
Efflux Pump-Binding 4(3-Aminocyclobutyl)Pyrimidin-2-Amines Are Colloidal Aggregators
 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. AcrA Protein Modeling
2.2. Virtual Screening
2.3. Antimicrobial Potentiation Assay
2.4. Cell Viability Studies
2.5. Protein Expression and Purification
2.6. Microscale Thermophoresis (MST)
2.7. Dynamic Light Scattering (DLS)
2.8. Nano-Differential Scanning Fluorimetry (nDSF)
3. Results
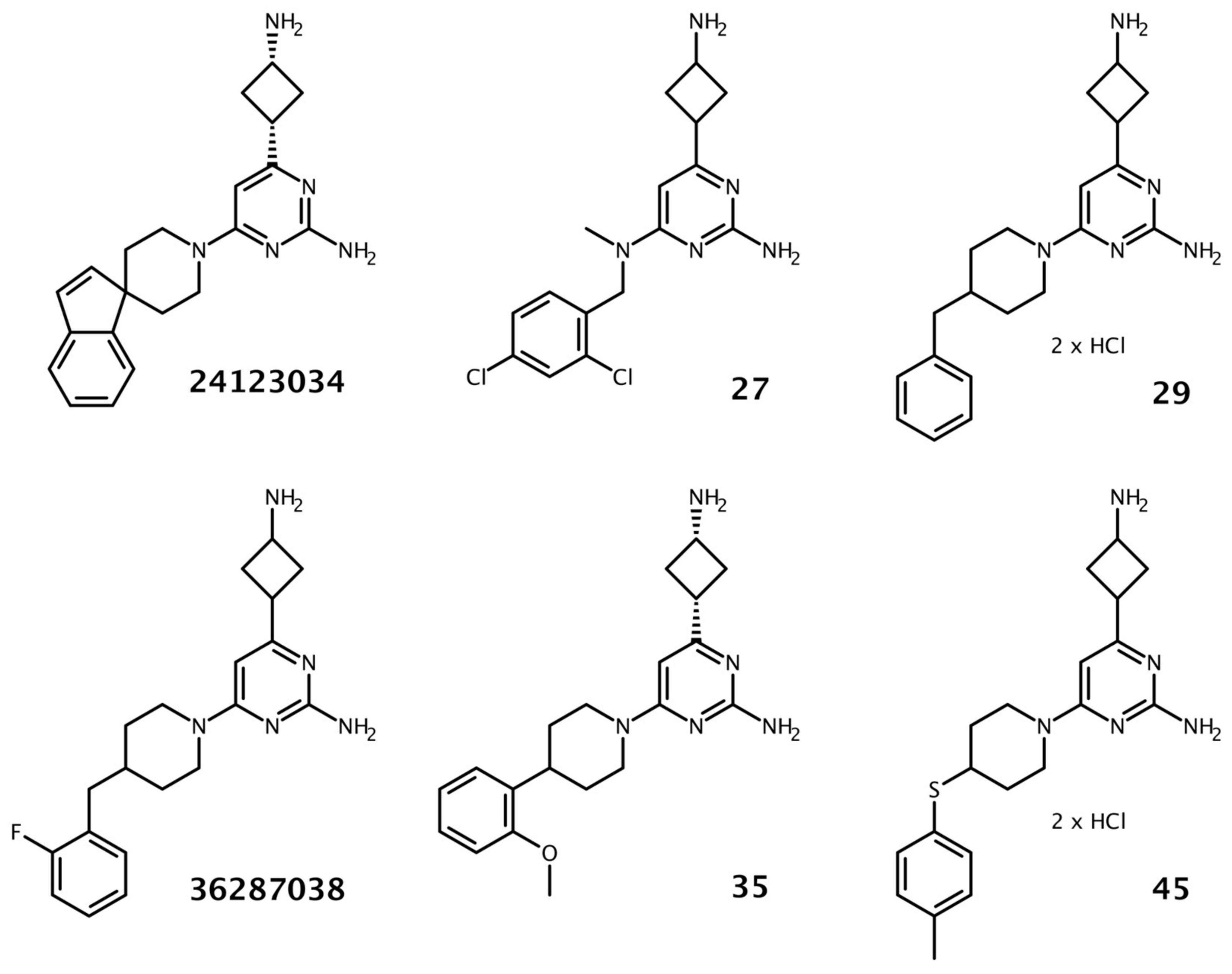
3.1. Identification and Validation of 4(3-Aminocyclobutyl)Pyrimidin-2-Amine Analogs
3.2. Cytotoxic Potential of 4(3-Aminocyclobutyl)Pyrimidin-2-Amine Analogs
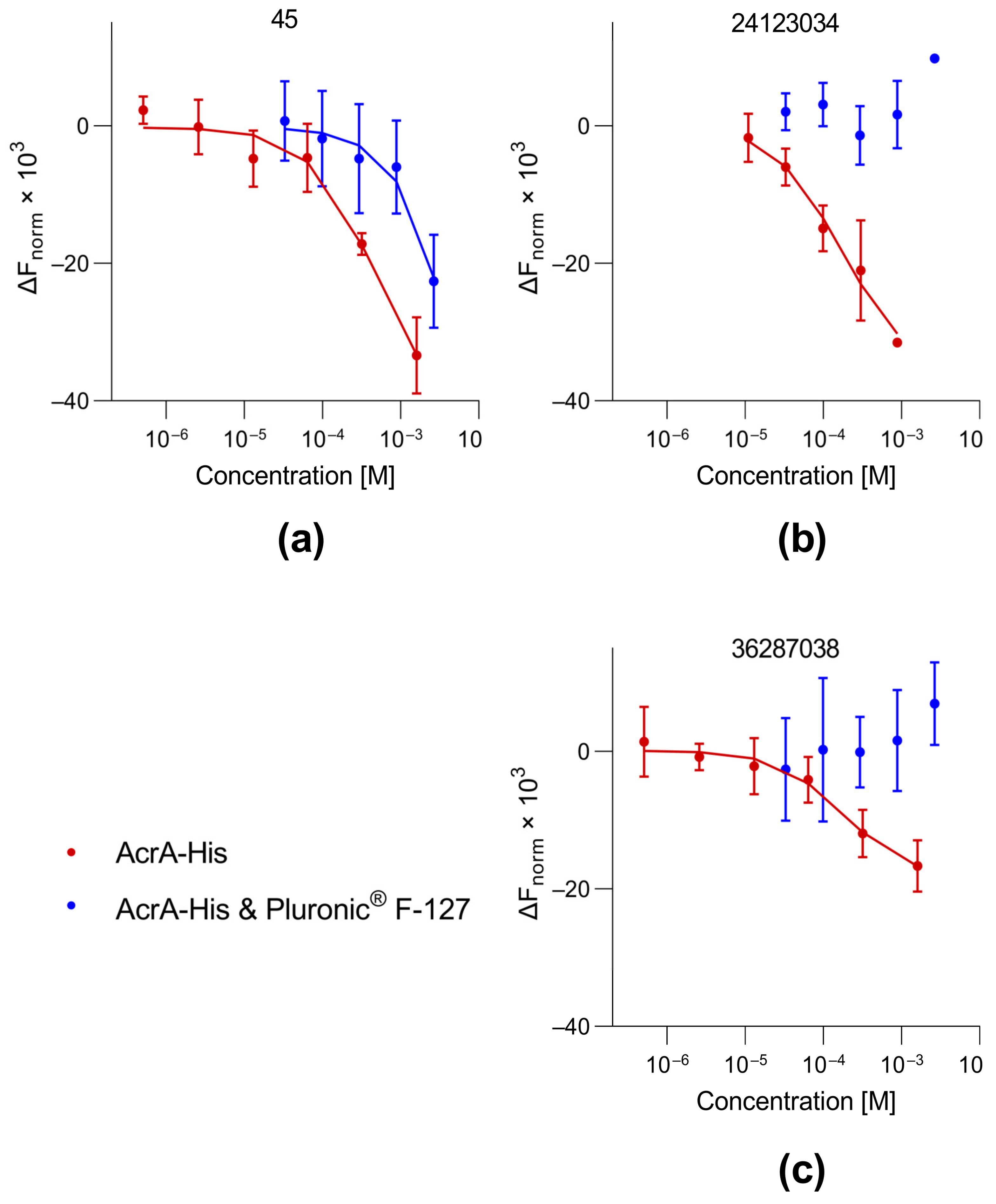
3.3. Protein Binding of Selected Compounds
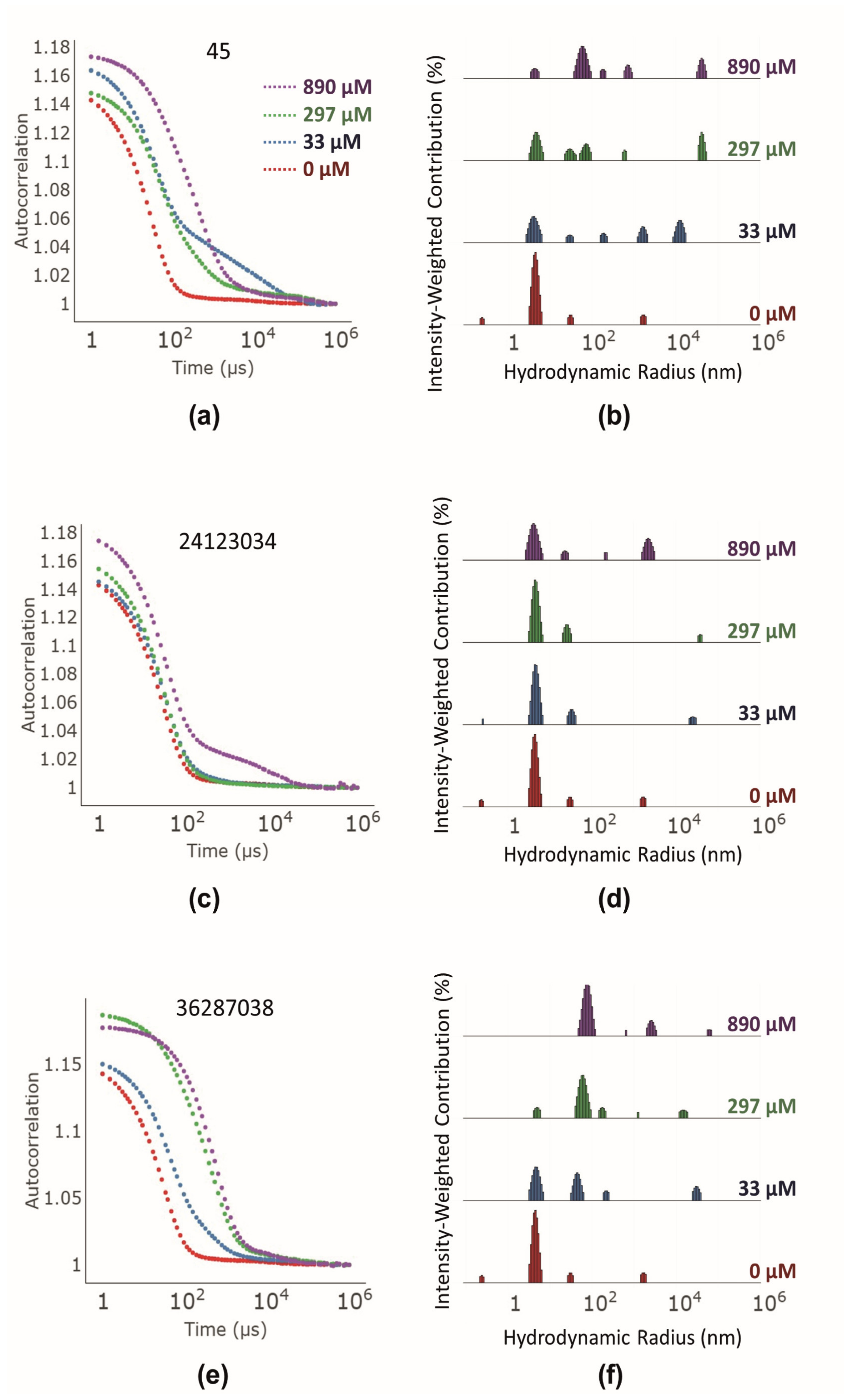
3.4. Investigation of Colloidal Aggregate Formation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ferri, M.; Ranucci, E.; Romagnoli, P.; Giaccone, V. Antimicrobial resistance: A global emerging threat to public health systems. Crit. Rev. Food Sci. Nutr. 2017, 57, 2857–2876. [Google Scholar] [CrossRef] [PubMed]
- Antimicrobial Resistance Collaborators. Global burden of bacterial antimicrobial resistance in 2019: A systematic analysis. Lancet 2022, 399, 629–655. [Google Scholar] [CrossRef] [PubMed]
- Tacconelli, E.; Carrara, E.; Savoldi, A.; Harbarth, S.; Mendelson, M.; Monnet, D.L.; Pulcini, C.; Kahlmeter, G.; Kluytmans, J.; Carmeli, Y.; et al. WHO Pathogens Priority List Working Group. Discovery, research, and development of new antibiotics: The WHO priority list of antibiotic-resistant bacteria and tuberculosis. Lancet Infect. Dis. 2018, 18, 318–327. [Google Scholar] [CrossRef]
- Mancuso, G.; Midiri, A.; Gerace, E.; Biondo, C. Bacterial Antibiotic Resistance: The Most Critical Pathogens. Pathogens 2021, 10, 1310. [Google Scholar] [CrossRef]
- De Oliveira, D.M.P.; Forde, B.M.; Kidd, T.J.; Harris, P.N.A.; Schembri, M.A.; Beatson, S.A.; Paterson, D.L.; Walker, M.J. Antimicrobial Resistance in ESKAPE Pathogens. Clin. Microbiol. Rev. 2020, 33, e00181-19. [Google Scholar] [CrossRef] [PubMed]
- Alav, I.; Kobylka, J.; Kuth, M.S.; Pos, K.M.; Picard, M.; Blair, J.M.A.; Bavro, V.N. Structure, Assembly, and Function of Tripartite Efflux and Type 1 Secretion Systems in Gram-Negative Bacteria. Chem. Rev. 2021, 121, 5479–5596. [Google Scholar] [CrossRef]
- Zwama, M.; Nishino, K. Ever-Adapting RND Efflux Pumps in Gram-Negative Multidrug-Resistant Pathogens: A Race against Time. Antibiotics 2021, 10, 774. [Google Scholar] [CrossRef]
- González-Bello, C. Antibiotic adjuvants—A strategy to unlock bacterial resistance to antibiotics. Bioorg. Med. Chem. Lett. 2017, 27, 4221–4228. [Google Scholar] [CrossRef]
- Thakur, V.; Uniyal, A.; Tiwari, V. A comprehensive review on pharmacology of efflux pumps and their inhibitors in antibiotic resistance. Eur. J. Pharmacol. 2021, 903, 174151. [Google Scholar] [CrossRef]
- Du, D.; Wang, Z.; James, N.R.; Voss, J.E.; Klimont, E.; Ohene-Agyei, T.; Venter, H.; Chiu, W.; Luisi, B.F. Structure of the AcrAB-TolC multidrug efflux pump. Nature 2014, 509, 512–515. [Google Scholar] [CrossRef] [Green Version]
- Compagne, N.; Vieira Da Cruz, A.; Müller, R.T.; Hartkoorn, R.C.; Flipo, M.; Pos, K.M. Update on the Discovery of Efflux Pump Inhibitors against Critical Priority Gram-Negative Bacteria. Antibiotics 2023, 12, 180. [Google Scholar] [CrossRef]
- Plé, C.; Tam, H.K.; Vieira Da Cruz, A.; Compagne, N.; Jiménez-Castellanos, J.C.; Müller, R.T.; Pradel, E.; Foong, W.E.; Malloci, G.; Ballée, A.; et al. Pyridylpiperazine-based allosteric inhibitors of RND-type multidrug efflux pumps. Nat. Commun. 2022, 13, 115. [Google Scholar] [CrossRef]
- Abdali, N.; Parks, J.M.; Haynes, K.M.; Chaney, J.L.; Green, A.T.; Wolloscheck, D.; Walker, J.K.; Rybenkov, V.V.; Baudry, J.; Smith, J.C.; et al. Reviving Antibiotics: Efflux Pump Inhibitors That Interact with AcrA, a Membrane Fusion Protein of the AcrAB-TolC Multidrug Efflux Pump. ACS Infect. Dis. 2017, 3, 89–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haynes, K.M.; Abdali, N.; Jhawar, V.; Zgurskaya, H.I.; Parks, J.M.; Green, A.T.; Baudry, J.; Rybenkov, V.V.; Smith, J.C.; Walker, J.K. Identification and Structure-Activity Relationships of Novel Compounds that Potentiate the Activities of Antibiotics in Escherichia coli. J. Med. Chem. 2017, 60, 6205–6219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Green, A.T.; Moniruzzaman, M.; Cooper, C.J.; Walker, J.K.; Smith, J.C.; Parks, J.M.; Zgurskaya, H.I. Discovery of multidrug efflux pump inhibitors with a novel chemical scaffold. Biochim. Biophys. Acta Gen. Subj. 2020, 1864, 129546. [Google Scholar] [CrossRef]
- Webb, B.; Sali, A. Protein Structure Modeling with MODELLER. Methods Mol. Biol. 2021, 2199, 239–255. [Google Scholar] [CrossRef] [PubMed]
- Sterling, T.; Irwin, J.J. ZINC 15-Ligand Discovery for Everyone. J. Chem. Inf. Model. 2015, 55, 2324–2337. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [Green Version]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Lomovskaya, O.; Warren, M.S.; Lee, A.; Galazzo, J.; Fronko, R.; Lee, M.; Blais, J.; Cho, D.; Chamberland, S.; Renau, T.; et al. Identification and characterization of inhibitors of multidrug resistance efflux pumps in Pseudomonas aeruginosa: Novel agents for combination therapy. Antimicrob. Agents Chemother. 2001, 45, 105–116. [Google Scholar] [CrossRef] [Green Version]
- Pos, K.M.; Diederichs, K. Purification, crystallization and preliminary diffraction studies of AcrB, an inner-membrane multi-drug efflux protein. Acta Crystallogr. D Biol. Crystallogr. 2002, 58, 1865–1867. [Google Scholar] [CrossRef]
- Burastero, O.; Niebling, S.; Defelipe, L.A.; Günther, C.; Struve, A.; Garcia Alai, M.M. eSPC: An online data-analysis platform for molecular biophysics. Acta Crystallogr. D Struct. Biol. 2021, 77, 1241–1250. [Google Scholar] [CrossRef] [PubMed]
- Paketurytė, V.; Petrauskas, V.; Zubrienė, A.; Abian, O.; Bastos, M.; Chen, W.Y.; Moreno, M.J.; Krainer, G.; Linkuvienė, V.; Sedivy, A.; et al. Uncertainty in protein-ligand binding constants: Asymmetric confidence intervals versus standard errors. Eur. Biophys. J. 2021, 50, 661–670. [Google Scholar] [CrossRef]
- Burastero, O.; Draper-Barr, G.; Raynal, B.; Chevreuil, M.; England, P.; Garcia-Alai, M.M. Raynals, an online tool for the analysis of dynamic light scattering. Acta Crystallogr. D Struct. Biol. 2023; in press. [Google Scholar]
- Kotov, V.; Mlynek, G.; Vesper, O.; Pletzer, M.; Wald, J.; Teixeira-Duarte, C.M.; Celia, H.; Garcia-Alai, M.; Nussberger, S.; Buchanan, S.K.; et al. In-depth interrogation of protein thermal unfolding data with MoltenProt. Protein Sci. 2021, 30, 201–217. [Google Scholar] [CrossRef]
- Yang, Z.-Y.; Yang, Z.-J.; Dong, J.; Wang, L.-L.; Zhang, L.X.; Ding, J.J.; Ding, X.Q.; Lu, A.P.; Hou, T.J.; Cao, D.S. Structural Analysis and Identification of Colloidal Aggregators in Drug Discovery. J. Chem. Inf. Model. 2019, 59, 3714–3726. [Google Scholar] [CrossRef]
- Auld, D.S.; Inglese, J.; Dahlin, J.L. Assay Interference by Aggregation. In Assay Guidance Manual [Internet]; Markossian, S., Grossman, A., Brimacombe, K., Eds.; Eli Lilly & Company and the National Center for Advancing Translational Sciences: Bethesda, MD, USA, 2017; pp. 1087–1112. Available online: https://www.ncbi.nlm.nih.gov/books/NBK442297/ (accessed on 3 January 2023).
- Ganesh, A.N.; Donders, E.N.; Shoichet, B.K.; Shoichet, M.S. Colloidal aggregation: From screening nuisance to formulation nuance. Nano Today 2018, 19, 188–200. [Google Scholar] [CrossRef]
- Giannetti, A.M.; Koch, B.D.; Browner, M.F. Surface plasmon resonance based assay for the detection and characterization of promiscuous inhibitors. J. Med. Chem. 2008, 51, 574–580. [Google Scholar] [CrossRef]
- Mustafi, D.; Smith, C.M.; Makinen, M.W.; Lee, R.C. Multi-block poloxamer surfactants suppress aggregation of denatured proteins. Biochim. Biophys. Acta Gen. Subj. 2008, 1780, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Coan, K.E.; Maltby, D.A.; Burlingame, A.L.; Shoichet, B.K. Promiscuous aggregate-based inhibitors promote enzyme unfolding. J. Med. Chem. 2009, 52, 2067–2075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venter, H.; Mowla, R.; Ohene-Agyei, T.; Ma, S. RND-type drug efflux pumps from Gram-negative bacteria: Molecular mechanism and inhibition. Front. Microbiol. 2015, 6, 377. [Google Scholar] [CrossRef]
- Moniruzzaman, M.; Cooper, C.J.; Uddin, M.R.; Walker, J.K. Analysis of Orthogonal Efflux and Permeation Properties of Compounds Leads to the Discovery of New Efflux Pump Inhibitors. ACS Infect. Dis. 2022, 8, 2149–2160. [Google Scholar] [CrossRef]
- Tambat, R.; Mahey, N.; Chandal, N.; Verma, D.K. A Microbe-Derived Efflux Pump Inhibitor of the Resistance-Nodulation-Cell Division Protein Restores Antibiotic Susceptibility in Escherichia coli and Pseudomonas aeruginosa. ACS Infect. Dis. 2022, 8, 255–270. [Google Scholar] [CrossRef]
- O’Donnell, H.R.; Tummino, T.A.; Bardine, C.; Craik, C.S. Colloidal Aggregators in Biochemical SARS-CoV-2 Repurposing Screens. J. Med. Chem. 2021, 64, 17530–17539. [Google Scholar] [CrossRef]
- Baell, J.B.; Nissink, J.W.M. Seven Year Itch: Pan-Assay Interference Compounds (PAINS) in 2017-Utility and Limitations. ACS Chem. Biol. 2018, 13, 36–44. [Google Scholar] [CrossRef] [Green Version]
- Donner, J.; Reck, M.; Bunk, B.; Jarek, M.; App, C.B.; Meier-Kolthoff, J.P.; Overmann, J.; Müller, R.; Kirschning, A.; Wagner-Döbler, I.; et al. The biofilm inhibitor carolacton enters gram-negative cells: Studies using a TolC-deficient strain of Escherichia coli. mSphere 2017, 2, e0037-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zgurskaya, H.I.; Krishnamoorthy, G.; Ntreh, A.; Lu, S. Mechanism and Function of the Outer Membrane Channel TolC in Multidrug Resistance and Physiology of Enterobacteria. Front. Microbiol. 2011, 2, 189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torosyan, H.; Shoichet, B.K. Protein Stability Effects in Aggregate-Based Enzyme Inhibition. J. Med. Chem. 2019, 62, 9593–9599. [Google Scholar] [CrossRef] [PubMed]
- Rothenaigner, I.; Hadian, K. Brief Guide: Experimental Strategies for High-Quality Hit Selection from Small-Molecule Screening Campaigns. SLAS Discov. 2021, 26, 851–854. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cpd. | AcrAHis (MST) (µM) a | AcrBHis (IF) (µM) b | AcrAHis (SPR) (µM) | AcrBS1043 (SPR) (µM) | TEVpHis (MST) (µM) c |
|---|---|---|---|---|---|
| 45 | 485 (204; 1440) | 301 (44.6; 1270) | N/A | N/A | 174 (71.8; 422) |
| 24123034 | 164 (42.4; 761) | 601 (475; 718) | 420 | 440 | 623 (23.9; 4550) |
| 36287038 | 183 (46.3; 762) | 402 (322; 493) | 230 | 1610 | 88.6 (11; 603) |
| Compound | MST (GST3C) a | |
|---|---|---|
| KD (µM) | CI 95% | |
| 45 | 903 | 582; 1420 |
| 24123034 | 334 | 111; 1070 |
| 36287038 | 619 | 231; 1700 |
| Compound | TM ±SD [°C] a | Cmeasured (µM) b |
|---|---|---|
| No compound | 50.4 ± 0.0 | - |
| 45 | 49.5 ± 0.1 | 2000 |
| 24123034 | 50.0 ± 0.2 | 2000 |
| 36287038 | 49.8 ± 0.2 | 2000 |
| Compound | TM ± SD [°C] a | Cmeasured (µM) b |
|---|---|---|
| No compound | 50.2 ± 0.1 | - |
| 2 | 50.1 ± 0.1 | 1000 c |
| 25 | 50.1 ± 0.0 | 2000 |
| 26 | 50.1 ± 0.0 | 2000 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Szal, T.; Chauhan, S.S.; Lewe, P.; Rachad, F.-Z.; Madre, M.; Paunina, L.; Witt, S.; Parthasarathi, R.; Windshügel, B. Efflux Pump-Binding 4(3-Aminocyclobutyl)Pyrimidin-2-Amines Are Colloidal Aggregators. Biomolecules 2023, 13, 1000. https://doi.org/10.3390/biom13061000
Szal T, Chauhan SS, Lewe P, Rachad F-Z, Madre M, Paunina L, Witt S, Parthasarathi R, Windshügel B. Efflux Pump-Binding 4(3-Aminocyclobutyl)Pyrimidin-2-Amines Are Colloidal Aggregators. Biomolecules. 2023; 13(6):1000. https://doi.org/10.3390/biom13061000
Chicago/Turabian StyleSzal, Tania, Shweta Singh Chauhan, Philipp Lewe, Fatima-Zahra Rachad, Marina Madre, Laura Paunina, Susanne Witt, Ramakrishnan Parthasarathi, and Björn Windshügel. 2023. "Efflux Pump-Binding 4(3-Aminocyclobutyl)Pyrimidin-2-Amines Are Colloidal Aggregators" Biomolecules 13, no. 6: 1000. https://doi.org/10.3390/biom13061000