
Skeletal Muscle Atrophy Induced by Diabetes Is Mediated by Non-Selective Channels and Prevented by Boldine
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
3. Results
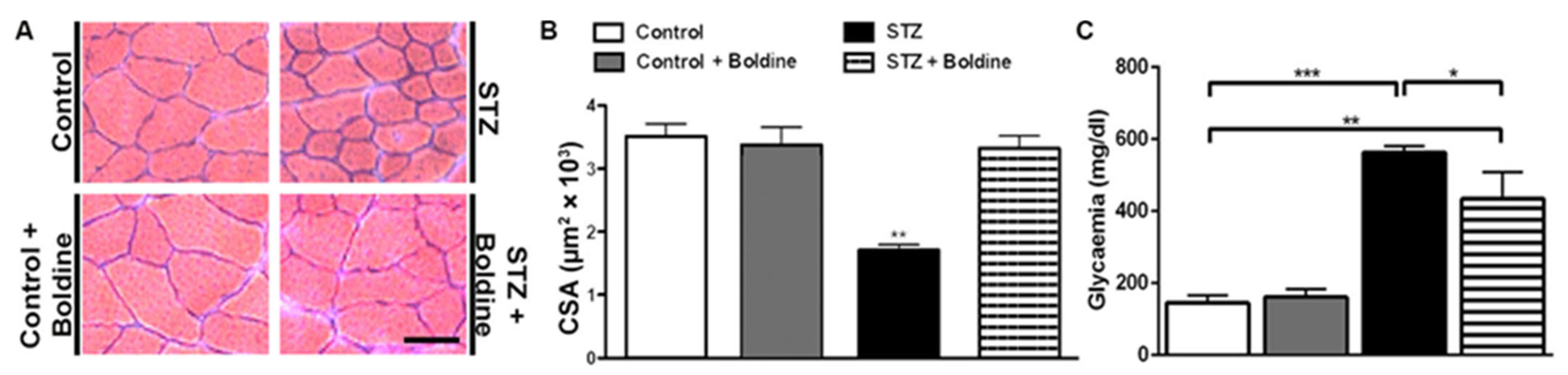
3.1. Boldine Prevents the Reduction in Cross-Sectional Area of Diabetic Skeletal Myofibers and Partially Reduced the Elevated Glycaemia
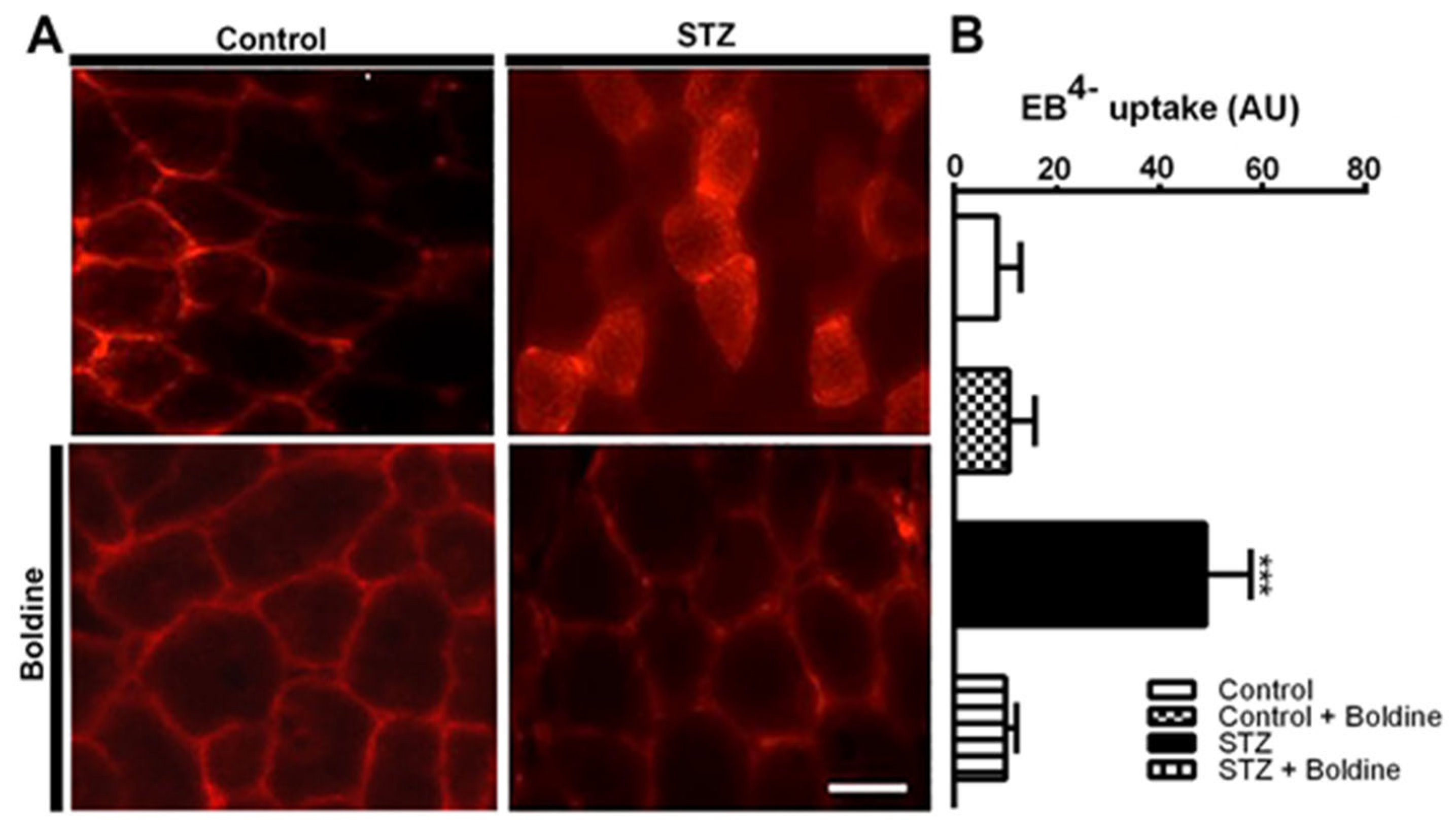
3.2. Boldine Prevents Diabetes-Induced Skeletal Myofiber Permeabilization
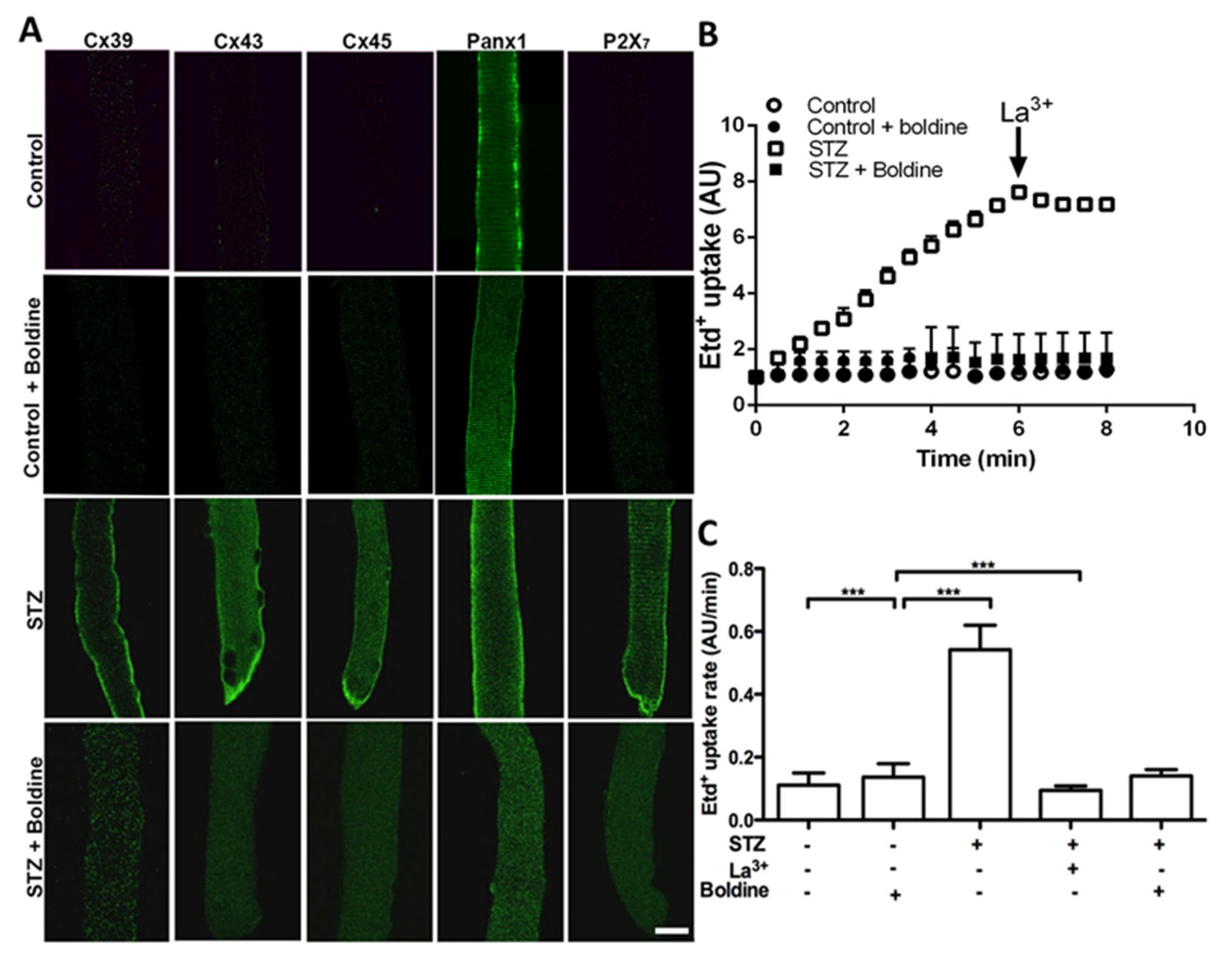
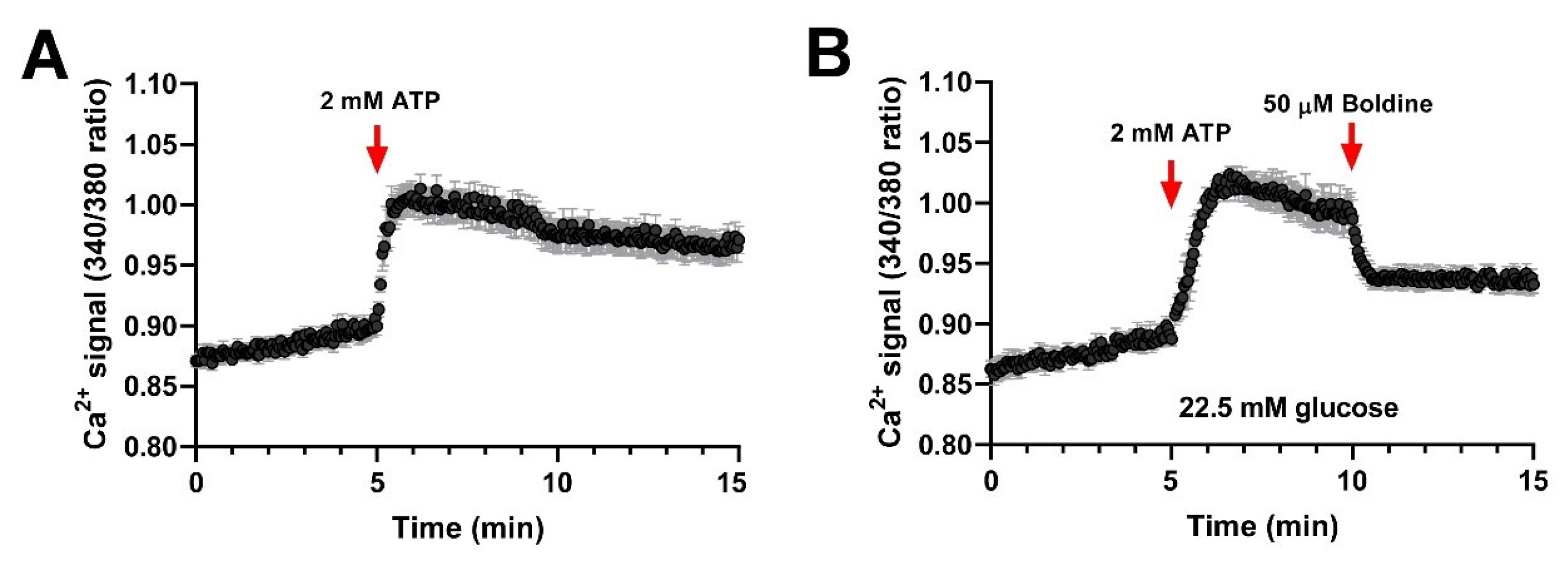
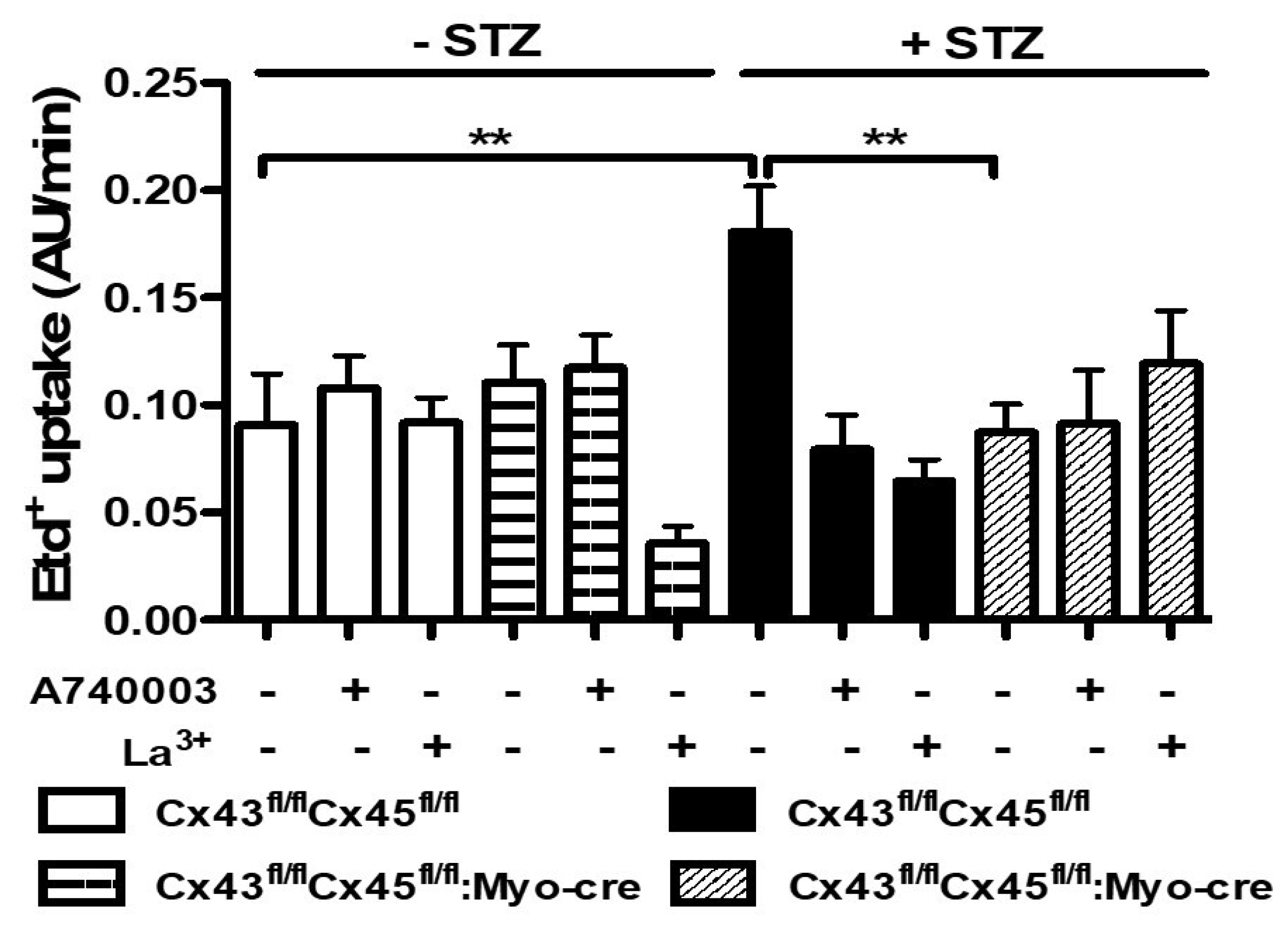
3.3. Boldine Reduces the Distribution and Functional Expression of Non-Selective Channels in Diabetic Skeletal Myofibers
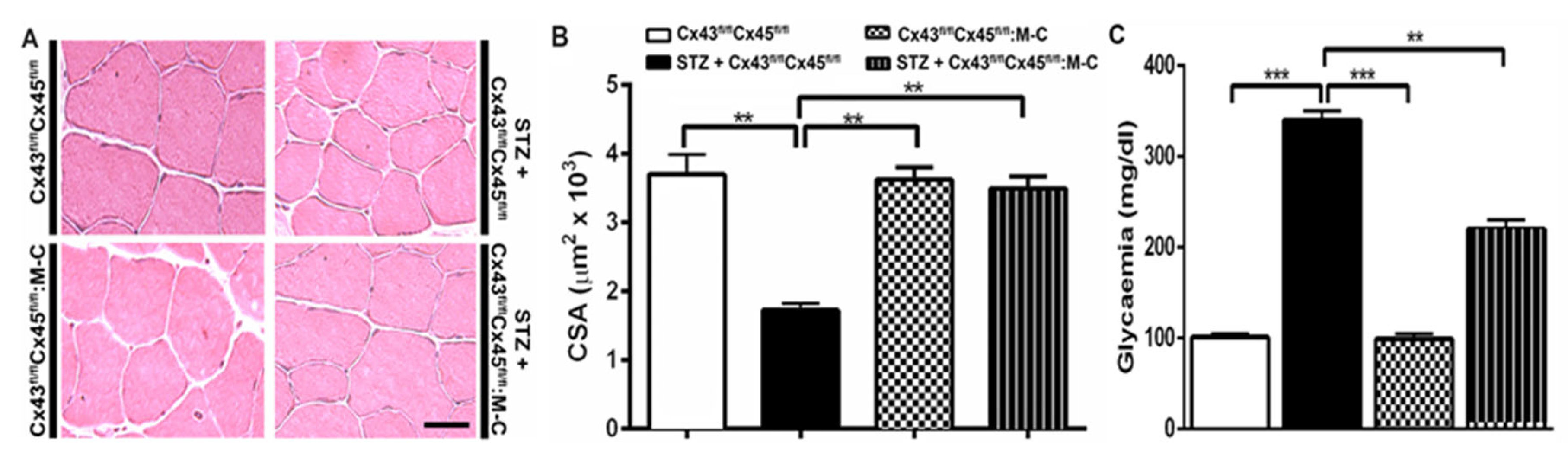
3.4. The Sarcolemma of Skeletal Myofibers Is Not Permeabilized in Diabetic Mice with Myofibers Deficient in Connexin43 and Connexin45 Expression
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Biondi-Zoccai, G.G.; Abbate, A.; Liuzzo, G.; Biasucci, L.M. Atherothrombosis, inflammation, and diabetes. J. Am. Coll. Cardiol. 2003, 41, 1071–1077. [Google Scholar] [CrossRef] [PubMed]
- Forbes, J.M.; Cooper, M.E. Mechanisms of diabetic complications. Physiol. Rev. 2013, 93, 137–188. [Google Scholar] [CrossRef] [PubMed]
- DeFronzo, R.A. Glucose intolerance and aging. Diabetes Care 1981, 4, 493–501. [Google Scholar] [CrossRef] [PubMed]
- D’Souza, D.M.; Al-Sajee, D.; Hawke, T.J. Diabetic myopathy: Impact of diabetes mellitus on skeletal muscle progenitor cells. Front. Physiol. 2013, 4, 379. [Google Scholar] [CrossRef]
- Cea, L.A.; Cisterna, B.A.; Puebla, C.; Frank, M.; Figueroa, X.F.; Cardozo, C.; Willecke, K.; Latorre, R.; Sáez, J.C. De novo expression of connexin hemichannels in denervated fast skeletal muscles leads to atrophy. Proc. Natl. Acad. Sci. USA 2013, 110, 16229–16234. [Google Scholar] [CrossRef]
- Cea, L.A.; Balboa, E.; Vargas, A.A.; Puebla, C.; Brañes, M.C.; Escamilla, R.; Regueira, T.; Sáez, J.C. De novo expression of functional connexins 43 and 45 hemichannels increases sarcolemmal permeability of skeletal myofibers during endotoxemia. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 2765–2773. [Google Scholar] [CrossRef]
- Cisterna, B.A.; Vargas, A.A.; Puebla, C.; Fernández, P.; Escamilla, R.; Lagos, C.F.; Matus, M.F.; Vilos, C.; Cea, L.A.; Barnafi, E.; et al. Active acetylcholine receptors prevent the atrophy of skeletal muscles and favor reinnervation. Nat. Commun. 2020, 11, 1073. [Google Scholar] [CrossRef]
- Balboa, E.; Saavedra-Leiva, F.; Cea, L.A.; Vargas, A.A.; Ramírez, V.; Escamilla, R.; Sáez, J.C.; Regueira, T. Sepsis-Induced Channelopathy in Skeletal Muscles is Associated with Expression of Non-Selective Channels. Shock 2018, 49, 221–228. [Google Scholar] [CrossRef]
- Peng, B.; Xu, C.; Wang, S.; Zhang, Y.; Li, W. The Role of Connexin Hemichannels in Inflammatory Diseases. Biology 2022, 11, 237. [Google Scholar] [CrossRef]
- Donath, M.Y.; Dinarello, C.A.; Mandrup-Poulsen, T. Targeting innate immune mediators in type 1 and type 2 diabetes. Nat. Rev. Immunol. 2019, 19, 734–746. [Google Scholar] [CrossRef]
- Hernández-Salinas, R.; Vielma, A.Z.; Arismendi, M.N.; Boric, M.P.; Sáez, J.C.; Velarde, V. Boldine prevents renal alterations in diabetic rats. J. Diabetes Res. 2013, 2013, 593672. [Google Scholar] [CrossRef]
- Yi, C.; Ezan, P.; Fernández, P.; Schmitt, J.; Sáez, J.C.; Giaume, C.; Koulakoff, A. Inhibition of glial hemichannels by boldine treatment reduces neuronal suffering in a murine model of Alzheimer’s disease. Glia 2017, 65, 1607–1625. [Google Scholar] [CrossRef]
- Koshimizu, T.; Koshimizu, M.; Stojilkovic, S.S. Contributions of the C-terminal domain to the control of P2X receptor desensitization. J. Biol. Chem. 1999, 274, 37651–37657. [Google Scholar] [CrossRef]
- Choi, E.J.; Palacios-Prado, N.; Sáez, J.C.; Lee, J. Identification of Cx45 as a Major Component of GJs in HeLa Cells. Biomolecules 2020. 10, 1389.
- Cea, L.A.; Puebla, C.; Cisterna, B.A.; Escamilla, R.; Vargas, A.A.; Frank, M.; Martínez-Montero, P.; Prior, C.; Molano, J.; Esteban-Rodríguez, I.; et al. Fast skeletal myofibers of mdx mouse, model of Duchenne muscular dystrophy, express connexin hemichannels that lead to apoptosis. Cell. Mol. Life Sci. 2016, 73, 2583–2599. [Google Scholar] [CrossRef]
- Cea, L.A.; Fernández, G.; Arias-Bravo, G.; Castillo-Ruiz, M.; Escamilla, R.; Brañes, M.C.; Sáez, J.C. Blockade of Hemichannels Normalizes the Differentiation Fate of Myoblasts and Features of Skeletal Muscles from Dysferlin-Deficient Mice. Int. J. Mol. Sci. 2020, 21, 6025. [Google Scholar] [CrossRef]
- Messemer, N.; Kunert, C.; Grohmann, M.; Sobottka, H.; Nieber, K.; Zimmermann, H.; Franke, H.; Nörenberg, W.; Straub, I.; Schaefer, M.; et al. P2X7 receptors at adult neural progenitor cells of the mouse subventricular zone. Neuropharmacology 2013, 73, 122–137. [Google Scholar] [CrossRef]
- Allsopp, R.C.; Dayl, S.; Dayel, A.B.; Schmid, R.; Evans, R.J. Mapping the Allosteric Action of Antagonists A740003 and A438079 Reveals a Role for the Left Flipper in Ligand Sensitivity at P2X7 Receptors. Mol. Pharmacol. 2018, 93, 553–562. [Google Scholar] [CrossRef]
- Cline, G.W.; Petersen, K.F.; Krssak, M.; Shen, J.; Hundal, R.S.; Trajanoski, Z.; Inzucchi, S.; Dresner, A.; Rothman, D.L.; Shulman, G.I. Impaired glucose transport as a cause of decreased insulin-stimulated muscle glycogen synthesis in type 2 diabetes. New Engl. J. Med. 1999, 341, 240–246. [Google Scholar] [CrossRef]
- Sáez, J.C.; Contreras-Duarte, S.; Labra, V.C.; Santibañez, C.A.; Mellado, L.A.; Inostroza, C.A.; Alvear, T.F.; Retamal, M.A.; Velarde, V.; Orellana, J.A. Interferon-γ and high glucose-induced opening of Cx43 hemichannels causes endothelial cell dysfunction and damage. Biochim. Biophys. Acta Mol. Cell Res. 2020, 1867, 118720. [Google Scholar] [CrossRef]
- Schalper KA, Sánchez HA, Lee SC, Altenberg GA, Nathanson MH, Sáez JC. Connexin 43 hemichannels mediate the Ca2+ influx induced by extracellular alkalinization.Am J Physiol Cell Physiol. 2010, 299, C1504–C1515.
- Liang, X.; Samways, D.S.K.; Wolf, K.; Bowles, E.A.; Richards, J.P.; Bruno, J.; Dutertre, S.; DiPaolo, R.J.; Egan, T.M. Quantifying Ca2+ Current and Permeability in ATP-gated P2X7 Receptors. J. Biol. Chem. 2015, 290, 7930–7942. [Google Scholar] [CrossRef] [PubMed]
- Balboa, E.; Saavedra, F.; Cea, L.A.; Ramírez, V.; Escamilla, R.; Vargas, A.A.; Regueira, T.; Sáez, J.C. Vitamin E Blocks Connexin Hemichannels and Prevents Deleterious Effects of Glucocorticoid Treatment on Skeletal Muscles. Int. J. Mol. Sci. 2020, 21, 4094. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; De Vuyst, E.; Ponsaerts, R.; Boengler, K.; Palacios-Prado, N.; Wauman, J.; Lai, C.P.; De Bock, M.; Decrock, E.; Bol, M.; et al. Selective inhibition of Cx43 hemichannels by Gap19 and its impact on myocardial ischemia/reperfusion injury. Basic Res. Cardiol. 2013, 108, 309. [Google Scholar] [CrossRef] [PubMed]
- Rusiecka, O.M.; Tournier, M.; Molica, F.; Kwak, B.R. Pannexin1 channels—A potential therapeutic target in inflammation. Front. Cell Dev. Biol. 2022, 10, 1020826. [Google Scholar] [CrossRef]
- Solini, A.; Novak, I. Role of the P2X7 receptor in the pathogenesis of type 2 diabetes and its microvascular complications. Curr. Opin. Pharmacol. 2019, 47, 75–81. [Google Scholar] [CrossRef]
- Giaume, C.; Naus, C.C.; Sáez, J.C.; Leybaert, L. Glial Connexins and Pannexins in the Healthy and Diseased Brain. Physiol. Rev. 2021, 101, 93–145. [Google Scholar] [CrossRef]
- De Vuyst, E.; Wang, N.; Decrock, E.; De Bock, M.; Vinken, M.; Van Moorhem, M.; Lai, C.; Culot, M.; Rogiers, V.; Cecchelli, R.; et al. Ca2+ regulation f connexin 43 hemichannels in C6 glioma and glial cells. Cell Calcium. 2009, 46, 176–187. [Google Scholar] [CrossRef]
- Araya, R.; Eckardt, D.; Maxeiner, S.; Krüger, O.; Theis, M.; Willecke, K.; Sáez, J.C. Expression of connexins during differentiation and regeneration of skeletal muscle: Functional relevance of connexin43. J. Cell Sci. 2005, 118, 27–37. [Google Scholar] [CrossRef]
- Anderson, C.; Catoe, H.; Werner, R. MIR-206 regulates connexin43 expression during skeletal muscle development. Nucleic Acids Res. 2006, 34, 5863–5871. [Google Scholar] [CrossRef]
- Cea, L.A.; Balboa, E.; Puebla, C.; Vargas, A.A.; Cisterna, B.A.; Escamilla, R.; Regueira, T.; Sáez, J.C. Dexamethasone-induced muscular atrophy is mediated by functional expression of connexin-based hemichannels. Biochim. Biophys. Acta 2016, 1862, 1891–1899. [Google Scholar] [CrossRef]
- Fernández, G.; Arias-Bravo, G.; Bevilacqua, J.A.; Castillo-Ruiz, M.; Caviedes, P.; Sáez, J.C.; Cea, L.A. Myofibers deficient in connexins 43 and 45 expression protect mice from skeletal muscle and systemic dysfunction promoted by a dysferlin mutation. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165800. [Google Scholar] [CrossRef]
- Kimura, M.; Kimura, I.; Nakamura, T.; Nojima, H. Diabetic state-induced modification of resting membrane potential and conductance in diaphragm muscle of alloxan and diabetic KK-CAy mice. Diabetologia 1988, 31, 103–107. [Google Scholar] [CrossRef]
- Hernández-Ochoa, E.O.; Banks, Q.; Schneider, M.F. Acute Elevated Glucose Promotes Abnormal Action Potential-Induced Ca2+ Transients in Cultured Skeletal Muscle Fibers. J. Diabetes Res. 2017, 2017, 1509048. [Google Scholar] [CrossRef]
- Retamal, M.A.; Cortés, C.J.; Reuss, L.; Bennett, M.V.; Sáez, J.C. S-nitrosylation and permeation through connexin 43 hemichannels in astrocytes: Induction by oxidant stress and reversal by reducing agents. Proc. Natl. Acad. Sci. USA 2006, 103, 4475–4480. [Google Scholar] [CrossRef]
- Retamal, M.A.; Schalper, K.A.; Shoji, K.F.; Bennett, M.V.; Sáez, J.C. Opening of connexin 43 hemichannels is increased by lowering intracellular redox potential. Proc. Natl. Acad. Sci. USA 2007, 104, 8322–8327. [Google Scholar] [CrossRef]
- Figueroa, X.F.; Lillo, M.A.; Gaete, P.S.; Riquelme, M.A.; Sáez, J.C. Diffusion of nitric oxide across cell membranes of the vascular wall requires specific connexin-based channels. Neuropharmacology 2013, 75, 471–478. [Google Scholar] [CrossRef]
- Orellana, J.A.; Díaz, E.; Schalper, K.A.; Vargas, A.A.; Bennett, M.V.; Sáez, J.C. Cation permeation through connexin 43 hemichannels is cooperative, competitive and saturable with parameters depending on the permeant species. Biochem. Biophys. Res. Commun. 2011, 409, 603–609. [Google Scholar] [CrossRef]
- Schalper, K.A.; Palacios-Prado, N.; Orellana, J.A.; Sáez, J.C. Currently used methods for identification and characterization of hemichannels. Cell Commun. Adhes. 2008, 15, 207–218. [Google Scholar] [CrossRef]
- Lee, H.M.; Kim, J.J.; Kim, H.J.; Shong, M.; Ku, B.J.; Jo, E.K. Upregulated NLRP3 inflammasome activation in patients with type 2 diabetes. Diabetes 2013, 62, 194–204. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cea, L.A.; Vásquez, W.; Hernández-Salinas, R.; Vielma, A.Z.; Castillo-Ruiz, M.; Velarde, V.; Salgado, M.; Sáez, J.C. Skeletal Muscle Atrophy Induced by Diabetes Is Mediated by Non-Selective Channels and Prevented by Boldine. Biomolecules 2023, 13, 708. https://doi.org/10.3390/biom13040708
Cea LA, Vásquez W, Hernández-Salinas R, Vielma AZ, Castillo-Ruiz M, Velarde V, Salgado M, Sáez JC. Skeletal Muscle Atrophy Induced by Diabetes Is Mediated by Non-Selective Channels and Prevented by Boldine. Biomolecules. 2023; 13(4):708. https://doi.org/10.3390/biom13040708
Chicago/Turabian StyleCea, Luis A., Walter Vásquez, Romina Hernández-Salinas, Alejandra Z. Vielma, Mario Castillo-Ruiz, Victoria Velarde, Magdiel Salgado, and Juan C. Sáez. 2023. "Skeletal Muscle Atrophy Induced by Diabetes Is Mediated by Non-Selective Channels and Prevented by Boldine" Biomolecules 13, no. 4: 708. https://doi.org/10.3390/biom13040708