
Identification of the Stapled α-Helical Peptide ATSP-7041 as a Substrate and Strong Inhibitor of OATP1B1 In Vitro


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Screening Experiments of ATSP-7041 Interactions
2.3. Evaluation of ATSP-7041 as a Substrate of OATP1B1
2.4. Evaluation of ATSP-7041 as an Inhibitor of OATP1B1
2.5. LC/MS Analysis
2.6. Data Analysis
3. Results
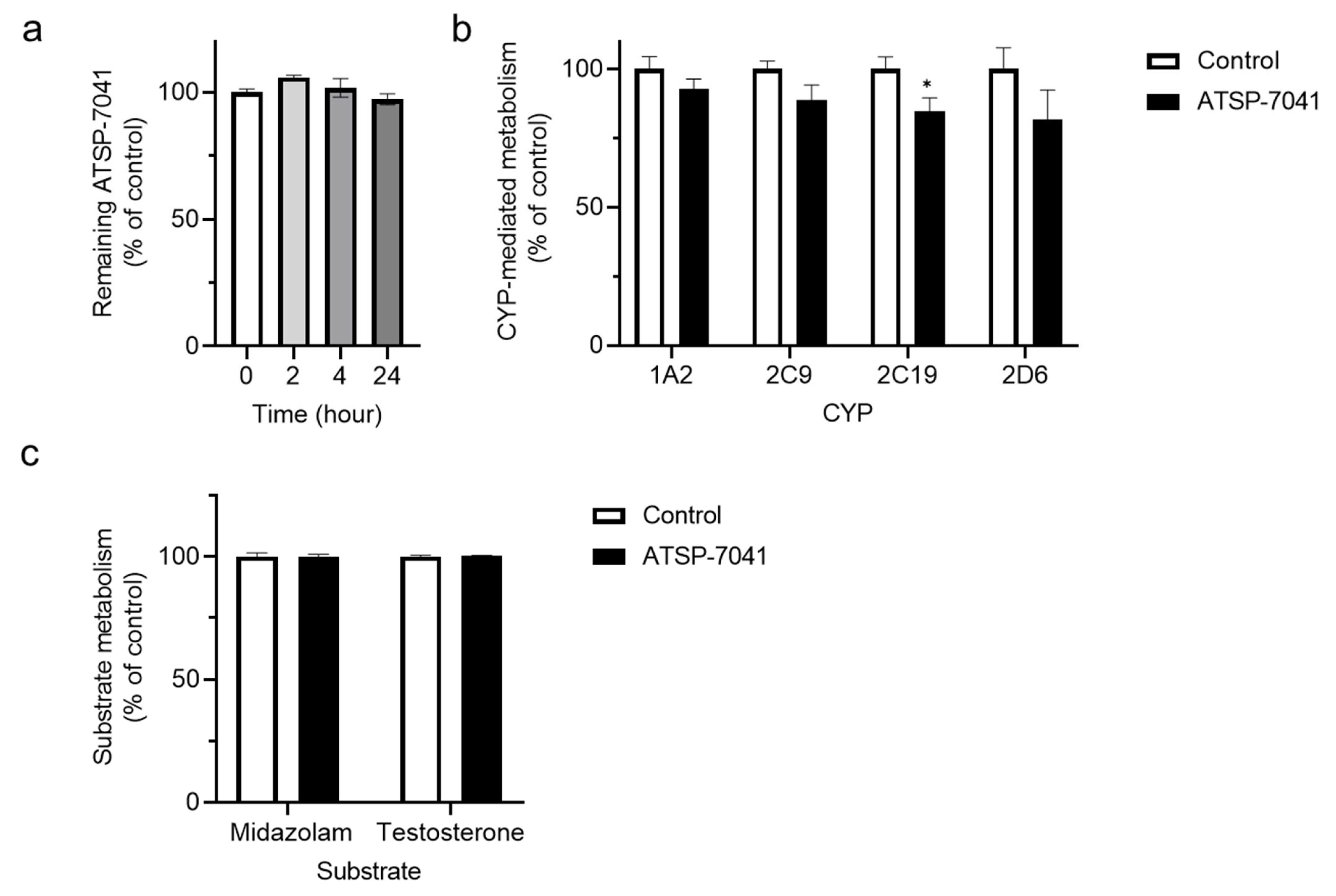
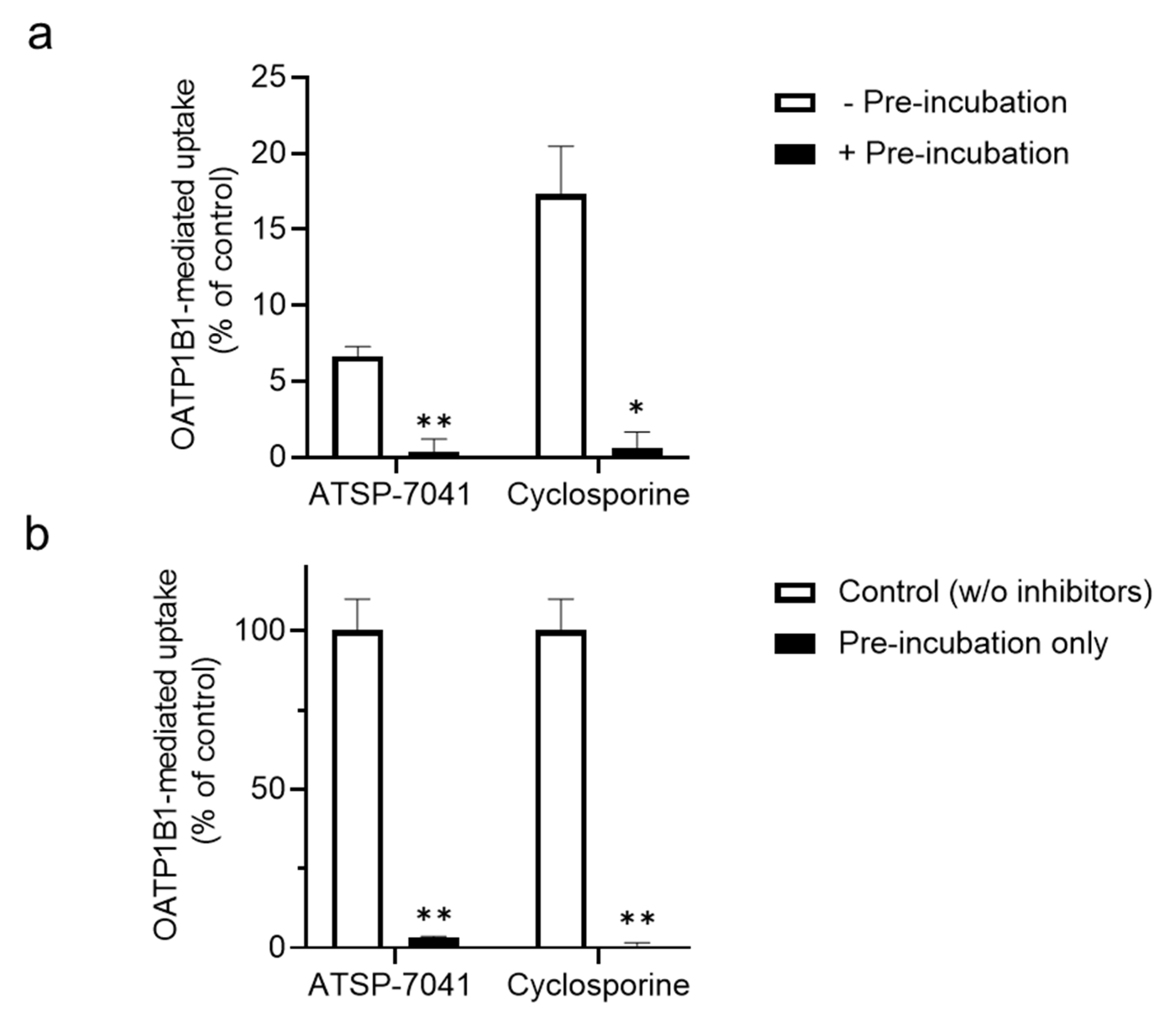
3.1. Screening for ATSP-7041 Interactions
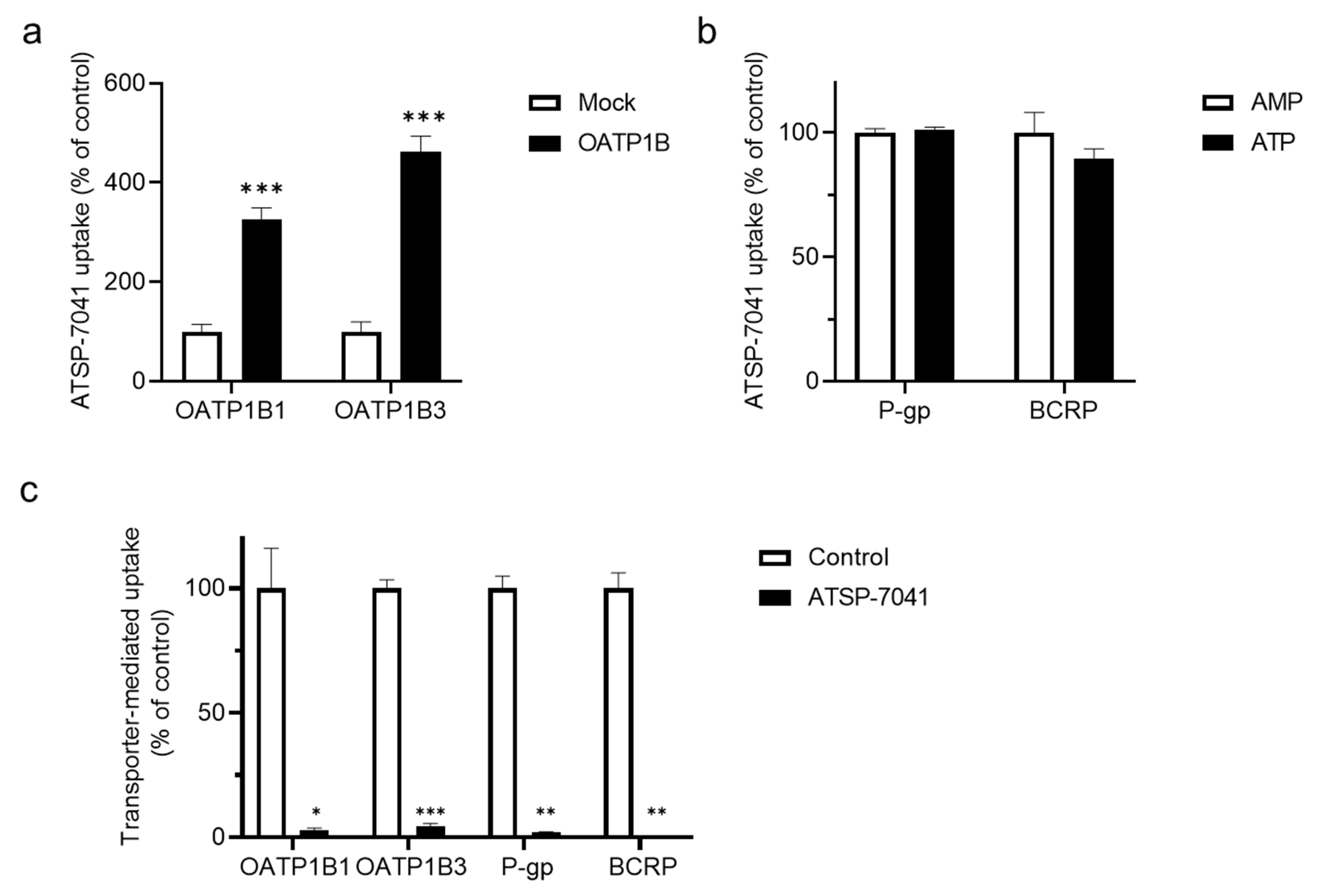
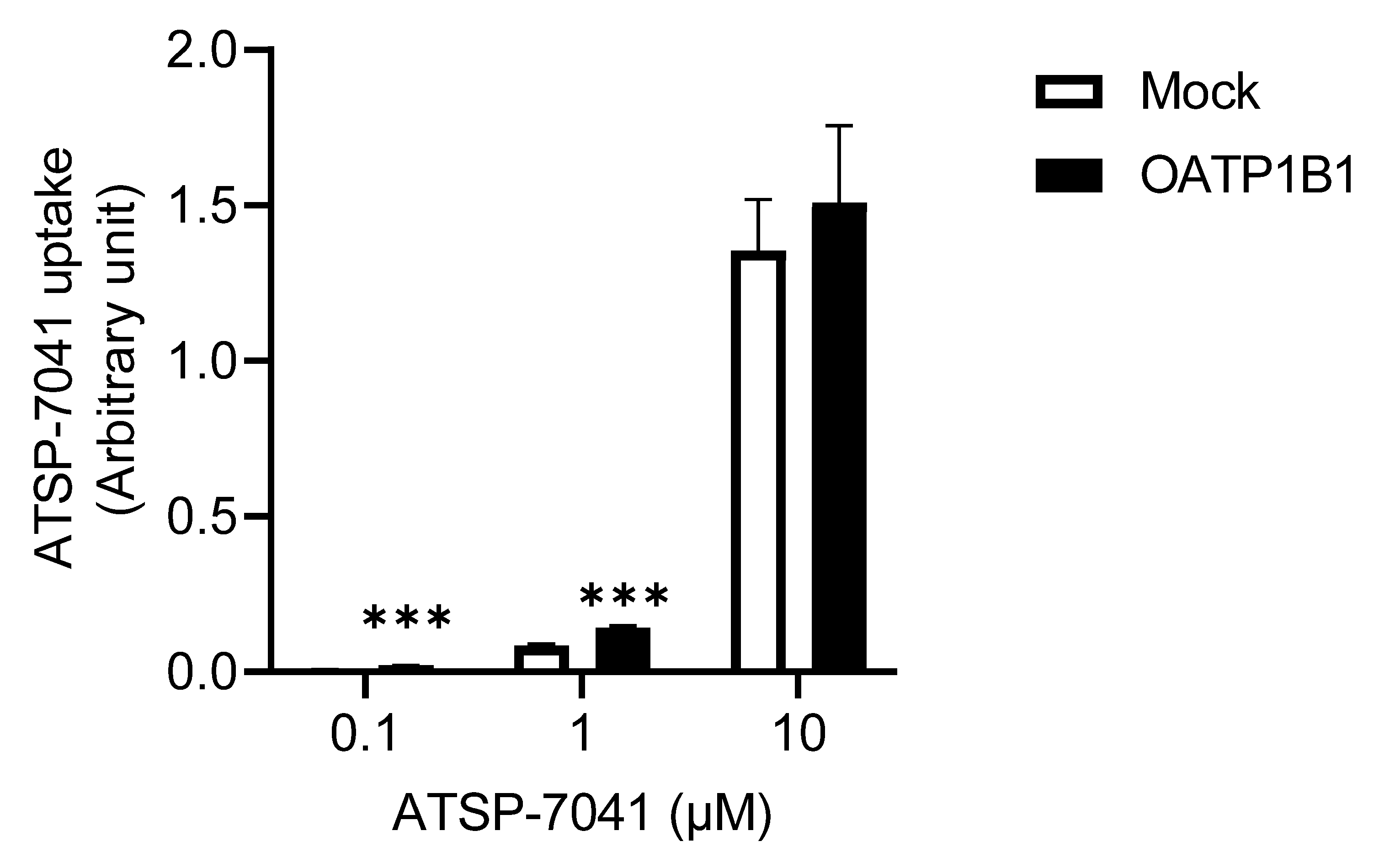
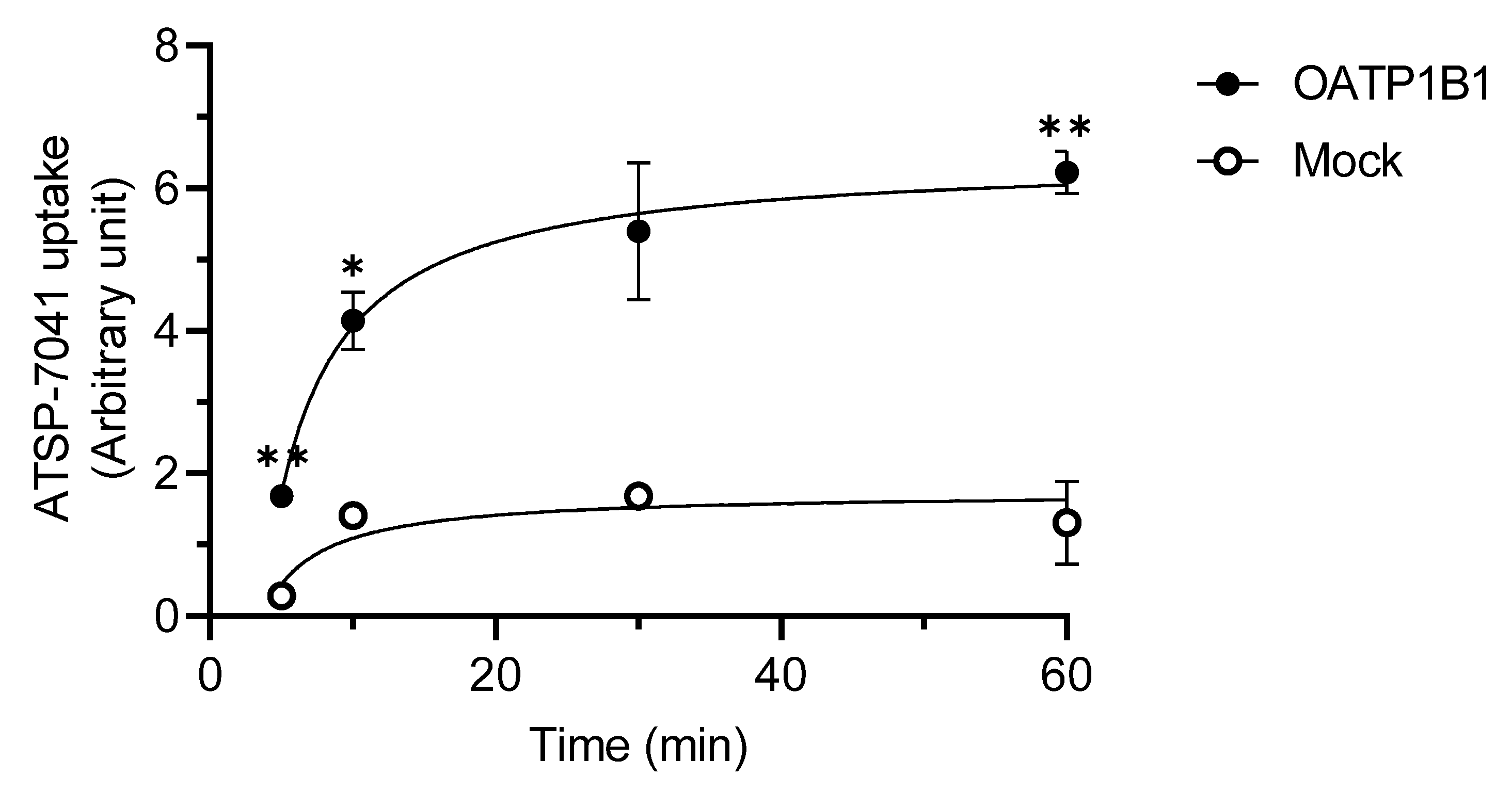
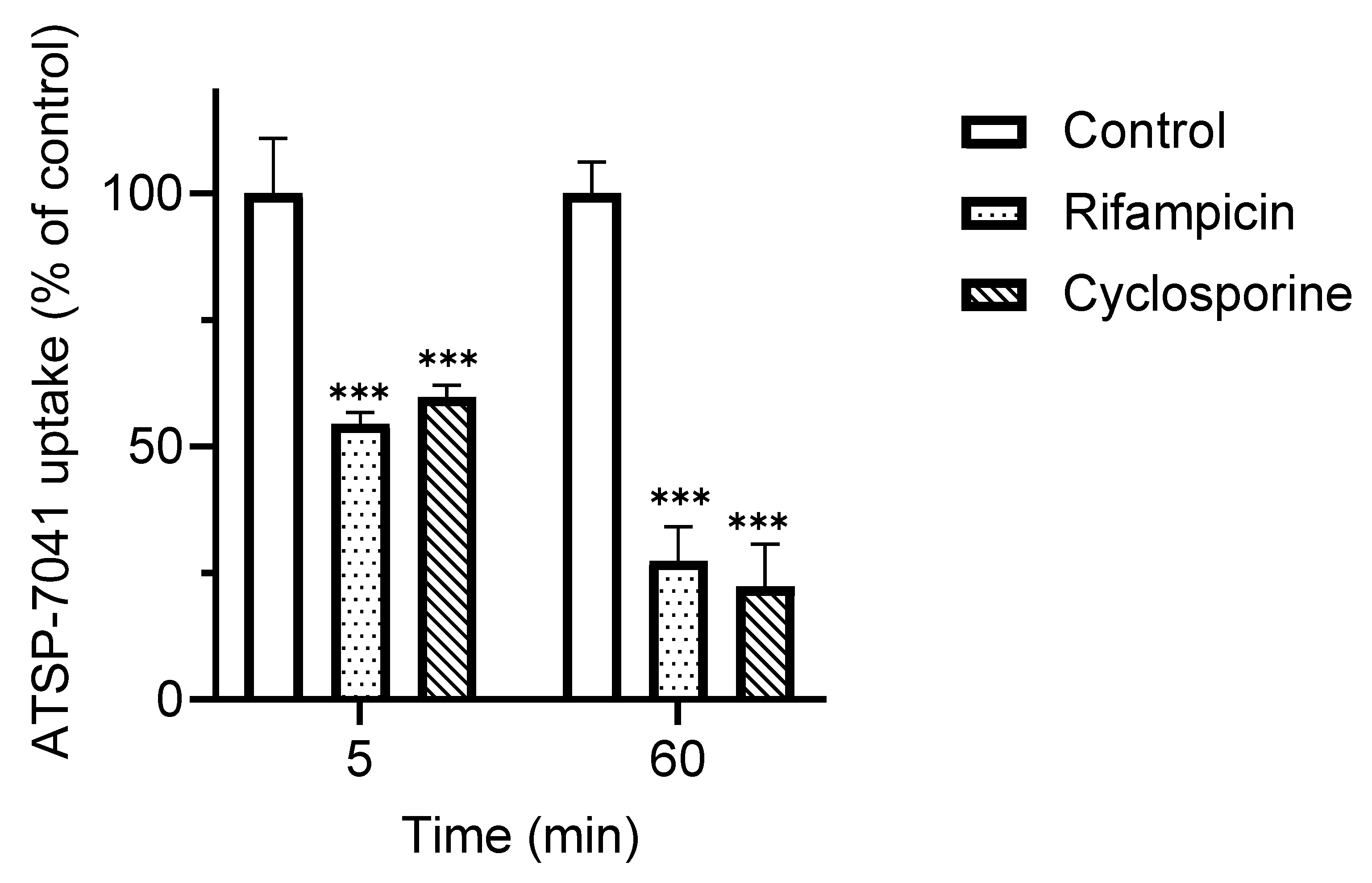
3.2. Characterization of ATSP-7041 as a Substrate of OATP1B1
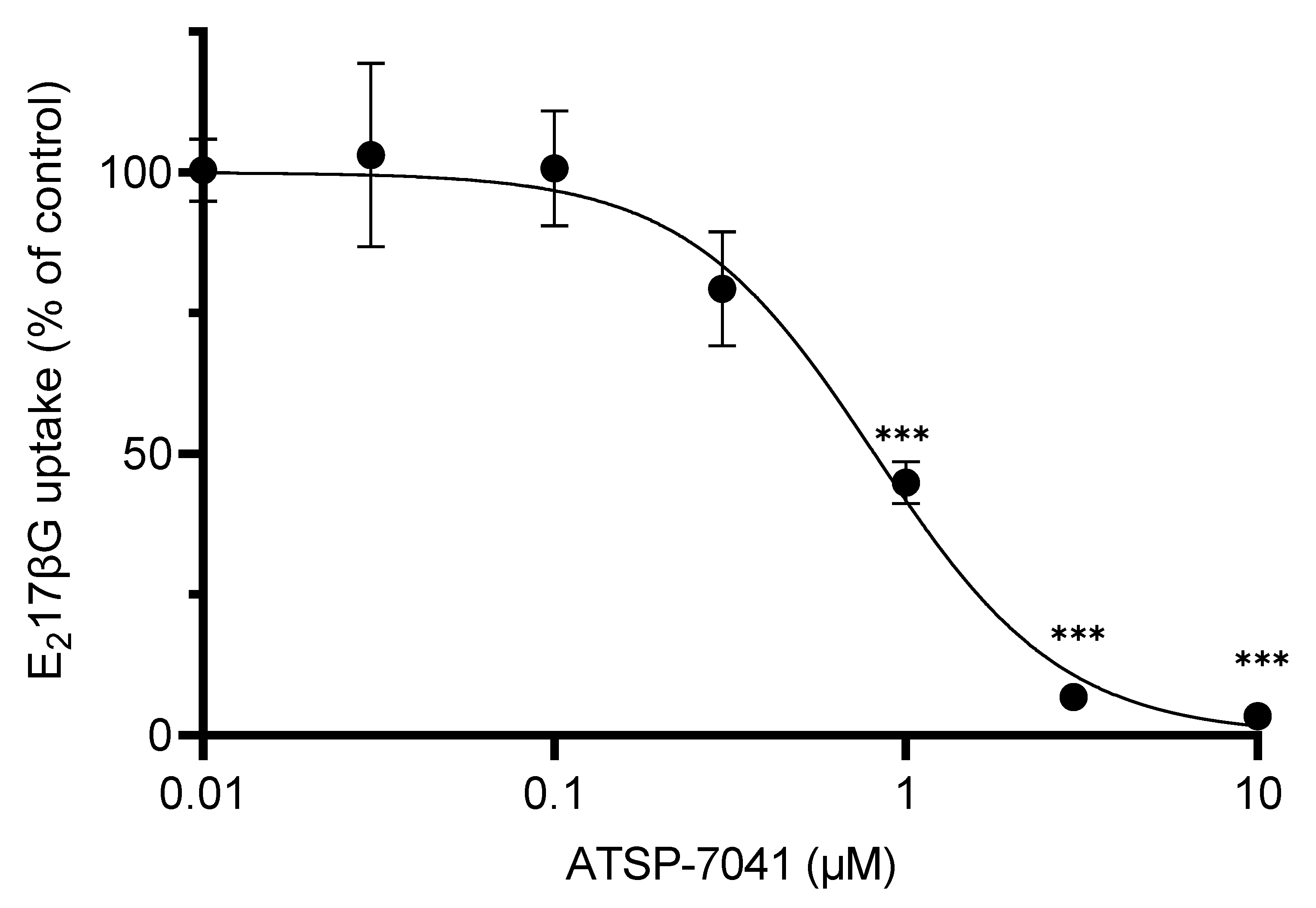
3.3. Characterization of ATSP-7041 as an Inhibitor of OATP1B1 Activity
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- White, A.M.; Craik, D.J. Discovery and optimization of peptide macrocycles. Expert Opin. Drug Discov. 2016, 11, 1151–1163. [Google Scholar] [CrossRef] [PubMed]
- Klein, M. Stabilized helical peptides: Overview of the technologies and its impact on drug discovery. Expert Opin. Drug Discov. 2017, 12, 1117–1125. [Google Scholar] [CrossRef] [PubMed]
- Schiller, P.W.; Nguyen, T.M.-D. Activity profiles of novel side-chain to side-chain cyclized opioid peptide analogs. Neuropeptides 1984, 5, 165–168. [Google Scholar] [CrossRef] [PubMed]
- Gilon, C.; Halle, D.; Chorev, M.; Selincer, Z.; Byk, G. Backbone cyclization: A new method for conferring conformational constraint on peptides. Biopolymers 1991, 31, 745–750. [Google Scholar] [CrossRef]
- De Planque, M.R.R.; Kruijtzer, J.A.W.; Liskamp, R.; Marsh, D.; Greathouse, D.V.; Ii, R.E.K.; de Kruijff, B.; Killian, J.A. Different Membrane Anchoring Positions of Tryptophan and Lysine in Synthetic Transmembrane α-Helical Peptides. J. Biol. Chem. 1999, 274, 20839–20846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schafmeister, C.E.; Po, J.; Verdine, G.L. An All-Hydrocarbon Cross-Linking System for Enhancing the Helicity and Metabolic Stability of Peptides. J. Am. Chem. Soc. 2000, 122, 5891–5892. [Google Scholar] [CrossRef]
- Walensky, L.D.; Kung, A.L.; Escher, I.; Malia, T.J.; Barbuto, S.; Wright, R.D.; Wagner, G.; Verdine, G.L.; Korsmeyer, S.J. Activation of Apoptosis in Vivo by a Hydrocarbon-Stapled BH3 Helix. Science 2004, 305, 1466–1470. [Google Scholar] [CrossRef] [Green Version]
- Sawyer, T.K.; Partridge, A.W.; Kaan, H.Y.K.; Juang, Y.-C.; Lim, S.; Johannes, C.; Yuen, T.Y.; Verma, C.; Kannan, S.; Aronica, P.; et al. Macrocyclic α helical peptide therapeutic modality: A perspective of learnings and challenges. Bioorganic Med. Chem. 2018, 26, 2807–2815. [Google Scholar] [CrossRef] [PubMed]
- Ali, A.M.; Atmaj, J.; Van Oosterwijk, N.; Groves, M.R.; Dömling, A. Stapled peptides inhibitors: A new window for target drug discovery. Comput. Struct. Biotechnol. J. 2019, 17, 263–281. [Google Scholar] [CrossRef]
- Bernal, F.; Tyler, A.F.; Korsmeyer, S.J.; Walensky, L.D.; Verdine, G.L. Reactivation of the p53 tumor suppressor pathway by a stapled p53 peptide. J. Am. Chem. Soc. 2007, 129, 2456–2457, Erratum in: J. Am. Chem. Soc. 2007, 129, 5298. [Google Scholar] [CrossRef]
- Bernal, F.; Wade, M.; Godes, M.; Davis, T.N.; Whitehead, D.G.; Kung, A.L.; Wahl, G.M.; Walensky, L.D. A stapled p53 helix overcomes HDMX-mediated suppression of p53. Cancer Cell. 2010, 18, 411–422. [Google Scholar] [CrossRef] [Green Version]
- Rezaei Araghi, R.; Bird, G.H.; Ryan, J.A.; Jenson, J.M.; Godes, M.; Pritz, J.R.; Grant, R.A.; Letai, A.; Walensky, L.D.; Keating, A.E. Iterative optimization yields Mcl-1-targeting stapled peptides with selective cytotoxicity to Mcl-1-dependent cancer cells. Proc. Natl. Acad. Sci. USA 2018, 115, E886–E895. [Google Scholar] [CrossRef] [Green Version]
- Phillips, C.; Roberts, L.R.; Schade, M.; Bazin, R.; Bent, A.; Davies, N.L.; Moore, R.; Pannifer, A.D.; Pickford, A.R.; Prior, S.H.; et al. Design and structure of stapled peptides binding to estrogen receptors. J. Am. Chem. Soc. 2011, 133, 9696–9699. [Google Scholar] [CrossRef]
- Dietrich, L.; Rathmer, B.; Ewan, K.; Bange, T.; Heinrichs, S.; Dale, T.C.; Schade, D.; Grossmann, T.N. Cell permeable stapled peptide inhibitor of Wnt signaling that targets β-catenin protein-protein interactions. Cell Chem. Biol. 2017, 24, 958–968.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, Y.S.; Graves, B.; Guerlavais, V.; Tovar, C.; Packman, K.; To, K.H.; Olson, K.A.; Kesavan, K.; Gangurde, P.; Mukherjee, A.; et al. Stapled α-helical peptide drug development: A potent dual inhibitor of MDM2 and MDMX for p53-dependent cancer therapy. Proc. Natl. Acad. Sci. USA 2013, 110, E3445–E3454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wachter, F.; Morgan, A.M.; Godes, M.; Mourtada, R.; Bird, G.H.; Walensky, L.D. Mechanistic validation of a clinical lead stapled peptide that reactivates p53 by dual HDM2 and HDMX targeting. Oncogene 2017, 36, 2184–2190. [Google Scholar] [CrossRef]
- Woodfield, S.E.; Shi, Y.; Patel, R.H.; Chen, Z.; Shah, A.P.; Srivastava, R.K.; Whitlock, R.S.; Ibarra, A.M.; Larson, S.R.; Sarabia, S.F.; et al. MDM4 inhibition: A novel therapeutic strategy to reactivate p53 in hepatoblastoma. Sci. Rep. 2021, 11, 2967. [Google Scholar] [CrossRef] [PubMed]
- Stolte, B.; Iniguez, A.B.; Dharia, N.V.; Robichaud, A.L.; Conway, A.S.; Morgan, A.M.; Alexe, G.; Schauer, N.J.; Liu, X.; Bird, G.H.; et al. Genome-scale CRISPR-Cas9 screen identifies druggable dependencies in TP53 wild-type Ewing sarcoma. J. Exp. Med. 2018, 215, 2137–2155. [Google Scholar] [CrossRef]
- Howard, T.P.; Arnoff, T.E.; Song, M.R.; Giacomelli, A.O.; Wang, X.; Hong, A.L.; Dharia, N.V.; Wang, S.; Vazquez, F.; Pham, M.T.; et al. MDM2 and MDM4 are therapeutic vulnerabilities in malignant rhabdoid tumors. Cancer Res. 2019, 79, 2404–2414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saleh, M.N.; Patel, M.R.; Bauer, T.M.; Goel, S.; Falchook, G.S.; Shapiro, G.I.; Chung, K.Y.; Infante, J.R.; Conry, R.M.; Rabinowits, G.; et al. Phase 1 Trial of ALRN-6924, a Dual Inhibitor of MDMX and MDM2, in Patients with Solid Tumors and Lymphomas Bearing Wild-type TP53. Clin. Cancer Res. 2021, 27, 5236–5247. [Google Scholar] [CrossRef] [PubMed]
- EMA Guideline on the Investigation of Drug Interactions. 2013. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-investigation-drug-interactions-revision-1_en.pdf (accessed on 10 April 2023).
- Ministry of Health, Labour and Welfare. Guideline on Drug Interaction for Drug Development and Appropriate Provision of Information. 2018. Available online: https://www.pmda.go.jp/files/000228122.pdf (accessed on 10 April 2023).
- FDA Guidance for Industry: In Vitro Drug Interaction Studies—Cytochrome P450 Enzyme- and Transporter-Mediated Drug Interactions. 2020. Available online: https://www.fda.gov/media/134582/download (accessed on 10 April 2023).
- Srinivas, M. Cytochrome P450 Enzymes, Drug Transporters and their Role in Pharmacokinetic Drug-Drug Interactions of Xenobiotics: A Comprehensive Review. Open. J. Chem. 2017, 3, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.; Petrie, I.D.; Levy, R.H.; Ragueneau-Majlessi, I. Mechanisms and clinical significance of pharmacokinetic-based drug-drug interactions with drugs approved by the U.S. Food and Drug Administration in 2017. Drug Metab. Dispos. 2019, 47, 135–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beck, T.C.; Beck, K.R.; Morningstar, J.; Benjamin, M.M.; Norris, R.A. Descriptors of cytochrome inhibitors and useful machine learning based methods for the design of safer drugs. Pharmaceuticals 2021, 14, 472. [Google Scholar] [CrossRef]
- Prachayasittikul, V.; Mandi, P.; Prachayasittikul, S.; Prachayasittikul, V.; Nantasenamat, C. Exploring the chemical space of P-glycoprotein interacting compounds. Mini Rev. Med. Chem. 2017, 17, 1332–1345. [Google Scholar] [CrossRef] [PubMed]
- Tuerkova, A.; Bongers, B.J.; Norinder, U.; Ungvári, O.; Székely, V.; Tarnovskiy, A.; Szakács, G.; Özvegy-Laczka, C.; van Westen, G.J.P.; Zdrazil, B. Identifying novel inhibitors for hepatic organic anion transporting polypeptides by machine learning-based virtual screening. J. Chem. Inf. Model. 2022, 62, 6323–6335. [Google Scholar] [CrossRef]
- Shitara, Y.; Itoh, T.; Sato, H.; Li, A.P.; Sugiyama, Y. Inhibition of transporter-mediated hepatic uptake as a mechanism for drug-drug interaction between cerivastatin and cyclosporin A. J. Pharmacol. Exp. Ther. 2003, 304, 610–616. [Google Scholar] [CrossRef] [Green Version]
- Simonson, S.G.; Raza, A.; Martin, P.D.; Mitchell, P.D.; Jarcho, J.A.; Brown, C.D.; Windass, A.S.; Schneck, D.W. Rosuvastatin pharmacokinetics in heart transplant recipients administered an antirejection regimen including cyclosporine. Clin. Pharmacol. Ther. 2004, 76, 167–177. [Google Scholar] [CrossRef] [PubMed]
- Fox, R.I.; Morgan, S.L.; Smith, H.T.; Robbins, B.A.; Choc, M.G.; Baggott, J.E. Combined oral cyclosporin and methotrexate therapy in patients with rheumatoid arthritis elevates methotrexate levels and reduces 7-hydroxymethotrexate levels when compared with methotrexate alone. Rheumatol 2003, 42, 989–994. [Google Scholar] [CrossRef] [Green Version]
- Bartlett, N.L.; Lum, B.L.; Fisher, G.A.; Brophy, N.A.; Ehsan, M.N.; Halsey, J.; Sikic, B.I. Phase I trial of doxorubicin with cyclosporine as a modulator of multidrug resistance. J. Clin. Oncol. 1994, 12, 835–842. [Google Scholar] [CrossRef] [PubMed]
- Rushing, D.A.; Raber, S.R.; Rodvold, K.A.; Piscitelli, S.C.; Plank, G.S.; Tewksbury, D.A. The effects of cyclosporine on the pharmacokinetics of doxorubicin in patients with small cell lung cancer. Cancer 1994, 74, 834–841. [Google Scholar] [CrossRef] [PubMed]
- Shitara, Y.; Takeuchi, K.; Nagamatsu, Y.; Wada, S.; Sugiyama, Y.; Horie, T. Long-lasting inhibitory effects of cyclosporin A, but not tacrolimus, on OATP1B1- and OATP1B3-mediated uptake. Drug. Metab. Pharmacokinet. 2012, 27, 368–378. [Google Scholar] [CrossRef] [Green Version]
- Izumi, S.; Nozaki, Y.; Maeda, K.; Komori, T.; Takenaka, O.; Kusuhara, H.; Sugiyama, Y. Investigation of the impact of substrate selection on in vitro organic anion transporting polypeptide 1B1 inhibition profiles for the prediction of drug-drug interactions. Drug Metab. Dispos. 2015, 43, 235–247. [Google Scholar] [CrossRef] [Green Version]
- Schnegelberger, R.D.; Steiert, B.; Sandoval, P.J.; Hagenbuch, B. Using a competitive counterflow assay to identify novel cationic substrates of OATP1B1 and OATP1B3. Front. Physiol. 2022, 13, 969363. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, T.; Masuo, Y.; Sakai, Y.; Kato, Y. Short-lasting inhibition of hepatic uptake transporter OATP1B1 by tyrosine kinase inhibitor pazopanib. Drug. Metab. Pharmacokinet. 2019, 34, 372–379. [Google Scholar] [CrossRef] [PubMed]
- Kalliokoski, A.; Niemi, M. Impact of OATP transporters on pharmacokinetics. Br. J. Pharmacol. 2009, 158, 693–705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirano, M.; Maeda, K.; Shitara, Y.; Sugiyama, Y. Drug-drug interaction between pitavastatin and various drugs via OATP1B1. Drug. Metab. Dispos. 2006, 34, 1229–1236. [Google Scholar] [CrossRef]
- Tirona, R.G.; Leake, B.F.; Wolkoff, A.W.; Kim, R.B. Human organic anion transporting polypeptide-C (SLC21A6) is a major determinant of rifampin-mediated pregnane X receptor activation. J. Pharmacol. Exp. Ther. 2003, 304, 223–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, R.H.; Tirona, R.G.; Leake, B.F.; Glaeser, H.; Lee, W.; Lemke, C.J.; Wang, Y.; Kim, R.B. Drug and bile acid transporters in rosuvastatin hepatic uptake: Function, expression, and pharmacogenetics. Gastroenterology 2006, 130, 1793–1806. [Google Scholar] [CrossRef]
- Kummar, S.; Chen, H.X.; Wright, J.; Holbeck, S.; Millin, M.D.; Tomaszewski, J.; Zweibel, J.; Collins, J.; Doroshow, J.H. Utilizing targeted cancer therapeutic agents in combination: Novel approaches and urgent requirements. Nat. Rev. Drug Discov. 2010, 9, 843–856. [Google Scholar] [CrossRef]
- Madani Tonekaboni, S.A.; Soltan Ghoraie, L.; Manem, V.S.K.; Haibe-Kains, B. Predictive approaches for drug combination discovery in cancer. Brief. Bioinform. 2018, 19, 263–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thakkar, N.; Lockhart, A.C.; Lee, W. Role of organic anion-transporting polypeptides (OATPs) in cancer therapy. AAPS J. 2015, 17, 535–545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, S.; Mathijssen, R.H.; de Bruijn, P.; Baker, S.D.; Sparreboom, A. Inhibition of OATP1B1 by tyrosine kinase inhibitors: In vitro-in vivo correlations. Br. J. Cancer 2014, 110, 894–898. [Google Scholar] [CrossRef] [PubMed]
- Fowler, H.; Belot, A.; Ellis, L.; Maringe, C.; Luque-Fernandez, M.A.; Njagi, E.N.; Navani, N.; Sarfati, D.; Rachet, B. Comorbidity prevalence among cancer patients: A population-based cohort study of four cancers. BMC Cancer 2020, 20, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ishikawa, R.; Saito, K.; Misawa, T.; Demizu, Y.; Saito, Y. Identification of the Stapled α-Helical Peptide ATSP-7041 as a Substrate and Strong Inhibitor of OATP1B1 In Vitro. Biomolecules 2023, 13, 1002. https://doi.org/10.3390/biom13061002
Ishikawa R, Saito K, Misawa T, Demizu Y, Saito Y. Identification of the Stapled α-Helical Peptide ATSP-7041 as a Substrate and Strong Inhibitor of OATP1B1 In Vitro. Biomolecules. 2023; 13(6):1002. https://doi.org/10.3390/biom13061002
Chicago/Turabian StyleIshikawa, Rika, Kosuke Saito, Takashi Misawa, Yosuke Demizu, and Yoshiro Saito. 2023. "Identification of the Stapled α-Helical Peptide ATSP-7041 as a Substrate and Strong Inhibitor of OATP1B1 In Vitro" Biomolecules 13, no. 6: 1002. https://doi.org/10.3390/biom13061002