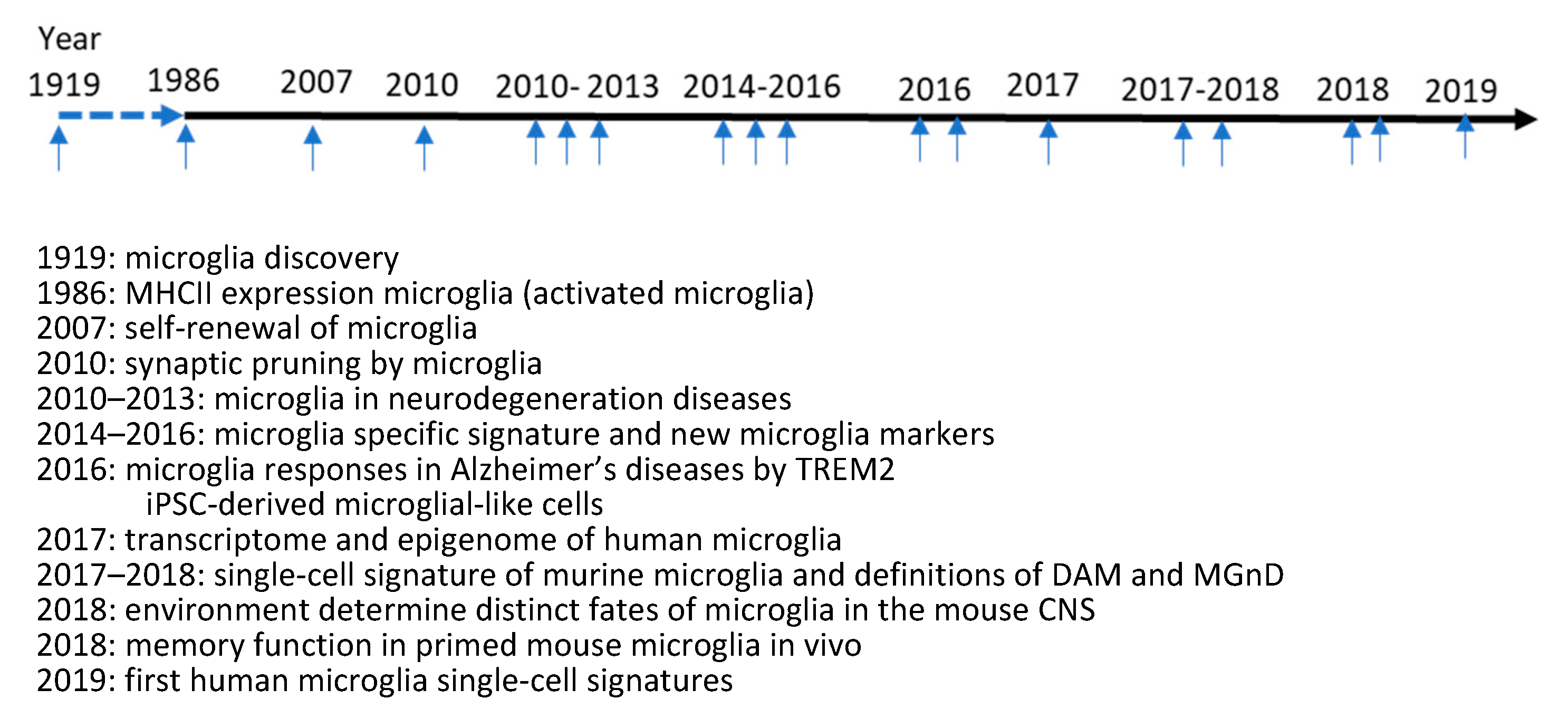
2.1. Updates on Microglia Biology
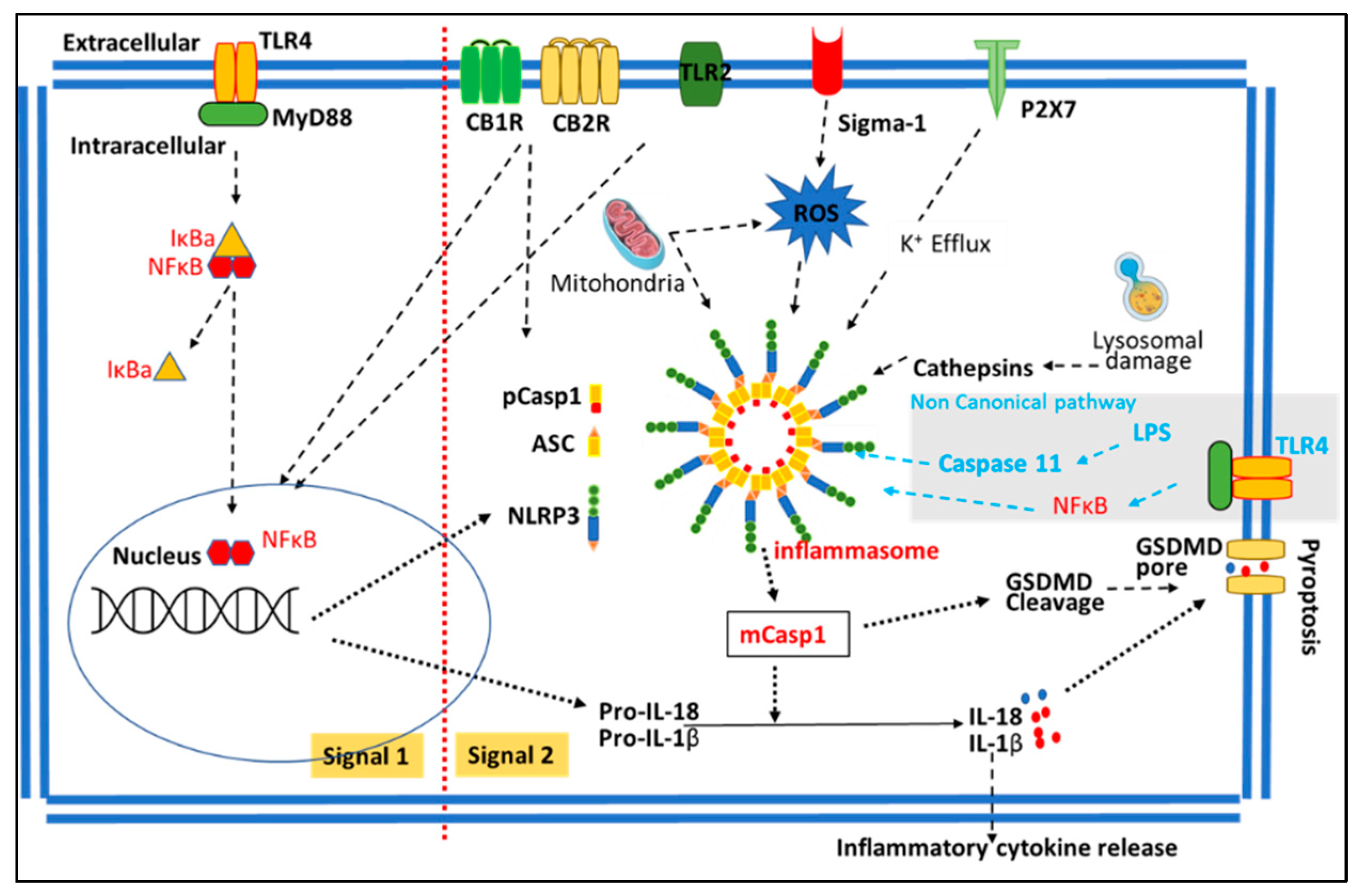
2.2. NLRP3 Inflammasome Pathway
2.3. SUDs and Neuroinflammation
3. The Effects of Abused Drugs on Microglia and Inflammasomes
3.1. The Effects of Cocaine on Neuroimmune Signaling and NLRP3 Inflammasome
3.2. The Effects of Meth on Neuroimmune Signaling and NLRP3 Inflammasome
3.3. The Effects of Alcohol on Neuroimmune Signaling and NLRP3 Inflammasome
3.4. The Effects of Marijuana on Neuroimmune Signaling and NLRP3 Inflammasome
3.5. The Effects of Opioids on Neuroimmune Signaling and NLRP3 Inflammasome
4. The Potential Therapeutic Effects of NLRP3 Inflammasome in SUDs
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lawson, L.J.; Perry, V.H.; Dri, P.; Gordon, S. Heterogeneity in the distribution and morphology of microglia in the normal adult mouse brain. Neuroscience 1990, 39, 151–170. [Google Scholar] [CrossRef] [PubMed]
- Mittelbronn, M.; Dietz, K.; Schluesener, H.J.; Meyermann, R. Local distribution of microglia in the normal adult human central nervous system differs by up to one order of magnitude. Acta Neuropathol. 2001, 101, 249–255. [Google Scholar] [CrossRef] [PubMed]
- Pelvig, D.P.; Pakkenberg, H.; Stark, A.K.; Pakkenberg, B. Neocortical glial cell numbers in human brains. Neurobiol. Aging 2008, 29, 1754–1762. [Google Scholar] [CrossRef] [PubMed]
- Prinz, M.; Jung, S.; Priller, J. Microglia Biology: One Century of Evolving Concepts. Cell 2019, 179, 292–311. [Google Scholar] [CrossRef]
- Salter, M.W.; Beggs, S. Sublime microglia: Expanding roles for the guardians of the CNS. Cell 2014, 158, 15–24. [Google Scholar] [CrossRef]
- Deczkowska, A.; Amit, I.; Schwartz, M. Microglial immune checkpoint mechanisms. Nat. Neurosci. 2018, 21, 779–786. [Google Scholar] [CrossRef]
- Hickman, S.; Izzy, S.; Sen, P.; Morsett, L.; El Khoury, J. Microglia in neurodegeneration. Nat. Neurosci. 2018, 21, 1359–1369. [Google Scholar] [CrossRef]
- Rong, G.; Hongrong, W.; Qingqi, L.; Jianfeng, Z. Roles of Microglia in AD Pathology. Curr. Alzheimer Res. 2023, 19, 854–869. [Google Scholar] [CrossRef]
- Jorfi, M.; Maaser-Hecker, A.; Tanzi, R.E. The neuroimmune axis of Alzheimer’s disease. Genome Med. 2023, 15, 6. [Google Scholar] [CrossRef]
- Xu, Y.; Li, Y.; Wang, C.; Han, T.; Liu, H.; Sun, L.; Hong, J.; Hashimoto, M.; Wei, J. The reciprocal interactions between microglia and T cells in Parkinson’s disease: A double-edged sword. J. Neuroinflamm. 2023, 20, 33. [Google Scholar] [CrossRef]
- Zhu, R.; Luo, Y.; Li, S.; Wang, Z. The role of microglial autophagy in Parkinson’s disease. Front. Aging Neurosci. 2022, 14, 1039780. [Google Scholar] [CrossRef]
- Wang, M.J.; Kang, L.; Wang, Y.Z.; Yang, B.R.; Zhang, C.; Lu, Y.F.; Kang, L. Microglia in motor neuron disease: Signaling evidence from last 10 years. Dev. Neurobiol. 2022, 82, 625–638. [Google Scholar] [CrossRef]
- McGrath, A.G.; Briand, L.A. A potential role for microglia in stress- and drug-induced plasticity in the nucleus accumbens: A mechanism for stress-induced vulnerability to substance use disorder. Neurosci. Biobehav. Rev. 2019, 107, 360–369. [Google Scholar] [CrossRef]
- Stellwagen, D.; Kemp, G.M.; Valade, S.; Chambon, J. Glial regulation of synaptic function in models of addiction. Curr. Opin. Neurobiol. 2019, 57, 179–185. [Google Scholar] [CrossRef]
- Strowig, T.; Henao-Mejia, J.; Elinav, E.; Flavell, R. Inflammasomes in health and disease. Nature 2012, 481, 278–286. [Google Scholar] [CrossRef]
- Zhan, X.; Li, Q.; Xu, G.; Xiao, X.; Bai, Z. The mechanism of NLRP3 inflammasome activation and its pharmacological inhibitors. Front. Immunol. 2022, 13, 1109938. [Google Scholar] [CrossRef]
- Butovsky, O.; Weiner, H.L. Microglial signatures and their role in health and disease. Nat. Rev. Neurosci. 2018, 19, 622–635. [Google Scholar] [CrossRef]
- De Biase, L.M.; Schuebel, K.E.; Fusfeld, Z.H.; Jair, K.; Hawes, I.A.; Cimbro, R.; Zhang, H.Y.; Liu, Q.R.; Shen, H.; Xi, Z.X.; et al. Local Cues Establish and Maintain Region-Specific Phenotypes of Basal Ganglia Microglia. Neuron 2017, 95, 341–356.e6. [Google Scholar] [CrossRef]
- Cserep, C.; Posfai, B.; Denes, A. Shaping Neuronal Fate: Functional Heterogeneity of Direct Microglia-Neuron Interactions. Neuron 2021, 109, 222–240. [Google Scholar] [CrossRef]
- Grabert, K.; Michoel, T.; Karavolos, M.H.; Clohisey, S.; Baillie, J.K.; Stevens, M.P.; Freeman, T.C.; Summers, K.M.; McColl, B.W. Microglial brain region-dependent diversity and selective regional sensitivities to aging. Nat. Neurosci. 2016, 19, 504–516. [Google Scholar] [CrossRef]
- Zia, S.; Hammond, B.P.; Zirngibl, M.; Sizov, A.; Baaklini, C.S.; Panda, S.P.; Ho, M.F.S.; Lee, K.V.; Mainali, A.; Burr, M.K.; et al. Single-cell microglial transcriptomics during demyelination defines a microglial state required for lytic carcass clearance. Mol. Neurodegener. 2022, 17, 82. [Google Scholar] [CrossRef] [PubMed]
- Bennett, J.P., Jr.; Keeney, P.M.; Brohawn, D.G. RNA Sequencing Reveals Small and Variable Contributions of Infectious Agents to Transcriptomes of Postmortem Nervous Tissues from Amyotrophic Lateral Sclerosis, Alzheimer’s Disease and Parkinson’s Disease Subjects, and Increased Expression of Genes From Disease-Activated Microglia. Front. Neurosci. 2019, 13, 235. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.J.; Au, N.P.B.; Ma, C.H.E. Functional and Phenotypic Diversity of Microglia: Implication for Microglia-Based Therapies for Alzheimer’s Disease. Front. Aging Neurosci. 2022, 14, 896852. [Google Scholar] [CrossRef] [PubMed]
- Keren-Shaul, H.; Spinrad, A.; Weiner, A.; Matcovitch-Natan, O.; Dvir-Szternfeld, R.; Ulland, T.K.; David, E.; Baruch, K.; Lara-Astaiso, D.; Toth, B.; et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 2017, 169, 1276–1290.e17. [Google Scholar] [CrossRef]
- Krasemann, S.; Madore, C.; Cialic, R.; Baufeld, C.; Calcagno, N.; El Fatimy, R.; Beckers, L.; O’Loughlin, E.; Xu, Y.; Fanek, Z.; et al. The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity 2017, 47, 566–581.e9. [Google Scholar] [CrossRef]
- Li, Q.; Cheng, Z.; Zhou, L.; Darmanis, S.; Neff, N.F.; Okamoto, J.; Gulati, G.; Bennett, M.L.; Sun, L.O.; Clarke, L.E.; et al. Developmental Heterogeneity of Microglia and Brain Myeloid Cells Revealed by Deep Single-Cell RNA Sequencing. Neuron 2019, 101, 207–223.e10. [Google Scholar] [CrossRef]
- Liu, X.; Yang, W.; Zhu, C.; Sun, S.; Wu, S.; Wang, L.; Wang, Y.; Ge, Z. Toll-like receptors and their role in neuropathic pain and migraine. Mol. Brain 2022, 15, 73. [Google Scholar] [CrossRef]
- Clark, A.K.; Malcangio, M. Fractalkine/CX3CR1 signaling during neuropathic pain. Front. Cell. Neurosci. 2014, 8, 121. [Google Scholar] [CrossRef]
- Wang, X.J.; Ye, M.; Zhang, Y.H.; Chen, S.D. CD200-CD200R regulation of microglia activation in the pathogenesis of Parkinson’s disease. J. Neuroimmune Pharmacol. 2007, 2, 259–264. [Google Scholar] [CrossRef]
- Fu, J.; Wu, H. Structural Mechanisms of NLRP3 Inflammasome Assembly and Activation. Annu. Rev. Immunol. 2022, 41, 301–316. [Google Scholar] [CrossRef]
- Erickson, E.K.; Grantham, E.K.; Warden, A.S.; Harris, R.A. Neuroimmune signaling in alcohol use disorder. Pharmacol. Biochem. Behav. 2019, 177, 34–60. [Google Scholar] [CrossRef]
- Cui, C.; Shurtleff, D.; Harris, R.A. Neuroimmune mechanisms of alcohol and drug addiction. Int. Rev. Neurobiol. 2014, 118, 1–12. [Google Scholar] [CrossRef]
- Rodrigues, L.C.; Gobira, P.H.; de Oliveira, A.C.; Pelicao, R.; Teixeira, A.L.; Moreira, F.A.; Campos, A.C. Neuroinflammation as a possible link between cannabinoids and addiction. Acta Neuropsychiatr. 2014, 26, 334–346. [Google Scholar] [CrossRef]
- Moreira, F.P.; Medeiros, J.R.; Lhullier, A.C.; Souza, L.D.; Jansen, K.; Portela, L.V.; Lara, D.R.; da Silva, R.A.; Wiener, C.D.; Oses, J.P. Cocaine abuse and effects in the serum levels of cytokines IL-6 and IL-10. Drug Alcohol Depend. 2016, 158, 181–185. [Google Scholar] [CrossRef]
- Narvaez, J.C.; Magalhaes, P.V.; Fries, G.R.; Colpo, G.D.; Czepielewski, L.S.; Vianna, P.; Chies, J.A.; Rosa, A.R.; Von Diemen, L.; Vieta, E.; et al. Peripheral toxicity in crack cocaine use disorders. Neurosci. Lett. 2013, 544, 80–84. [Google Scholar] [CrossRef]
- Namba, M.D.; Leyrer-Jackson, J.M.; Nagy, E.K.; Olive, M.F.; Neisewander, J.L. Neuroimmune Mechanisms as Novel Treatment Targets for Substance Use Disorders and Associated Comorbidities. Front. Neurosci. 2021, 15, 650785. [Google Scholar] [CrossRef]
- Jones, J.D. Potential of Glial Cell Modulators in the Management of Substance Use Disorders. CNS Drugs 2020, 34, 697–722. [Google Scholar] [CrossRef]
- Liao, K.; Guo, M.; Niu, F.; Yang, L.; Callen, S.E.; Buch, S. Cocaine-mediated induction of microglial activation involves the ER stress-TLR2 axis. J. Neuroinflamm. 2016, 13, 33. [Google Scholar] [CrossRef]
- Karimi-Haghighi, S.; Chavoshinezhad, S.; Mozafari, R.; Noorbakhsh, F.; Borhani-Haghighi, A.; Haghparast, A. Neuroinflammatory Response in Reward-Associated Psychostimulants and Opioids: A Review. Cell. Mol. Neurobiol. 2023, 43, 649–682. [Google Scholar] [CrossRef]
- Northcutt, A.L.; Hutchinson, M.R.; Wang, X.; Baratta, M.V.; Hiranita, T.; Cochran, T.A.; Pomrenze, M.B.; Galer, E.L.; Kopajtic, T.A.; Li, C.M.; et al. DAT isn’t all that: Cocaine reward and reinforcement require Toll-like receptor 4 signaling. Mol. Psychiatry 2015, 20, 1525–1537. [Google Scholar] [CrossRef]
- Periyasamy, P.; Liao, K.; Kook, Y.H.; Niu, F.; Callen, S.E.; Guo, M.L.; Buch, S. Cocaine-Mediated Downregulation of miR-124 Activates Microglia by Targeting KLF4 and TLR4 Signaling. Mol. Neurobiol. 2018, 55, 3196–3210. [Google Scholar] [CrossRef] [PubMed]
- Guo, M.L.; Periyasamy, P.; Liao, K.; Kook, Y.H.; Niu, F.; Callen, S.E.; Buch, S. Cocaine-mediated downregulation of microglial miR-124 expression involves promoter DNA methylation. Epigenetics 2016, 11, 819–830. [Google Scholar] [CrossRef] [PubMed]
- Guo, M.L.; Liao, K.; Periyasamy, P.; Yang, L.; Cai, Y.; Callen, S.E.; Buch, S. Cocaine-mediated microglial activation involves the ER stress-autophagy axis. Autophagy 2015, 11, 995–1009. [Google Scholar] [CrossRef] [PubMed]
- Xu, E.; Liu, J.; Wang, X.; Xiong, H. Inflammasome in drug abuse. Int. J. Physiol. Pathophysiol. Pharmacol. 2017, 9, 165–177. [Google Scholar]
- Atluri, V.S.; Pilakka-Kanthikeel, S.; Garcia, G.; Jayant, R.D.; Sagar, V.; Samikkannu, T.; Yndart, A.; Nair, M. Effect of Cocaine on HIV Infection and Inflammasome Gene Expression Profile in HIV Infected Macrophages. Sci. Rep. 2016, 6, 27864. [Google Scholar] [CrossRef]
- Chivero, E.T.; Thangaraj, A.; Tripathi, A.; Periyasamy, P.; Guo, M.L.; Buch, S. NLRP3 Inflammasome Blockade Reduces Cocaine-Induced Microglial Activation and Neuroinflammation. Mol. Neurobiol. 2021, 58, 2215–2230. [Google Scholar] [CrossRef]
- Guo, M.L.; Chivero, E.T.; Callen, S.E.; Buch, S. NLRP3 Inflammasome Is Involved in Cocaine-Mediated Potentiation on Behavioral Changes in CX3CR1-Deficient Mice. J. Pers. Med. 2021, 11, 963. [Google Scholar] [CrossRef]
- Ge, Y.; Wang, L.; Wang, C.; Chen, J.; Dai, M.; Yao, S.; Lin, Y. CX3CL1 inhibits NLRP3 inflammasome-induced microglial pyroptosis and improves neuronal function in mice with experimentally-induced ischemic stroke. Life Sci. 2022, 300, 120564. [Google Scholar] [CrossRef]
- Palamar, J.J.; Han, B.H.; Keyes, K.M. Trends in characteristics of individuals who use methamphetamine in the United States, 2015–2018. Drug Alcohol Depend. 2020, 213, 108089. [Google Scholar] [CrossRef]
- Shi, S.; Chen, T.; Zhao, M. The Crosstalk between Neurons and Glia in Methamphetamine-Induced Neuroinflammation. Neurochem. Res. 2022, 47, 872–884. [Google Scholar] [CrossRef]
- Kim, B.; Yun, J.; Park, B. Methamphetamine-Induced Neuronal Damage: Neurotoxicity and Neuroinflammation. Biomol. Ther. 2020, 28, 381–388. [Google Scholar] [CrossRef]
- Shaerzadeh, F.; Streit, W.J.; Heysieattalab, S.; Khoshbouei, H. Methamphetamine neurotoxicity, microglia, and neuroinflammation. J. Neuroinflamm. 2018, 15, 341. [Google Scholar] [CrossRef]
- Kousik, S.M.; Napier, T.C.; Carvey, P.M. The effects of psychostimulant drugs on blood brain barrier function and neuroinflammation. Front. Pharmacol. 2012, 3, 121. [Google Scholar] [CrossRef]
- Xiao, Y.; Jin, J.; Chang, M.; Chang, J.H.; Hu, H.; Zhou, X.; Brittain, G.C.; Stansberg, C.; Torkildsen, O.; Wang, X.; et al. Peli1 promotes microglia-mediated CNS inflammation by regulating Traf3 degradation. Nat. Med. 2013, 19, 595–602. [Google Scholar] [CrossRef]
- Huang, X.P.; Peng, J.H.; Pang, J.W.; Tian, X.C.; Li, X.S.; Wu, Y.; Li, Y.; Jiang, Y.; Sun, X.C. Peli1 Contributions in Microglial Activation, Neuroinflammatory Responses and Neurological Deficits Following Experimental Subarachnoid Hemorrhage. Front. Mol. Neurosci. 2017, 10, 398. [Google Scholar] [CrossRef]
- Yang, T.; Zang, S.; Wang, Y.; Zhu, Y.; Jiang, L.; Chen, X.; Zhang, X.; Cheng, J.; Gao, R.; Xiao, H.; et al. Methamphetamine induced neuroinflammation in mouse brain and microglial cell line BV2: Roles of the TLR4/TRIF/Peli1 signaling axis. Toxicol. Lett. 2020, 333, 150–158. [Google Scholar] [CrossRef]
- Namyen, J.; Permpoonputtana, K.; Nopparat, C.; Tocharus, J.; Tocharus, C.; Govitrapong, P. Protective Effects of Melatonin on Methamphetamine-Induced Blood-Brain Barrier Dysfunction in Rat Model. Neurotox. Res. 2020, 37, 640–660. [Google Scholar] [CrossRef]
- Yu, G.; Song, Y.; Xie, C.; Tao, L.; Wan, F.; Jiang, L.; Wang, J.; Tang, J. MiR-142a-3p and miR-155-5p reduce methamphetamine-induced inflammation: Role of the target protein Peli1. Toxicol. Appl. Pharmacol. 2019, 370, 145–153. [Google Scholar] [CrossRef]
- Robson, M.J.; Turner, R.C.; Naser, Z.J.; McCurdy, C.R.; Huber, J.D.; Matsumoto, R.R. SN79, a sigma receptor ligand, blocks methamphetamine-induced microglial activation and cytokine upregulation. Exp. Neurol. 2013, 247, 134–142. [Google Scholar] [CrossRef]
- Xu, E.; Liu, J.; Liu, H.; Wang, X.; Xiong, H. Inflammasome Activation by Methamphetamine Potentiates Lipopolysaccharide Stimulation of IL-1beta Production in Microglia. J. Neuroimmune Pharmacol. 2018, 13, 237–253. [Google Scholar] [CrossRef] [PubMed]
- Du, L.; Shen, K.; Bai, Y.; Chao, J.; Hu, G.; Zhang, Y.; Yao, H. Involvement of NLRP3 inflammasome in methamphetamine-induced microglial activation through miR-143/PUMA axis. Toxicol. Lett. 2019, 301, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Mahmoudiasl, G.R.; Abbaszadeh, H.A.; Rezaei-Tavirani, M.; Abdollahifar, M.A.; Khoramgah, M.S.; Niknazar, S.; Darabi, S.; Roozbahany, N.A. Nod-like receptor protein 3 and nod-like receptor protein 1 inflammasome activation in the hippocampal region of postmortem methamphetamine chronic user. Bratisl. Lek. Listy 2019, 120, 769–776. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.; Shen, L.; Ye, Y.; Hu, S.; Ren, Z.; Liu, T.; Dai, J.; Li, Z.; Wang, J.; Luo, Y.; et al. Inflammasome Inhibition Prevents Motor Deficit and Cerebellar Degeneration Induced by Chronic Methamphetamine Administration. Front. Mol. Neurosci. 2022, 15, 861340. [Google Scholar] [CrossRef]
- Zhao, J.; Shen, S.; Dai, Y.; Chen, F.; Wang, K. Methamphetamine Induces Intestinal Inflammatory Injury via Nod-Like Receptor 3 Protein (NLRP3) Inflammasome Overexpression In Vitro and In Vivo. Med. Sci. Monit. 2019, 25, 8515–8526. [Google Scholar] [CrossRef]
- Fan, R.; Shen, Y.; Li, X.; Luo, H.; Zhang, P.; Liu, Y.; Si, Z.; Zhou, W.; Liu, Y. The effect of the NLRP1 inflammasome on methamphetamine-induced cognitive impairment in rats. Drug Alcohol Depend. 2022, 237, 109537. [Google Scholar] [CrossRef]
- Crews, F.T.; Lawrimore, C.J.; Walter, T.J.; Coleman, L.G., Jr. The role of neuroimmune signaling in alcoholism. Neuropharmacology 2017, 122, 56–73. [Google Scholar] [CrossRef]
- Melbourne, J.K.; Thompson, K.R.; Peng, H.; Nixon, K. Its complicated: The relationship between alcohol and microglia in the search for novel pharmacotherapeutic targets for alcohol use disorders. Prog. Mol. Biol. Transl. Sci. 2019, 167, 179–221. [Google Scholar] [CrossRef]
- Melbourne, J.K.; Chandler, C.M.; Van Doorn, C.E.; Bardo, M.T.; Pauly, J.R.; Peng, H.; Nixon, K. Primed for addiction: A critical review of the role of microglia in the neurodevelopmental consequences of adolescent alcohol drinking. Alcohol. Clin. Exp. Res. 2021, 45, 1908–1926. [Google Scholar] [CrossRef]
- Lippai, D.; Bala, S.; Csak, T.; Kurt-Jones, E.A.; Szabo, G. Chronic alcohol-induced microRNA-155 contributes to neuroinflammation in a TLR4-dependent manner in mice. PLoS ONE 2013, 8, e70945. [Google Scholar] [CrossRef]
- Zhang, Y.; Wei, G.; Di, Z.; Zhao, Q. miR-339-5p inhibits alcohol-induced brain inflammation through regulating NF-kappaB pathway. Biochem. Biophys. Res. Commun. 2014, 452, 450–456. [Google Scholar] [CrossRef]
- Coleman, L.G., Jr.; Zou, J.; Crews, F.T. Microglial-derived miRNA let-7 and HMGB1 contribute to ethanol-induced neurotoxicity via TLR7. J. Neuroinflamm. 2017, 14, 22. [Google Scholar] [CrossRef] [PubMed]
- Mizuo, K.; Katada, R.; Okazaki, S.; Tateda, K.; Watanabe, S.; Matsumoto, H. Epigenetic regulation of MIR-124 under ethanol dependence and withdrawal. Nihon Arukoru Yakubutsu Igakkai Zasshi 2012, 47, 155–163. [Google Scholar] [PubMed]
- Leon, B.E.; Kang, S.; Franca-Solomon, G.; Shang, P.; Choi, D.S. Alcohol-Induced Neuroinflammatory Response and Mitochondrial Dysfunction on Aging and Alzheimer’s Disease. Front. Behav. Neurosci. 2021, 15, 778456. [Google Scholar] [CrossRef]
- Zou, J.; Walter, T.J.; Barnett, A.; Rohlman, A.; Crews, F.T.; Coleman, L.G., Jr. Ethanol Induces Secretion of Proinflammatory Extracellular Vesicles That Inhibit Adult Hippocampal Neurogenesis Through G9a/GLP-Epigenetic Signaling. Front. Immunol. 2022, 13, 866073. [Google Scholar] [CrossRef]
- Montesinos, J.; Alfonso-Loeches, S.; Guerri, C. Impact of the Innate Immune Response in the Actions of Ethanol on the Central Nervous System. Alcohol. Clin. Exp. Res. 2016, 40, 2260–2270. [Google Scholar] [CrossRef]
- Singh, S.; Jha, S. NLRs as Helpline in the Brain: Mechanisms and Therapeutic Implications. Mol. Neurobiol. 2018, 55, 8154–8178. [Google Scholar] [CrossRef]
- Lowe, P.P.; Cho, Y.; Tornai, D.; Coban, S.; Catalano, D.; Szabo, G. Inhibition of the Inflammasome Signaling Cascade Reduces Alcohol Consumption in Female But Not Male Mice. Alcohol. Clin. Exp. Res. 2020, 44, 567–578. [Google Scholar] [CrossRef]
- Hoyt, L.R.; Randall, M.J.; Ather, J.L.; DePuccio, D.P.; Landry, C.C.; Qian, X.; Janssen-Heininger, Y.M.; van der Vliet, A.; Dixon, A.E.; Amiel, E.; et al. Mitochondrial ROS induced by chronic ethanol exposure promote hyper-activation of the NLRP3 inflammasome. Redox Biol. 2017, 12, 883–896. [Google Scholar] [CrossRef]
- De Filippis, L.; Halikere, A.; McGowan, H.; Moore, J.C.; Tischfield, J.A.; Hart, R.P.; Pang, Z.P. Ethanol-mediated activation of the NLRP3 inflammasome in iPS cells and iPS cells-derived neural progenitor cells. Mol. Brain 2016, 9, 51. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Chu, G.; Yang, Z.; Sun, Y.; Zhou, H.; Li, M.; Shi, J.; Tian, B.; Zhang, C.; Meng, X. Ethanol directly induced HMGB1 release through NOX2/NLRP1 inflammasome in neuronal cells. Toxicology 2015, 334, 104–110. [Google Scholar] [CrossRef]
- Alfonso-Loeches, S.; Urena-Peralta, J.; Morillo-Bargues, M.J.; Gomez-Pinedo, U.; Guerri, C. Ethanol-Induced TLR4/NLRP3 Neuroinflammatory Response in Microglial Cells Promotes Leukocyte Infiltration Across the BBB. Neurochem. Res. 2016, 41, 193–209. [Google Scholar] [CrossRef] [PubMed]
- de Carvalho Ribeiro, M.; Szabo, G. Role of the Inflammasome in Liver Disease. Annu. Rev. Pathol. 2022, 17, 345–365. [Google Scholar] [CrossRef] [PubMed]
- Torres, S.; Segales, P.; Garcia-Ruiz, C.; Fernandez-Checa, J.C. Mitochondria and the NLRP3 Inflammasome in Alcoholic and Nonalcoholic Steatohepatitis. Cells 2022, 11, 1475. [Google Scholar] [CrossRef]
- Mainz, R.E.; Albers, S.; Haque, M.; Sonntag, R.; Treichel, N.S.; Clavel, T.; Latz, E.; Schneider, K.M.; Trautwein, C.; Otto, T. NLRP6 Inflammasome Modulates Disease Progression in a Chronic-Plus-Binge Mouse Model of Alcoholic Liver Disease. Cells 2022, 11, 182. [Google Scholar] [CrossRef]
- Ji, X.; Li, L.; Lu, P.; Li, X.; Tian, D.; Liu, M. NLRP6 exerts a protective role via NF-kB with involvement of CCL20 in a mouse model of alcoholic hepatitis. Biochem. Biophys. Res. Commun. 2020, 528, 485–492. [Google Scholar] [CrossRef]
- DeSantis, D.A.; Ko, C.W.; Liu, Y.; Liu, X.; Hise, A.G.; Nunez, G.; Croniger, C.M. Alcohol-induced liver injury is modulated by Nlrp3 and Nlrc4 inflammasomes in mice. Mediat. Inflamm. 2013, 2013, 751374. [Google Scholar] [CrossRef]
- Martins, S.S.; Levy, N.S.; Bruzelius, E.; Segura, L.E. Cannabis legalization in the U.S. Where do we go from here? Trends Psychiatry Psychother. 2022, 44 (Suppl. 1), e20220001. [Google Scholar] [CrossRef]
- Klumpers, L.E.; Thacker, D.L. A Brief Background on Cannabis: From Plant to Medical Indications. J. AOAC Int. 2019, 102, 412–420. [Google Scholar] [CrossRef]
- Calapai, F.; Cardia, L.; Sorbara, E.E.; Navarra, M.; Gangemi, S.; Calapai, G.; Mannucci, C. Cannabinoids, Blood-Brain Barrier, and Brain Disposition. Pharmaceutics 2020, 12, 265. [Google Scholar] [CrossRef]
- Blithikioti, C.; Miquel, L.; Batalla, A.; Rubio, B.; Maffei, G.; Herreros, I.; Gual, A.; Verschure, P.; Balcells-Olivero, M. Cerebellar alterations in cannabis users: A systematic review. Addict. Biol. 2019, 24, 1121–1137. [Google Scholar] [CrossRef]
- Katona, I.; Freund, T.F. Multiple functions of endocannabinoid signaling in the brain. Annu. Rev. Neurosci. 2012, 35, 529–558. [Google Scholar] [CrossRef] [PubMed]
- Bie, B.; Wu, J.; Foss, J.F.; Naguib, M. An overview of the cannabinoid type 2 receptor system and its therapeutic potential. Curr. Opin. Anaesthesiol. 2018, 31, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Cutando, L.; Busquets-Garcia, A.; Puighermanal, E.; Gomis-Gonzalez, M.; Delgado-Garcia, J.M.; Gruart, A.; Maldonado, R.; Ozaita, A. Microglial activation underlies cerebellar deficits produced by repeated cannabis exposure. J. Clin. Investig. 2013, 123, 2816–2831. [Google Scholar] [CrossRef] [PubMed]
- Suryavanshi, S.V.; Zaiachuk, M.; Pryimak, N.; Kovalchuk, I.; Kovalchuk, O. Cannabinoids Alleviate the LPS-Induced Cytokine Storm via Attenuating NLRP3 Inflammasome Signaling and TYK2-Mediated STAT3 Signaling Pathways In Vitro. Cells 2022, 11, 1391. [Google Scholar] [CrossRef] [PubMed]
- Suryavanshi, S.V.; Kovalchuk, I.; Kovalchuk, O. Cannabinoids as Key Regulators of Inflammasome Signaling: A Current Perspective. Front. Immunol. 2020, 11, 613613. [Google Scholar] [CrossRef]
- Dos-Santos-Pereira, M.; Guimaraes, F.S.; Del-Bel, E.; Raisman-Vozari, R.; Michel, P.P. Cannabidiol prevents LPS-induced microglial inflammation by inhibiting ROS/NF-kappaB-dependent signaling and glucose consumption. Glia 2020, 68, 561–573. [Google Scholar] [CrossRef]
- Rizzo, M.D.; Crawford, R.B.; Bach, A.; Sermet, S.; Amalfitano, A.; Kaminski, N.E. Delta(9)-Tetrahydrocannabinol Suppresses Monocyte-Mediated Astrocyte Production of Monocyte Chemoattractant Protein 1 and Interleukin-6 in a Toll-Like Receptor 7-Stimulated Human Coculture. J. Pharmacol. Exp. Ther. 2019, 371, 191–201. [Google Scholar] [CrossRef]
- Borgonetti, V.; Benatti, C.; Governa, P.; Isoldi, G.; Pellati, F.; Alboni, S.; Tascedda, F.; Montopoli, M.; Galeotti, N.; Manetti, F.; et al. Non-psychotropic Cannabis sativa L. phytocomplex modulates microglial inflammatory response through CB2 receptors-, endocannabinoids-, and NF-kappaB-mediated signaling. Phytother. Res. 2022, 36, 2246–2263. [Google Scholar] [CrossRef]
- Rimmerman, N.; Juknat, A.; Kozela, E.; Levy, R.; Bradshaw, H.B.; Vogel, Z. The non-psychoactive plant cannabinoid, cannabidiol affects cholesterol metabolism-related genes in microglial cells. Cell. Mol. Neurobiol. 2011, 31, 921–930. [Google Scholar] [CrossRef]
- Shao, B.Z.; Wei, W.; Ke, P.; Xu, Z.Q.; Zhou, J.X.; Liu, C. Activating cannabinoid receptor 2 alleviates pathogenesis of experimental autoimmune encephalomyelitis via activation of autophagy and inhibiting NLRP3 inflammasome. CNS Neurosci. Ther. 2014, 20, 1021–1028. [Google Scholar] [CrossRef]
- Qi, X.; Lin, W.; Wu, Y.; Li, Q.; Zhou, X.; Li, H.; Xiao, Q.; Wang, Y.; Shao, B.; Yuan, Q. CBD Promotes Oral Ulcer Healing via Inhibiting CMPK2-Mediated Inflammasome. J. Dent. Res. 2022, 101, 206–215. [Google Scholar] [CrossRef]
- Hedegaard, H.; Minino, A.M.; Warner, M. Drug Overdose Deaths in the United States, 1999–2019. NCHS Data Brief. 2020, 1–8. [Google Scholar] [CrossRef]
- Qiu, S.; Feng, Y.; LeSage, G.; Zhang, Y.; Stuart, C.; He, L.; Li, Y.; Caudle, Y.; Peng, Y.; Yin, D. Chronic morphine-induced microRNA-124 promotes microglial immunosuppression by modulating P65 and TRAF6. J. Immunol. 2015, 194, 1021–1030. [Google Scholar] [CrossRef]
- Peng, J.; Pan, J.; Wang, H.; Mo, J.; Lan, L.; Peng, Y. Morphine-induced microglial immunosuppression via activation of insufficient mitophagy regulated by NLRX1. J. Neuroinflamm. 2022, 19, 87. [Google Scholar] [CrossRef]
- Terminel, M.N.; Bassil, C.; Rau, J.; Trevino, A.; Ruiz, C.; Alaniz, R.; Hook, M.A. Morphine-induced changes in the function of microglia and macrophages after acute spinal cord injury. BMC Neurosci. 2022, 23, 58. [Google Scholar] [CrossRef]
- Yang, Y.; Sun, Y.; Hu, R.; Yan, J.; Wang, Z.; Li, W.; Jiang, H. Morphine promotes microglial activation by upregulating the EGFR/ERK signaling pathway. PLoS ONE 2021, 16, e0256870. [Google Scholar] [CrossRef]
- Wang, X.; Loram, L.C.; Ramos, K.; de Jesus, A.J.; Thomas, J.; Cheng, K.; Reddy, A.; Somogyi, A.A.; Hutchinson, M.R.; Watkins, L.R.; et al. Morphine activates neuroinflammation in a manner parallel to endotoxin. Proc. Natl. Acad. Sci. USA 2012, 109, 6325–6330. [Google Scholar] [CrossRef]
- Zhang, P.; Yang, M.; Chen, C.; Liu, L.; Wei, X.; Zeng, S. Toll-like Receptor 4 (TLR4)/Opioid Receptor Pathway Crosstalk and Impact on Opioid Analgesia, Immune Function, and Gastrointestinal Motility. Front. Immunol. 2020, 11, 1455. [Google Scholar] [CrossRef]
- Sil, S.; Periyasamy, P.; Guo, M.L.; Callen, S.; Buch, S. Morphine-Mediated Brain Region-Specific Astrocytosis Involves the ER Stress-Autophagy Axis. Mol. Neurobiol. 2018, 55, 6713–6733. [Google Scholar] [CrossRef]
- Sil, S.; Singh, S.; Chemparathy, D.T.; Chivero, E.T.; Gordon, L.; Buch, S. Astrocytes & Astrocyte derived Extracellular Vesicles in Morphine Induced Amyloidopathy: Implications for Cognitive Deficits in Opiate Abusers. Aging Dis. 2021, 12, 1389–1408. [Google Scholar] [CrossRef]
- Cai, Y.; Kong, H.; Pan, Y.B.; Jiang, L.; Pan, X.X.; Hu, L.; Qian, Y.N.; Jiang, C.Y.; Liu, W.T. Procyanidins alleviates morphine tolerance by inhibiting activation of NLRP3 inflammasome in microglia. J. Neuroinflamm. 2016, 13, 53. [Google Scholar] [CrossRef] [PubMed]
- Grace, P.M.; Strand, K.A.; Galer, E.L.; Urban, D.J.; Wang, X.; Baratta, M.V.; Fabisiak, T.J.; Anderson, N.D.; Cheng, K.; Greene, L.I.; et al. Morphine paradoxically prolongs neuropathic pain in rats by amplifying spinal NLRP3 inflammasome activation. Proc. Natl. Acad. Sci. USA 2016, 113, E3441–E3450. [Google Scholar] [CrossRef] [PubMed]
- Grace, P.M.; Strand, K.A.; Galer, E.L.; Rice, K.C.; Maier, S.F.; Watkins, L.R. Protraction of neuropathic pain by morphine is mediated by spinal damage associated molecular patterns (DAMPs) in male rats. Brain Behav. Immun. 2018, 72, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhang, Y.; Ma, X.; Wang, W.; Xu, X.; Huang, M.; Xu, L.; Shi, H.; Yuan, T.; Jiang, W.; et al. Spinal TLR4/P2X7 Receptor-Dependent NLRP3 Inflammasome Activation Contributes to the Development of Tolerance to Morphine-Induced Antinociception. J. Inflamm. Res. 2020, 13, 571–582. [Google Scholar] [CrossRef]
- Wang, H.; Huang, M.; Wang, W.; Zhang, Y.; Ma, X.; Luo, L.; Xu, X.; Xu, L.; Shi, H.; Xu, Y.; et al. Microglial TLR4-induced TAK1 phosphorylation and NLRP3 activation mediates neuroinflammation and contributes to chronic morphine-induced antinociceptive tolerance. Pharmacol. Res. 2021, 165, 105482. [Google Scholar] [CrossRef]
- Carranza-Aguilar, C.J.; Hernandez-Mendoza, A.; Mejias-Aponte, C.; Rice, K.C.; Morales, M.; Gonzalez-Espinosa, C.; Cruz, S.L. Morphine and Fentanyl Repeated Administration Induces Different Levels of NLRP3-Dependent Pyroptosis in the Dorsal Raphe Nucleus of Male Rats via Cell-Specific Activation of TLR4 and Opioid Receptors. Cell. Mol. Neurobiol. 2022, 42, 677–694. [Google Scholar] [CrossRef]
- Chen, Q.L.; Yin, H.R.; He, Q.Y.; Wang, Y. Targeting the NLRP3 inflammasome as new therapeutic avenue for inflammatory bowel disease. Biomed. Pharmacother. 2021, 138, 111442. [Google Scholar] [CrossRef]
- He, W.; Hu, Z.; Zhong, Y.; Wu, C.; Li, J. The Potential of NLRP3 Inflammasome as a Therapeutic Target in Neurological Diseases. Mol. Neurobiol. 2023, 60, 2520–2538. [Google Scholar] [CrossRef]
- Feng, T.; Ma, Z.; Pan, C.; Yu, P. Pentoxifylline decreases the activity of the nucleotide-binding oligomerization domain-like receptor protein 3 pathway: Potential role for preventing arteriovenous fistula stenosis. J. Vasc. Access 2022, 10, 11297298221124730. [Google Scholar] [CrossRef]
- Lu, Y.; Xiao, G.; Luo, W. Minocycline Suppresses NLRP3 Inflammasome Activation in Experimental Ischemic Stroke. Neuroimmunomodulation 2016, 23, 230–238. [Google Scholar] [CrossRef]
- Li, X.; Zou, Y.; Fu, Y.Y.; Xing, J.; Wang, K.Y.; Wan, P.Z.; Wang, M.; Zhai, X.Y. Ibudilast Attenuates Folic Acid-Induced Acute Kidney Injury by Blocking Pyroptosis Through TLR4-Mediated NF-kappaB and MAPK Signaling Pathways. Front. Pharmacol. 2021, 12, 650283. [Google Scholar] [CrossRef]
- Zhang, Y.L.; Wang, R.B.; Li, W.Y.; Xia, F.Z.; Liu, L. Pioglitazone ameliorates retinal ischemia/reperfusion injury via suppressing NLRP3 inflammasome activities. Int. J. Ophthalmol. 2017, 10, 1812–1818. [Google Scholar] [CrossRef]
- Liu, X.; Liu, H.; Lu, X.; Zhao, S. N-acetylcysteine alleviates ocular surface damage in STZ-induced diabetic mice by inhibiting the ROS/NLRP3/Caspase-1/IL-1beta signaling pathway. Exp. Eye Res. 2021, 209, 108654. [Google Scholar] [CrossRef]
- Liu, L.; Chen, M.; Lin, K.; Xiang, X.; Zheng, Y.; Zhu, S. Inhibiting Caspase-12 Mediated Inflammasome Activation protects against Oxygen-Glucose Deprivation Injury in Primary Astrocytes. Int. J. Med. Sci. 2020, 17, 1936–1945. [Google Scholar] [CrossRef]
- Panicker, N.; Kam, T.I.; Wang, H.; Neifert, S.; Chou, S.C.; Kumar, M.; Brahmachari, S.; Jhaldiyal, A.; Hinkle, J.T.; Akkentli, F.; et al. Neuronal NLRP3 is a parkin substrate that drives neurodegeneration in Parkinson’s disease. Neuron 2022, 110, 2422–2437.e9. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Drugs | Mechanisms of Action | Opioids | Psychostimulants | Alcohol | Cannabis | NLRP3 Inhibition |
|---|---|---|---|---|---|---|
| Minocycline | microglial inhibitor | positive effects  | positive effects | positive effects (-) | N/A | yes |
| craving (-) | ||||||
| Ibudilast | TNFα inhibitor | positive effects (-) | N/A | positive effects (-) | N/A | yes |
| withdrawal | craving | |||||
| Pioglitazone | cytokine inhibitor | positive effects (-) | reinforcing effects | N/A | N/A | yes |
| craving | craving | |||||
| N-Acetylcysteine | GLT-1 upregulation | N/A | positive effects | N/A | craving | yes |
| ROS scavenger | craving | abstinence (-) | ||||
| abstinence | ||||||
| Pentoxifylline | cytokine inhibitor | N/A | abstinence (-) | N/A | N/A | yes |
| Cocaine | Meth | Alcohol | Marijuana | Morphine | |
|---|---|---|---|---|---|
| TLR/NF-кB | Up | Up | Up | Down | Up |
| ROS | Up | Up | Up | No test | Up |
| NLRP3 | Up | Up | Up | Down | Up |
| miR-124 | Down | Down | Down | No test | Up |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guo, M.-L.; Roodsari, S.K.; Cheng, Y.; Dempsey, R.E.; Hu, W. Microglia NLRP3 Inflammasome and Neuroimmune Signaling in Substance Use Disorders. Biomolecules 2023, 13, 922. https://doi.org/10.3390/biom13060922
Guo M-L, Roodsari SK, Cheng Y, Dempsey RE, Hu W. Microglia NLRP3 Inflammasome and Neuroimmune Signaling in Substance Use Disorders. Biomolecules. 2023; 13(6):922. https://doi.org/10.3390/biom13060922
Chicago/Turabian StyleGuo, Ming-Lei, Soheil Kazemi Roodsari, Yan Cheng, Rachael Elizabeth Dempsey, and Wenhui Hu. 2023. "Microglia NLRP3 Inflammasome and Neuroimmune Signaling in Substance Use Disorders" Biomolecules 13, no. 6: 922. https://doi.org/10.3390/biom13060922