
Monoamine Oxidase A Contributes to Serotonin—But Not Norepinephrine-Dependent Damage of Rat Ventricular Myocytes
 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethical Concerns
2.2. Cell Isolation
2.3. Samples from Ageing and Hypertensive Rats
2.4. Mitochondria Isolation
2.5. Detection of ROS
2.6. Cardiomyocytes Contraction
2.7. Cell Structure
2.8. Western Blot
2.9. PCR Analysis
2.10. Statistics
3. Results
3.1. Effect of Pressure Overload on the Expression of MAOA and MAOB in Rats Hearts
3.2. Cardiac Expression of MAO-A and MAO-B in Rats and Mice
3.3. Effect of 5-HT on Load-Free Cell Shortening in Mouse Myocytes and ARVM
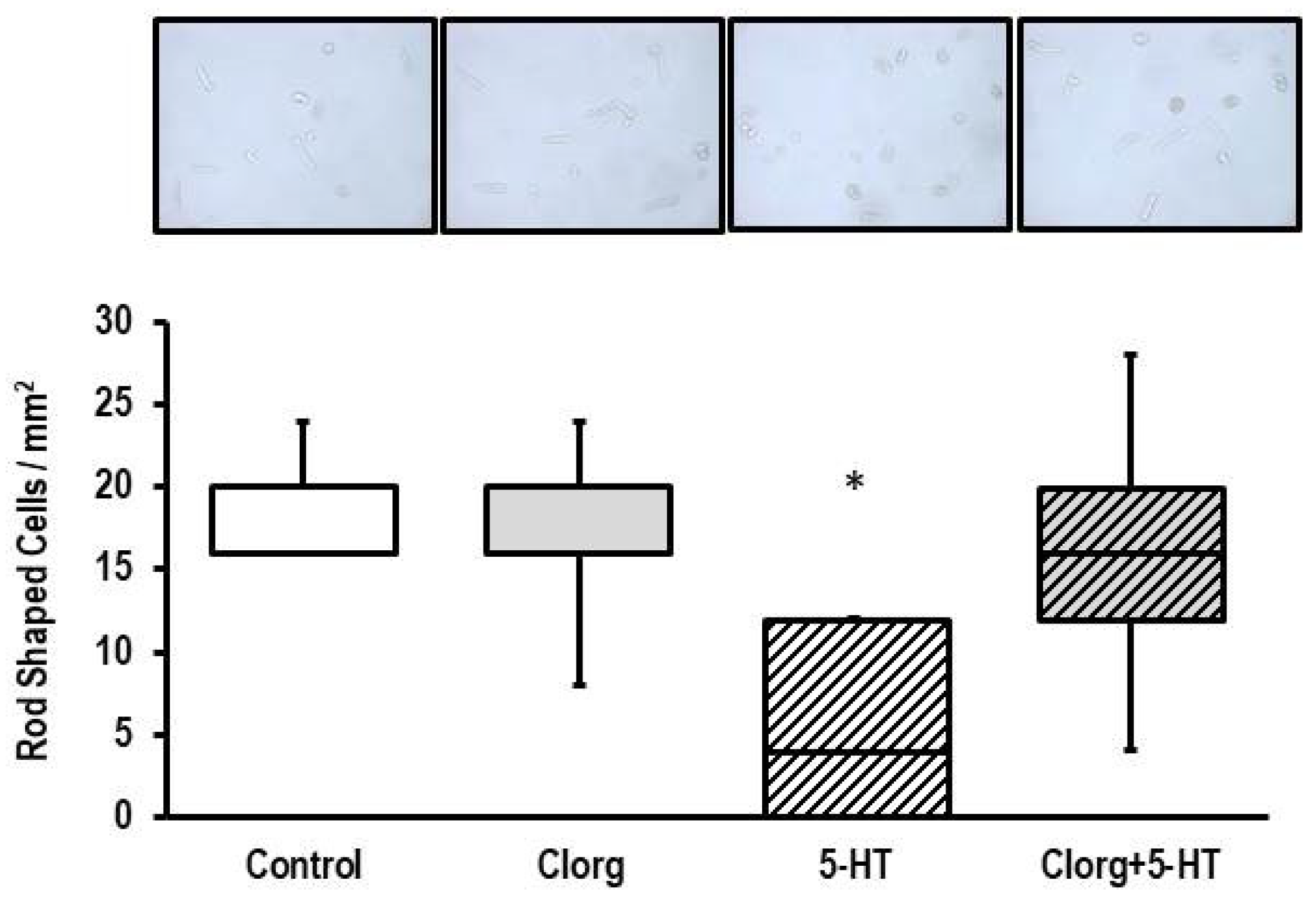
3.4. Effect of 5-HT on Structural Integrity of ARVM
3.5. Effect of 5-HT on ARVM Isolated from Either the Right or Left Ventricle
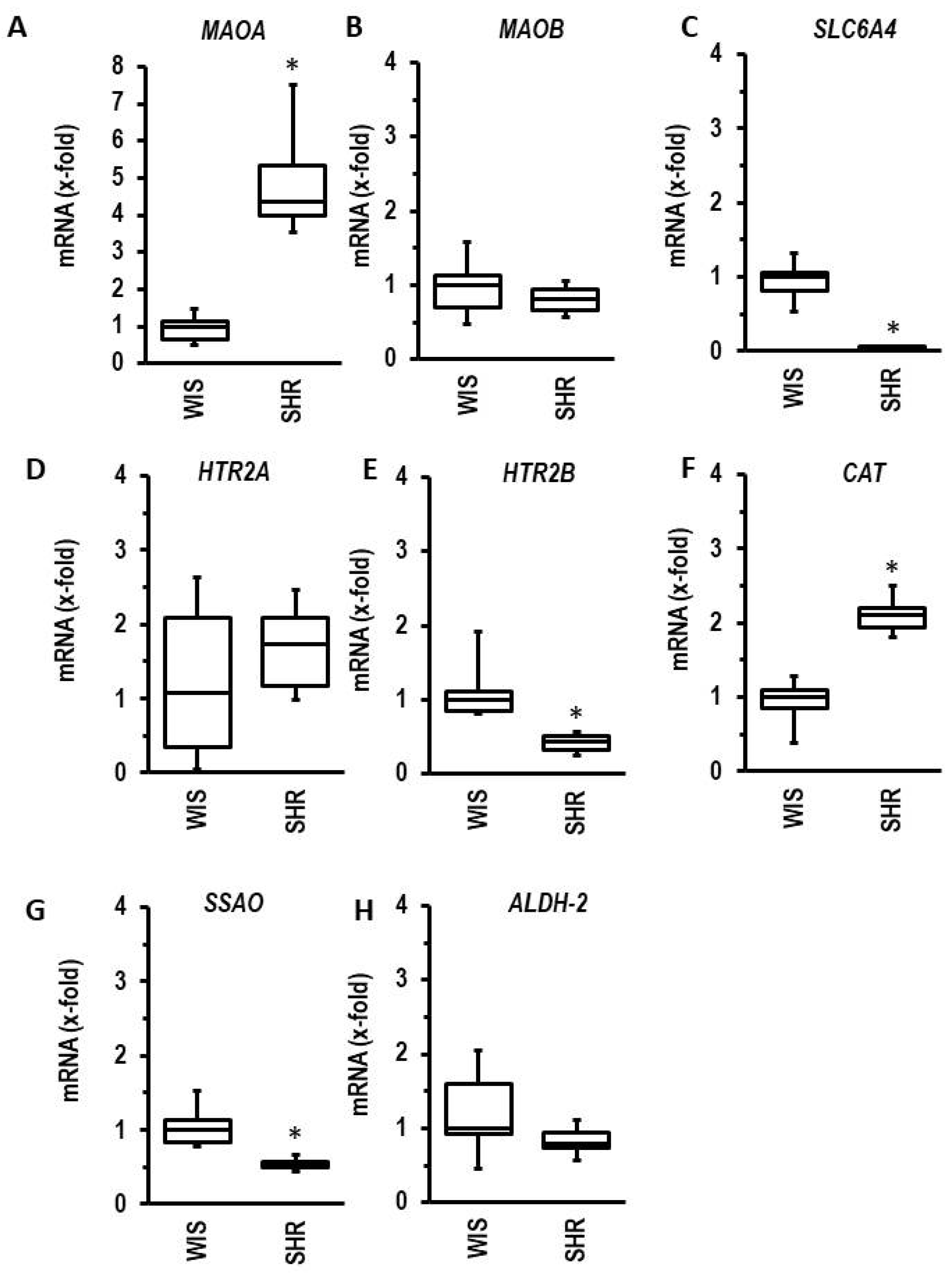
3.6. Effect of Ageing and Hypertension on the Expression of MAOA and Genes Required for 5-HT Metabolism
3.7. Effect of MAO-A on Norepinephrine-Dependent Cell Damages
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Peroutka, S.J. 5-Hydroxytryptamine receptors in vertebrates and invertebrates: Why are there so many? Neurochem. Int. 1994, 25, 533–536. [Google Scholar] [CrossRef] [PubMed]
- Saxena, P.R.; Lawang, A. A comparison of cardiovascular and smooth muscle effects of 5-hydroxytryptamine and 5-carboxyamidotryptamine, a selective agonist of 5-HT1 receptors. Arch. Int. Pharmacodyn. Ther. 1985, 277, 235–252. [Google Scholar] [PubMed]
- Mammadova-Bach, E.; Mauler, M.; Braun, A.; Duerschmied, D. Autocrine and paracrine regulatory functions of platelet serotonin. Platelets 2018, 29, 541–548. [Google Scholar] [CrossRef] [PubMed]
- Ursin, R. Serotonin and sleep. Sleep Med. Rev. 2002, 6, 55–69. [Google Scholar] [CrossRef] [Green Version]
- Curzon, G. Serotonin and appetite. Ann. N. Y. Acad. Sci. 1990, 600, 521–530. [Google Scholar] [CrossRef]
- Richardson, B.P. Serotonin and nociception. Ann. N. Y. Acad. Sci. 1990, 600, 511–519. [Google Scholar] [CrossRef]
- Malcolm, B.; Thomas, K. Serotonin toxicity and serotonergic psychedelics. Psychopharmacology 2022, 239, 1881–1891. [Google Scholar] [CrossRef]
- Kaludercic, N.; Mialet-Perez, J.; Paolocci, N.; Parini, A.; di Lisa, F. Monoamine oxidases as source of oxidants in the heart. J. Mol. Cell. Cardiol. 2014, 73, 34–42. [Google Scholar] [CrossRef] [Green Version]
- Nebigil, C.G.; Etienne, N.; Messaddeq, N.; Maroteaux, L. Serotonin is a novel survival factor of cardiomyocytes: Mitochondria as a target of 5-HT 2B receptor signaling. FASEB J. 2003, 17, 1373–1375. [Google Scholar] [CrossRef] [Green Version]
- Gao, W.; Guo, N.; Zhao, S.; Chen, Z.; Zhang, W.; Yan, F.; Liao, H.; Chi, K. HTR2A promotes the development of cardiac hypertrophy by activating PI3K-PDK1-Akt-mTOR signaling. Cell Stress Chaperons 2020, 25, 899–908. [Google Scholar] [CrossRef]
- Mialet-Perez, J.; Bianchi, P.; Kunduzova, O.; Parini, A. New insights in receptor-dependent and monoamine oxidase-dependent effects of serotonin in the heart. J. Neural Transm. 2007, 114, 823–827. [Google Scholar] [CrossRef]
- Mialet-Perez, J.; Parini, A. Cardiac monoamine oxidases: At the heart of mitochondrial dysfunction. Cell Death Dis. 2020, 11, 54. [Google Scholar] [CrossRef] [Green Version]
- Villeneuve, C.; Caudriller, A.; Ordener, C.; Pizzinat, N.; Parini, A.; Mialet-Perez, J. Dose-dependent activation of distinct hypertrophic pathways by serotonin in cardiac cells. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H821–H828. [Google Scholar] [CrossRef]
- Pönicke, K.; Gergs, U.; Buchwalow, I.B.; Hauptmann, S.; Neumann, J. On the presence of serotonin in mammalian cardiomyocytes. Mol. Cell. Biochem. 2012, 365, 301–312. [Google Scholar] [CrossRef]
- Gergs, U.; Jung, F.; Buchwalow, I.B.; Hofmann, B.; Simm, A.; Treede, H.; Neumann, J. Pharmacological and physiological assessment of serotonin formation and degradation in isolated preparations from mouse and human hearts. Am. J. Physiol. Heart Circ. Physiol. 2017, 313, H1087–H1097. [Google Scholar] [CrossRef] [Green Version]
- Monassier, L.; Laplante, M.A.; Ayadi, T.; Doly, S.; Maroteaux, L. Contribution of gene-modified mice and rats to our understanding of the cardiovascular pharmacology of serotonin. Pharmacol. Therap. 2010, 128, 559–567. [Google Scholar] [CrossRef]
- Fischer, Y.; Thomas, J.; Kamp, J.; Jüngling, E.; Rose, H.; Carpene, C.; Kammermeier, H. 5-Hydroxytryptamine stimulates glucose transport in cardiomyocytes via a monoamine oxidase-dependent reaction. Biochem. J. 1995, 311, 575–583. [Google Scholar] [CrossRef] [Green Version]
- Kutsche, H.S.; Schreckenberg, R.; Weber, M.; Hirschhäuser, C.; Rohrbach, S.; Li, L.; Niemann, B.; Schulz, R.; Schlüter, K.-D. Alterations in glucose metabolism during the transition to heart failure: The contribution of UCP2. Cells 2020, 9, 552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwang, A.B.; Lee, S.-J. Regulation of life span by mitochondrial respiration: The HIF-1 and ROS connection. Aging 2011, 3, 304–310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michiels, C.; Minet, E.; Michel, G.; Mottet, D.; Piret, J.P.; Raes, M. HIF-1 and AP-1 cooperate to increase gene expression in hypoxia: Role of MAP kinases. IUBMB Life 2001, 52, 49–53. [Google Scholar] [CrossRef] [PubMed]
- Santin, Y.; Sicard, P.; Vigneron, F.; Guilbeau-Frugier, C.; Dutaur, M.; Lairez, O.; Couderc, B.; Manni, D.; Korolchuk, V.I.; Lezoualc’h, F.; et al. Oxidative stress by monomine oxidase-A impairs transcription factor EB activation and autophagosome clearance, leading to cardiomyocyte necrosis and heart failure. Antioxid. Redox Signal. 2016, 25, 10–27. [Google Scholar] [CrossRef]
- Villeneuve, C.; Guilbeau-Frugier, C.; Sicard, P.; Lairez, O.; Ordener, C.; Duparc, T.; de Paulis, D.; Couderc, B.; Spreux-Varoquaux, O.; Tortosa, F.; et al. p53-PGC-1α pathway mediated oxidative mitochondrial damage and cardiomyocyte necrosis induced by monoamine oxidase-A upregulation: Role in chronic left ventricular dysfunction in mice. Antioxid. Redox Signal. 2013, 18, 5–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaludercic, N.; Takimoto, E.; Nagayama, T.; Feng, N.; Lai, E.W.; Bedja, D.; Chen, K.; Gabrielson, K.L.; Blakely, R.D.; Shih, J.C.; et al. Monoamine oxidase A mediated enhanced catabolism of norepinephrine contributes to adverse remodeling and pump failure in hearts with pressure overload. Circ. Res. 2010, 106, 193–202. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhao, M.; Shi, Q.; Xu, B.; Zhu, C.; Li, M.; Mir, V.; Bers, D.M.; Xiang, Y.K. Monoamine oxidase desensitize intracellular β1AR signaling in heart failure. Circ. Res. 2021, 129, 965–967. [Google Scholar] [CrossRef]
- Song, Y.; Xu, C.; Liu, J.; Li, Y.; Wang, H.; Shan, D.; Wainer, I.W.; Hu, X.; Zhang, Y.; Woo, A.Y.-H.; et al. Heterodimerization with 5-HT2BR is indispensable for β2AR-mediated cardioprotection. Circ. Res. 2021, 128, 262377. [Google Scholar] [CrossRef]
- Heger, J.; Hirschhäuser, C.; Bornbaum, J.; Sydykov, A.; Dempfle, A.; Schneider, A.; Braun, T.; Schlüter, K.-D.; Schulz, R. Cardiomyocytes-specific deletion of monoamine oxidase B reduces irreversible myocardial ischemia/reperfusion injury. Free Radic. Biol. Med. 2021, 165, 14–23. [Google Scholar] [CrossRef]
- Knapp, F.; Niemann, B.; Li, L.; Molenda, N.; Kracht, M.; Schulz, R.; Rohrbach, S. Differential effects of right and left heart failure on skeletal muscle in rats. J. Cachexia Sarcopenia Muscle 2020, 11, 1830–1849. [Google Scholar] [CrossRef]
- Nippert, F.; Schreckenberg, R.; Schlüter, K.-D. Isolation and cultivation of adult rat cardiomyocytes. J. Vis. Exp. 2017, 128, e56634. [Google Scholar] [CrossRef]
- Bøtker, H.E.; Hausenloy, D.; Andreadou, I.; Antonucci, S.; Boengler, K.; Davidson, S.M.; Deshwal, S.; Devaux, Y.; di Lisa, F.; di Sante, M.; et al. Practical guidelines for rigor and reproducibility in preclinical and clinical studies on cardioprotection. Basic Res. Cardiol. 2018, 113, 39. [Google Scholar] [CrossRef] [Green Version]
- da Costa Rebelo, R.M.; Schreckenberg, R.; Schlüter, K.-D. Adverse cardiac remodeling in spontaneously hypertensive rats: Acceleration by high aerobic exercise intensity. J. Physiol. 2012, 12, 5389–5400. [Google Scholar] [CrossRef]
- Schreckenberg, R.; Horn, A.-M.; da Costa Rebelo, R.M.; Simsekyilmaz, S.; Niemann, B.; Li, L.; Rohrbach, S.; Schlüter, K.-D. Effect of 6-months’ exercise on cardiac function, structure and metabolism in female hypertensive rats—The decisive role of lysyl oxidase and collagen III. Front. Physiol. 2017, 8, 556. [Google Scholar] [CrossRef] [Green Version]
- Wolf, A.; Kutsche, H.S.; Atmanspacher, F.; Karadedeli, M.S.; Schreckenberg, R.; Schlüter, K.-D. Untypical metabolic adaptations in spontaneously hypertensive rats to free running wheel activity includes uncoupling protein-3 (UCP-3) and proprotein convertase subtilisin/kexin type 9 (PCSK9) expression. Front. Physiol. 2021, 12, 598723. [Google Scholar] [CrossRef]
- Langer, M.; Lüttecke, D.; Schlüter, K.-D. Mechanism of the positive contractile effect of nitric oxide on rat ventricular cardiomyocytes with positive force/frequency relationship. Pflug. Arch. 2003, 447, 289. [Google Scholar] [CrossRef] [PubMed]
- Nippert, F.; Schreckenberg, R.; Hess, A.; Weber, M.; Schlüter, K.-D. The effects of swiprosin-1 on formation of pseudopodia-like structures and β-adrenoceptor coupling in cultured adult rat ventricular cardiomyocytes. PLoS ONE 2016, 11, e0167655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of gene expression data using real-time quantitative PCR and the 2-ΔΔCT. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-F.; Cowley, A.W.; Zou, A.-P. Increased H2O2 counteracts the vasodilator and natriuretic effects of superoxide dismutation by tempol in renal medulla. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2003, 285, R827–R833. [Google Scholar] [CrossRef] [Green Version]
- Hahnova, K.; Brabcova, I.; Neckar, J.; Weissova, R.; Svatonova, A.; Novakova, O.; Zurmanova, J.; Kalous, M.; Silhavy, J.; Pravenec, M.; et al. Beta-Adrenergic signaling, monoamine oxidase A and antioxidant defence in the myocardium of SHR and SHR-mtBN conplastic rat strains: The effect of chronic hypoxia. J. Physiol. Sci. 2018, 68, 441–454. [Google Scholar] [CrossRef] [Green Version]
- Havlenova, T.; Skaroupkova, P.; Miklovic, M.; Behounek, M.; Chmel, M.; Jarkovska, D.; Sviglerova, J.; Stengl, M.; Kolar, M.; Novotny, J.; et al. Right versus left ventricular remodeling in heart failure due to chronic volume overload. Sci. Rep. 2021, 11, 17136. [Google Scholar] [CrossRef]
- Maggiorani, D.; Manzella, N.; Edmondson, D.E.; Mattevi, A.; Parini, A.; Binda, C.; Mialet-Perez, J. Monoamine oxidase, oxidative stress, and altered mitochondrial dynamics in cardiac ageing. Oxid. Med. Cell. Longev. 2017, 2017, 3017947. [Google Scholar] [CrossRef] [Green Version]
- Van Eif, V.W.W.; Bogaards, S.J.P.; van der Laarse, W.J. Intrinsic cardiac adrenergic (ICA) cell density and MAO-A activity in failing rat hearts. J. Muscle Res. Cell Motil. 2014, 35, 47–53. [Google Scholar] [CrossRef]
- Sun, X.-Q.; Peters, E.L.; Schalij, I.; Axelsen, J.B.; Andersen, S.; Kurakula, K.; Gomez-Puerto, M.C.; Szulcek, R.; Pan, X.; da Silva Goncalves Bos, D.; et al. Increased MAO-A activity promotes progression of pulmonary arterial hypertension. Am. J. Respir. Cell Mol. Biol. 2021, 64, 331–343. [Google Scholar] [CrossRef] [PubMed]
- Pino, R.; Failli, P.; Mazzetti, L.; Buffoni, F. Monoamine oxidase and semicarbazide-sensitive amine oxidase activities in isolated cardiomyocytes of spontenaously hypertensive rats. Biochem. Mol. Med. 1997, 62, 188–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schulz, R.; Schlüter, K.-D. Importance of mitochondria in cardiac pathologies: Focus on uncoupling proteins and monoamine oxidase. Int. J. Mol. Sci. 2023, 24, 6459. [Google Scholar] [CrossRef] [PubMed]
- Lairez, O.; Calise, D.; Bianchi, P.; Ordener, C.; Spreux-Varoquaux, O.; Guilbeau-Frugier, C.; Escourrou, G.; Seif, I.; Roncalli, J.; Pizzinat, N.; et al. Genetic deletion of MAO-A promotes serotonin-dependent ventricular hypertrophy by pressure overload. J. Mol. Cell. Cardiol. 2009, 46, 587–595. [Google Scholar] [CrossRef]
- Maurel, A.; Hernandez, C.; Kunduzova, O.; Bompart, G.; Cambon, C.; Parini, A.; Frances, B. Age-dependent increase in hydrogen peroxide production by cardiac monoamine oxidase A in rats. Am. J. Physiol. Heart Circ. Physiol. 2003, 284, H1460–H1467. [Google Scholar] [CrossRef]
- Schreckenberg, R.; Rebelo, M.; Deten, A.; Weber, M.; Rohrbach, S.; Pipicz, M.; Csonka, C.; Ferdinandy, P.; Schulz, R.; Schlüter, K.-D. Specific mechanisms underlying right heart failure: The missing upregulation of superoxide dismutase-2 and its decisive role in antioxidative defense. Antioxid. Redox Signal. 2015, 23, 1220–1232. [Google Scholar] [CrossRef]
- Manzella, N.; Santin, Y.; Maggiorani, D.; Martini, H.; Douin-Echinard, V.; Passos, J.F.; Lezoualc’h, F.; Binda, C.; Parini, A.; Mialet-Perez, J. Monoamino oxidase-A is a novel driver of stress-induced premature senescence through inhibition of parkin-mediated mitophagy. Aging Cell 2018, 17, e12811. [Google Scholar] [CrossRef] [Green Version]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Knittel, J.; Itani, N.; Schreckenberg, R.; Heger, J.; Rohrbach, S.; Schulz, R.; Schlüter, K.-D. Monoamine Oxidase A Contributes to Serotonin—But Not Norepinephrine-Dependent Damage of Rat Ventricular Myocytes. Biomolecules 2023, 13, 1013. https://doi.org/10.3390/biom13061013
Knittel J, Itani N, Schreckenberg R, Heger J, Rohrbach S, Schulz R, Schlüter K-D. Monoamine Oxidase A Contributes to Serotonin—But Not Norepinephrine-Dependent Damage of Rat Ventricular Myocytes. Biomolecules. 2023; 13(6):1013. https://doi.org/10.3390/biom13061013
Chicago/Turabian StyleKnittel, Jonas, Nadja Itani, Rolf Schreckenberg, Jacqueline Heger, Susanne Rohrbach, Rainer Schulz, and Klaus-Dieter Schlüter. 2023. "Monoamine Oxidase A Contributes to Serotonin—But Not Norepinephrine-Dependent Damage of Rat Ventricular Myocytes" Biomolecules 13, no. 6: 1013. https://doi.org/10.3390/biom13061013