
Cellular Senescence in Intervertebral Disc Aging and Degeneration: Molecular Mechanisms and Potential Therapeutic Opportunities

 ,
,
Abstract
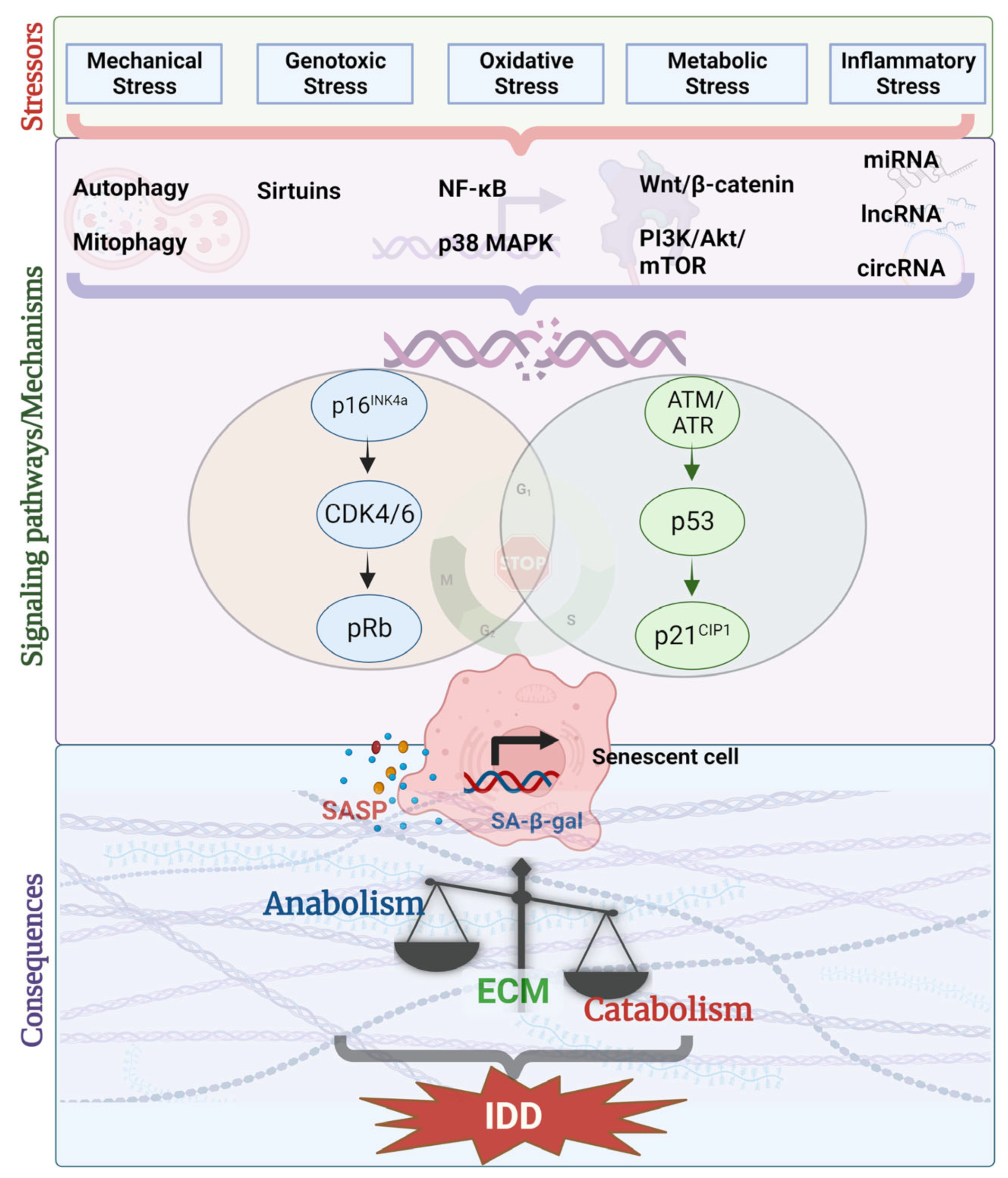
:1. Introduction
2. Overview of Cellular Senescence
3. Cellular Senescence in Aging and Degenerating Disc
4. Disc Cell Senescence under Various Stressors
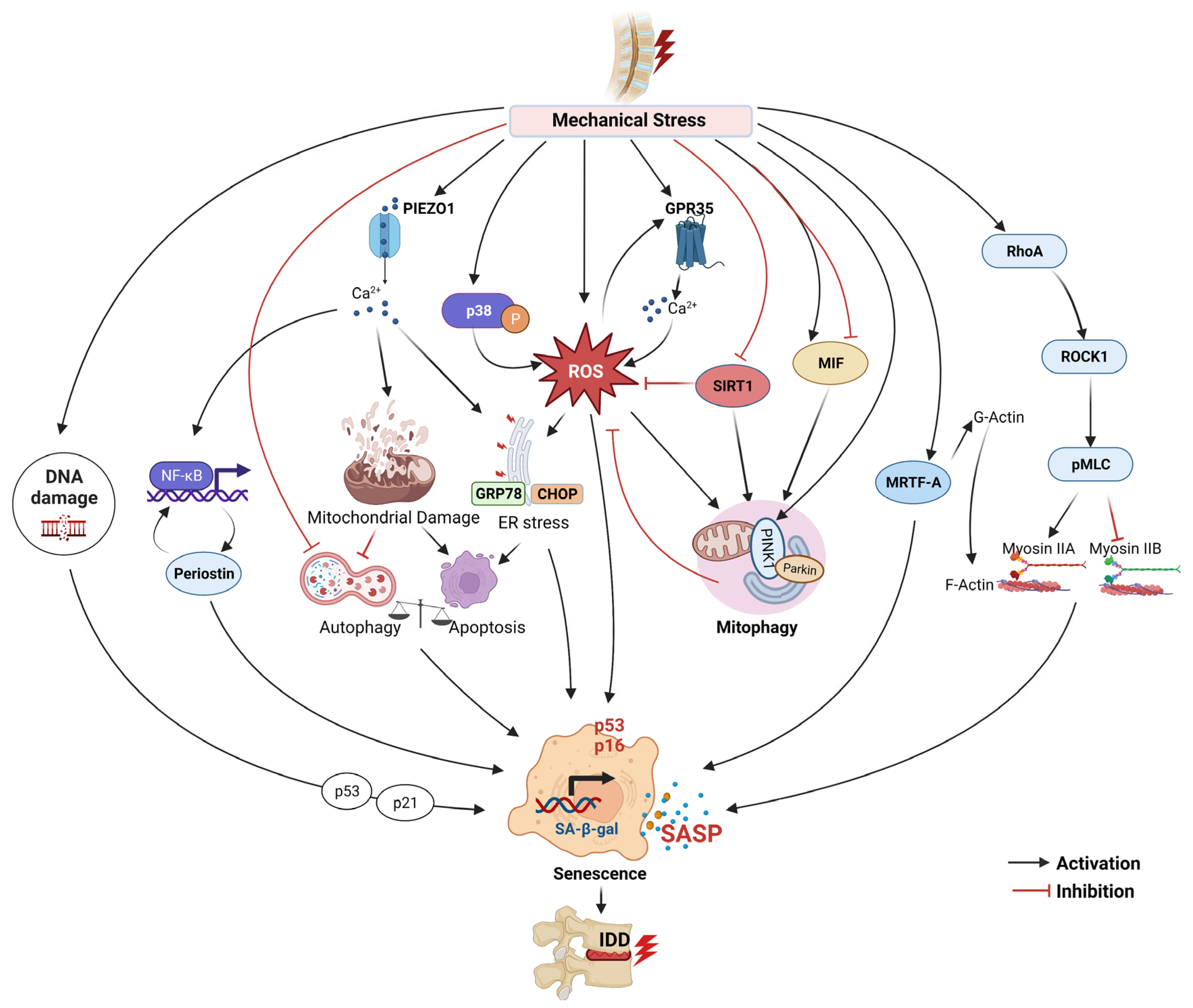
4.1. Mechanical Stress
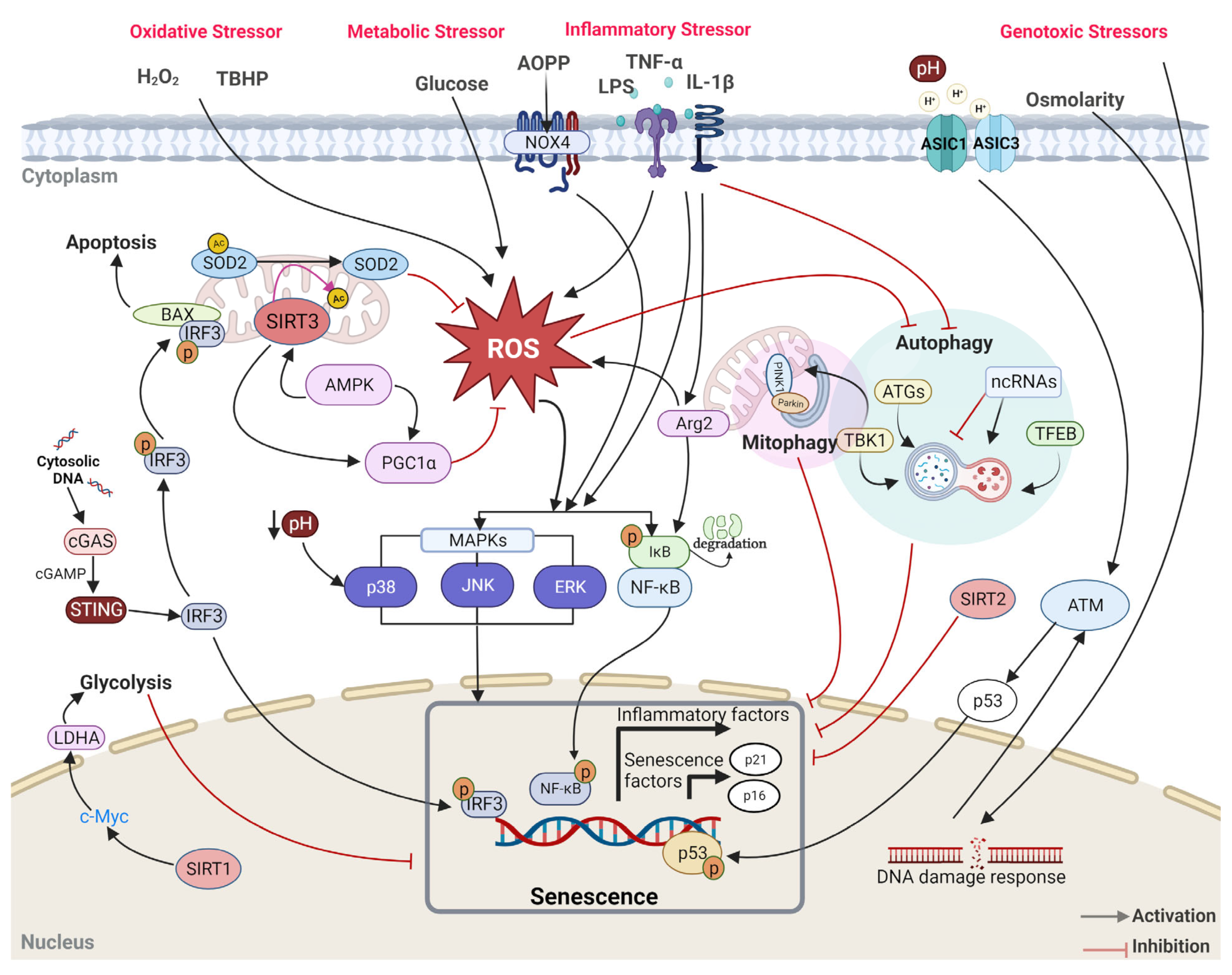
4.2. Genotoxic Stress
4.3. Oxidative Stress
4.4. Inflammatory Stress
4.5. Metabolic Stress
5. Signaling Molecules/Pathways
5.1. p53-p21CIP1-Rb and p16INK4a-Rb Pathways
5.2. MAPK and NF-κB Signaling
5.3. Sirtuins
5.4. Wnt/β Catenin Signaling
5.5. PI3K/Akt/mTOR Signaling
5.6. Non-Coding RNAs and Epigenetic Mechanisms
5.7. Autophagy/Mitophagy
6. Therapeutics
6.1. Senolytics
6.2. Other Therapeutic Possibilities
6.2.1. Natural and Synthetic Compounds
6.2.2. Genetic Alterations and Other Interventions
7. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Patil, P.; Dong, Q.; Wang, D.; Chang, J.; Wiley, C.; Demaria, M.; Lee, J.; Kang, J.; Niedernhofer, L.J.; Robbins, P.D.; et al. Systemic clearance of p16(INK4a) -positive senescent cells mitigates age-associated intervertebral disc degeneration. Aging Cell 2019, 18, e12927. [Google Scholar] [CrossRef]
- Patil, P.; Niedernhofer, L.J.; Robbins, P.D.; Lee, J.; Sowa, G.; Vo, N. Cellular senescence in intervertebral disc aging and degeneration. Curr. Mol. Biol. Rep. 2018, 4, 180–190. [Google Scholar] [CrossRef] [PubMed]
- Alessio, N.; Acar, M.B.; Squillaro, T.; Aprile, D.; Ayaz-Guner, S.; Di Bernardo, G.; Peluso, G.; Ozcan, S.; Galderisi, U. Progression of irradiated mesenchymal stromal cells from early to late senescence: Changes in SASP composition and anti-tumour properties. Cell Prolif. 2023, e13401. [Google Scholar] [CrossRef] [PubMed]
- Herranz, N.; Gil, J. Mechanisms and functions of cellular senescence. J. Clin. Investig. 2018, 128, 1238–1246. [Google Scholar] [CrossRef] [PubMed]
- Ott, C.; Jung, T.; Grune, T.; Hohn, A. SIPS as a model to study age-related changes in proteolysis and aggregate formation. Mech. Ageing Dev. 2018, 170, 72–81. [Google Scholar] [CrossRef] [PubMed]
- Gorgoulis, V.; Adams, P.D.; Alimonti, A.; Bennett, D.C.; Bischof, O.; Bishop, C.; Campisi, J.; Collado, M.; Evangelou, K.; Ferbeyre, G.; et al. Cellular Senescence: Defining a Path Forward. Cell 2019, 179, 813–827. [Google Scholar] [CrossRef]
- Vo, N.; Seo, H.Y.; Robinson, A.; Sowa, G.; Bentley, D.; Taylor, L.; Studer, R.; Usas, A.; Huard, J.; Alber, S.; et al. Accelerated aging of intervertebral discs in a mouse model of progeria. J. Orthop. Res. 2010, 28, 1600–1607. [Google Scholar] [CrossRef]
- Le Maitre, C.L.; Freemont, A.J.; Hoyland, J.A. Accelerated cellular senescence in degenerate intervertebral discs: A possible role in the pathogenesis of intervertebral disc degeneration. Arthritis Res. Ther. 2007, 9, R45. [Google Scholar] [CrossRef]
- Gruber, H.E.; Ingram, J.A.; Norton, H.J.; Hanley, E.N., Jr. Senescence in cells of the aging and degenerating intervertebral disc: Immunolocalization of senescence-associated beta-galactosidase in human and sand rat discs. Spine (Phila Pa 1976) 2007, 32, 321–327. [Google Scholar] [CrossRef]
- Roberts, S.; Evans, E.H.; Kletsas, D.; Jaffray, D.C.; Eisenstein, S.M. Senescence in human intervertebral discs. Eur. Spine J. 2006, 15 (Suppl. 3), S312–S316. [Google Scholar] [CrossRef]
- Chung, S.A.; Wei, A.Q.; Connor, D.E.; Webb, G.C.; Molloy, T.; Pajic, M.; Diwan, A.D. Nucleus pulposus cellular longevity by telomerase gene therapy. Spine (Phila Pa 1976) 2007, 32, 1188–1196. [Google Scholar] [CrossRef]
- Hartman, R.; Patil, P.; Tisherman, R.; St Croix, C.; Niedernhofer, L.J.; Robbins, P.D.; Ambrosio, F.; Van Houten, B.; Sowa, G.; Vo, N. Age-dependent changes in intervertebral disc cell mitochondria and bioenergetics. Eur. Cell Mater. 2018, 36, 171–183. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Luo, R.; Zhang, W.; He, S.; Wang, B.; Liang, H.; Song, Y.; Ke, W.; Shi, Y.; Feng, X.; et al. m6A hypomethylation of DNMT3B regulated by ALKBH5 promotes intervertebral disc degeneration via E4F1 deficiency. Clin. Transl. Med. 2022, 12, e765. [Google Scholar] [CrossRef] [PubMed]
- Vamvakas, S.S.; Mavrogonatou, E.; Kletsas, D. Human nucleus pulposus intervertebral disc cells becoming senescent using different treatments exhibit a similar transcriptional profile of catabolic and inflammatory genes. Eur. Spine J. 2017, 26, 2063–2071. [Google Scholar] [CrossRef]
- van Deursen, J.M. The role of senescent cells in ageing. Nature 2014, 509, 439–446. [Google Scholar] [CrossRef]
- Shi, J.; Pang, L.; Jiao, S. The response of nucleus pulposus cell senescence to static and dynamic compressions in a disc organ culture. Biosci. Rep. 2018, 38, BSR20180064. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Wang, C.; Xiao, C.; Gu, Q.; Long, L.; Wang, X.; Xu, H.; Li, S. Role of the mechanosensitive piezo1 channel in intervertebral disc degeneration. Clin. Physiol. Funct. Imaging 2023, 43, 59–70. [Google Scholar] [CrossRef]
- Shi, S.; Kang, X.J.; Zhou, Z.; He, Z.M.; Zheng, S.; He, S.S. Excessive mechanical stress-induced intervertebral disc degeneration is related to Piezo1 overexpression triggering the imbalance of autophagy/apoptosis in human nucleus pulpous. Arthritis Res. Ther. 2022, 24, 119. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Chen, Y.; Liao, Z.; Liu, H.; Zhang, S.; Zhong, D.; Qiu, X.; Chen, T.; Su, D.; Ke, X.; et al. Self-amplifying loop of NF-kappaB and periostin initiated by PIEZO1 accelerates mechano-induced senescence of nucleus pulposus cells and intervertebral disc degeneration. Mol. Ther. 2022, 30, 3241–3256. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Ke, W.; Wang, K.; Li, G.; Ma, L.; Lu, S.; Xiang, Q.; Liao, Z.; Luo, R.; Song, Y.; et al. Mechanosensitive Ion Channel Piezo1 Activated by Matrix Stiffness Regulates Oxidative Stress-Induced Senescence and Apoptosis in Human Intervertebral Disc Degeneration. Oxid. Med. Cell Longev. 2021, 2021, 8884922. [Google Scholar] [CrossRef]
- Wang, Y.; Hu, Y.; Wang, H.; Liu, N.; Luo, L.; Zhao, C.; Zhou, D.; Tong, H.; Li, P.; Zhou, Q. Deficiency of MIF Accentuates Overloaded Compression-Induced Nucleus Pulposus Cell Oxidative Damage via Depressing Mitophagy. Oxid. Med. Cell Longev. 2021, 2021, 6192498. [Google Scholar] [CrossRef] [PubMed]
- Ke, W.; Wang, B.; Hua, W.; Song, Y.; Lu, S.; Luo, R.; Li, G.; Wang, K.; Liao, Z.; Xiang, Q.; et al. The distinct roles of myosin IIA and IIB under compression stress in nucleus pulposus cells. Cell Prolif. 2021, 54, e12987. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, H.; Zhuo, Y.; Hu, Y.; Zhang, Z.; Ye, J.; Liu, L.; Luo, L.; Zhao, C.; Zhou, Q.; et al. SIRT1 alleviates high-magnitude compression-induced senescence in nucleus pulposus cells via PINK1-dependent mitophagy. Aging 2020, 12, 16126–16141. [Google Scholar] [CrossRef] [PubMed]
- Niu, M.; Ma, F.; Qian, J.; Li, J.; Wang, T.; Gao, Y.; Jin, J. N-cadherin attenuates nucleus pulposus cell senescence under high-magnitude compression. Mol. Med. Rep. 2018, 17, 2879–2884. [Google Scholar] [CrossRef]
- Feng, C.; Yang, M.; Zhang, Y.; Lan, M.; Huang, B.; Liu, H.; Zhou, Y. Cyclic mechanical tension reinforces DNA damage and activates the p53-p21-Rb pathway to induce premature senescence of nucleus pulposus cells. Int. J. Mol. Med. 2018, 41, 3316–3326. [Google Scholar] [CrossRef]
- Chen, Z.; Jiao, Y.; Zhang, Y.; Wang, Q.; Wu, W.; Zheng, J.; Li, J. G-Protein Coupled Receptor 35 Induces Intervertebral Disc Degeneration by Mediating the Influx of Calcium Ions and Upregulating Reactive Oxygen Species. Oxid. Med. Cell Longev. 2022, 2022, 5469220. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.; Peng, Y.; Li, Z.; Chen, S.; Deng, X.; Shao, Z.; Ma, K. Compression-induced senescence of nucleus pulposus cells by promoting mitophagy activation via the PINK1/PARKIN pathway. J. Cell Mol. Med. 2020, 24, 5850–5864. [Google Scholar] [CrossRef]
- Zhao, L.; Tian, B.; Xu, Q.; Zhang, C.; Zhang, L.; Fang, H. Extensive mechanical tension promotes annulus fibrosus cell senescence through suppressing cellular autophagy. Biosci. Rep. 2019, 39, BSR20190163. [Google Scholar] [CrossRef]
- Li, P.; Hou, G.; Zhang, R.; Gan, Y.; Xu, Y.; Song, L.; Zhou, Q. High-magnitude compression accelerates the premature senescence of nucleus pulposus cells via the p38 MAPK-ROS pathway. Arthritis Res. Ther. 2017, 19, 209. [Google Scholar] [CrossRef]
- Franco-Obregon, A.; Cambria, E.; Greutert, H.; Wernas, T.; Hitzl, W.; Egli, M.; Sekiguchi, M.; Boos, N.; Hausmann, O.; Ferguson, S.J.; et al. TRPC6 in simulated microgravity of intervertebral disc cells. Eur. Spine J. 2018, 27, 2621–2630. [Google Scholar] [CrossRef]
- Han, Y.; Zhou, C.M.; Shen, H.; Tan, J.; Dong, Q.; Zhang, L.; McGowan, S.J.; Zhao, J.; Sowa, G.A.; Kang, J.D.; et al. Attenuation of ataxia telangiectasia mutated signalling mitigates age-associated intervertebral disc degeneration. Aging Cell 2020, 19, e13162. [Google Scholar] [CrossRef]
- Nasto, L.A.; Wang, D.; Robinson, A.R.; Clauson, C.L.; Ngo, K.; Dong, Q.; Roughley, P.; Epperly, M.; Huq, S.M.; Pola, E.; et al. Genotoxic stress accelerates age-associated degenerative changes in intervertebral discs. Mech. Ageing Dev. 2013, 134, 35–42. [Google Scholar] [CrossRef]
- Gao, C.; Ning, B.; Sang, C.; Zhang, Y. Rapamycin prevents the intervertebral disc degeneration via inhibiting differentiation and senescence of annulus fibrosus cells. Aging 2018, 10, 131–143. [Google Scholar] [CrossRef]
- Zhong, J.; Chen, J.; Oyekan, A.A.; Epperly, M.W.; Greenberger, J.S.; Lee, J.Y.; Sowa, G.A.; Vo, N.V. Ionizing Radiation Induces Disc Annulus Fibrosus Senescence and Matrix Catabolism via MMP-Mediated Pathways. Int. J. Mol. Sci. 2022, 23, 4014. [Google Scholar] [CrossRef] [PubMed]
- Kouroumalis, A.; Mavrogonatou, E.; Savvidou, O.D.; Papagelopoulos, P.J.; Pratsinis, H.; Kletsas, D. Major traits of the senescent phenotype of nucleus pulposus intervertebral disc cells persist under the specific microenvironmental conditions of the tissue. Mech. Ageing Dev. 2019, 177, 118–127. [Google Scholar] [CrossRef]
- Wen, P.; Zheng, B.; Zhang, B.; Ma, T.; Hao, L.; Zhang, Y. The role of ageing and oxidative stress in intervertebral disc degeneration. Front. Mol. Biosci. 2022, 9, 1052878. [Google Scholar] [CrossRef] [PubMed]
- Cheng, F.; Yang, H.; Cheng, Y.; Liu, Y.; Hai, Y.; Zhang, Y. The role of oxidative stress in intervertebral disc cellular senescence. Front. Endocrinol. 2022, 13, 1038171. [Google Scholar] [CrossRef]
- Dimozi, A.; Mavrogonatou, E.; Sklirou, A.; Kletsas, D. Oxidative stress inhibits the proliferation, induces premature senescence and promotes a catabolic phenotype in human nucleus pulposus intervertebral disc cells. Eur. Cell Mater. 2015, 30, 89–102. [Google Scholar] [CrossRef]
- Wang, J.; Xia, D.; Lin, Y.; Xu, W.; Wu, Y.; Chen, J.; Chu, J.; Shen, P.; Weng, S.; Wang, X.; et al. Oxidative stress-induced circKIF18A downregulation impairs MCM7-mediated anti-senescence in intervertebral disc degeneration. Exp. Mol. Med. 2022, 54, 285–297. [Google Scholar] [CrossRef]
- Li, F.; Sun, X.; Zheng, B.; Sun, K.; Zhu, J.; Ji, C.; Lin, F.; Huan, L.; Luo, X.; Yan, C.; et al. Arginase II Promotes Intervertebral Disc Degeneration Through Exacerbating Senescence and Apoptosis Caused by Oxidative Stress and Inflammation via the NF-kappaB Pathway. Front. Cell Dev. Biol. 2021, 9, 737809. [Google Scholar] [CrossRef] [PubMed]
- Che, H.; Li, J.; Li, Y.; Ma, C.; Liu, H.; Qin, J.; Dong, J.; Zhang, Z.; Xian, C.J.; Miao, D.; et al. p16 deficiency attenuates intervertebral disc degeneration by adjusting oxidative stress and nucleus pulposus cell cycle. Elife 2020, 9, e52570. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Du, J.; Wu, X.; Xu, C.; Liu, J.; Jiang, L.; Cheng, X.; Ge, G.; Chen, L.; Pang, Q.; et al. SIRT3 mitigates intervertebral disc degeneration by delaying oxidative stress-induced senescence of nucleus pulposus cells. J. Cell Physiol. 2021, 236, 6441–6456. [Google Scholar] [CrossRef]
- Wu, T.; Jia, X.; Zhu, Z.; Guo, K.; Wang, Q.; Gao, Z.; Li, X.; Huang, Y.; Wu, D. Inhibition of miR-130b-3p restores autophagy and attenuates intervertebral disc degeneration through mediating ATG14 and PRKAA1. Apoptosis 2022, 27, 409–425. [Google Scholar] [CrossRef]
- Xie, C.; Shi, Y.; Chen, Z.; Zhou, X.; Luo, P.; Hong, C.; Tian, N.; Wu, Y.; Zhou, Y.; Lin, Y.; et al. Apigenin Alleviates Intervertebral Disc Degeneration via Restoring Autophagy Flux in Nucleus Pulposus Cells. Front. Cell Dev. Biol. 2021, 9, 787278. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Nisar, M.; Huang, C.; Pan, X.; Lin, D.; Zheng, G.; Jin, H.; Chen, D.; Tian, N.; Huang, Q.; et al. Small molecule natural compound agonist of SIRT3 as a therapeutic target for the treatment of intervertebral disc degeneration. Exp. Mol. Med. 2018, 50, 1–14. [Google Scholar] [CrossRef]
- Guo, Q.; Zhu, D.; Wang, Y.; Miao, Z.; Chen, Z.; Lin, Z.; Lin, J.; Huang, C.; Pan, L.; Wang, L.; et al. Targeting STING attenuates ROS induced intervertebral disc degeneration. Osteoarthr. Cartil. 2021, 29, 1213–1224. [Google Scholar] [CrossRef]
- Ashraf, S.; Chatoor, K.; Chong, J.; Pilliar, R.; Santerre, P.; Kandel, R. Transforming Growth Factor beta Enhances Tissue Formation by Passaged Nucleus Pulposus Cells In Vitro. J. Orthop. Res. 2020, 38, 438–449. [Google Scholar] [CrossRef] [PubMed]
- Liao, C.R.; Wang, S.N.; Zhu, S.Y.; Wang, Y.Q.; Li, Z.Z.; Liu, Z.Y.; Jiang, W.S.; Chen, J.T.; Wu, Q. Advanced oxidation protein products increase TNF-alpha and IL-1beta expression in chondrocytes via NADPH oxidase 4 and accelerate cartilage degeneration in osteoarthritis progression. Redox Biol. 2020, 28, 101306. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Chen, Y.; Yu, Z.; Liao, C.; Liu, Z.; Chen, J.; Wu, Q. Advanced oxidation protein products induce annulus fibrosus cell senescence through a NOX4-dependent, MAPK-mediated pathway and accelerate intervertebral disc degeneration. PeerJ 2022, 10, e13826. [Google Scholar] [CrossRef] [PubMed]
- Lyu, F.J.; Cui, H.; Pan, H.; Mc Cheung, K.; Cao, X.; Iatridis, J.C.; Zheng, Z. Painful intervertebral disc degeneration and inflammation: From laboratory evidence to clinical interventions. Bone Res. 2021, 9, 7. [Google Scholar] [CrossRef]
- Chen, C.C.; Chen, J.; Wang, W.L.; Xie, L.; Shao, C.Q.; Zhang, Y.X. Inhibition of the P53/P21 Pathway Attenuates the Effects of Senescent Nucleus Pulposus Cell-Derived Exosomes on the Senescence of Nucleus Pulposus Cells. Orthop. Surg. 2021, 13, 583–591. [Google Scholar] [CrossRef]
- Shao, Z.; Wang, B.; Shi, Y.; Xie, C.; Huang, C.; Chen, B.; Zhang, H.; Zeng, G.; Liang, H.; Wu, Y.; et al. Senolytic agent Quercetin ameliorates intervertebral disc degeneration via the Nrf2/NF-kappaB axis. Osteoarthr. Cartil. 2021, 29, 413–422. [Google Scholar] [CrossRef]
- Xie, J.; Li, B.; Zhang, P.; Wang, L.; Lu, H.; Song, X. Osteogenic protein-1 attenuates the inflammatory cytokine-induced NP cell senescence through regulating the ROS/NF-kappaB pathway. Biomed. Pharmacother. 2018, 99, 431–437. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Xie, J.J.; Jin, M.Y.; Gu, Y.T.; Wu, C.C.; Guo, W.J.; Yan, Y.Z.; Zhang, Z.J.; Wang, J.L.; Zhang, X.L.; et al. Sirt6 overexpression suppresses senescence and apoptosis of nucleus pulposus cells by inducing autophagy in a model of intervertebral disc degeneration. Cell Death Dis. 2018, 9, 56. [Google Scholar] [CrossRef] [PubMed]
- Ashraf, S.; Santerre, P.; Kandel, R. Induced senescence of healthy nucleus pulposus cells is mediated by paracrine signaling from TNF-alpha-activated cells. FASEB J. 2021, 35, e21795. [Google Scholar] [CrossRef] [PubMed]
- Feng, C.; Zhang, Y.; Yang, M.; Lan, M.; Liu, H.; Wang, J.; Zhou, Y.; Huang, B. The matrikine N-acetylated proline-glycine-proline induces premature senescence of nucleus pulposus cells via CXCR1-dependent ROS accumulation and DNA damage and reinforces the destructive effect of these cells on homeostasis of intervertebral discs. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 220–230. [Google Scholar] [CrossRef] [PubMed]
- Krock, E.; Rosenzweig, D.H.; Currie, J.B.; Bisson, D.G.; Ouellet, J.A.; Haglund, L. Toll-like Receptor Activation Induces Degeneration of Human Intervertebral Discs. Sci. Rep. 2017, 7, 17184. [Google Scholar] [CrossRef]
- Klawitter, M.; Hakozaki, M.; Kobayashi, H.; Krupkova, O.; Quero, L.; Ospelt, C.; Gay, S.; Hausmann, O.; Liebscher, T.; Meier, U.; et al. Expression and regulation of toll-like receptors (TLRs) in human intervertebral disc cells. Eur. Spine J. 2014, 23, 1878–1891. [Google Scholar] [CrossRef]
- Mannarino, M.; Cherif, H.; Li, L.; Sheng, K.; Rabau, O.; Jarzem, P.; Weber, M.H.; Ouellet, J.A.; Haglund, L. Toll-like receptor 2 induced senescence in intervertebral disc cells of patients with back pain can be attenuated by o-vanillin. Arthritis Res. Ther. 2021, 23, 117. [Google Scholar] [CrossRef] [PubMed]
- Zhong, H.; Zhou, Z.; Guo, L.; Liu, F.; Zheng, B.; Bi, S.; Tian, C. The miR-623/CXCL12 axis inhibits LPS-induced nucleus pulposus cell apoptosis and senescence. Mech. Ageing Dev. 2021, 194, 111417. [Google Scholar] [CrossRef]
- Han, Y.; Yuan, F.; Deng, C.; He, F.; Zhang, Y.; Shen, H.; Chen, Z.; Qian, L. Metformin decreases LPS-induced inflammatory response in rabbit annulus fibrosus stem/progenitor cells by blocking HMGB1 release. Aging 2019, 11, 10252–10265. [Google Scholar] [CrossRef] [PubMed]
- Wiley, C.D.; Campisi, J. The metabolic roots of senescence: Mechanisms and opportunities for intervention. Nat. Metab. 2021, 3, 1290–1301. [Google Scholar] [CrossRef] [PubMed]
- Bohme, I.; Bosserhoff, A. Extracellular acidosis triggers a senescence-like phenotype in human melanoma cells. Pigment Cell Melanoma Res. 2020, 33, 41–51. [Google Scholar] [CrossRef]
- Rider, S.M.; Mizuno, S.; Kang, J.D. Molecular Mechanisms of Intervertebral Disc Degeneration. Spine Surg. Relat. Res. 2019, 3, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Nachemson, A. Intradiscal measurements of pH in patients with lumbar rhizopathies. Acta Orthop. Scand. 1969, 40, 23–42. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, H.T.J.; Hodson, N.; Baird, P.; Richardson, S.M.; Hoyland, J.A. Acidic pH promotes intervertebral disc degeneration: Acid-sensing ion channel -3 as a potential therapeutic target. Sci. Rep. 2016, 6, 37360. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.; Zhang, R.; Li, H.; Ji, Q.; Cheng, X.; Thorne, R.F.; Hondermarck, H.; Liu, X.; Shen, C. ASIC1 and ASIC3 mediate cellular senescence of human nucleus pulposus mesenchymal stem cells during intervertebral disc degeneration. Aging 2021, 13, 10703–10723. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Yu, W.; Jiang, D. Acidic pH promotes nucleus pulposus cell senescence through activating the p38 MAPK pathway. Biosci. Rep. 2018, 38, BSR20181451. [Google Scholar] [CrossRef] [PubMed]
- Mavrogonatou, E.; Kletsas, D. High osmolality activates the G1 and G2 cell cycle checkpoints and affects the DNA integrity of nucleus pulposus intervertebral disc cells triggering an enhanced DNA repair response. DNA Repair 2009, 8, 930–943. [Google Scholar] [CrossRef]
- Kong, J.G.; Park, J.B.; Lee, D.; Park, E.Y. Effect of high glucose on stress-induced senescence of nucleus pulposus cells of adult rats. Asian Spine J. 2015, 9, 155–161. [Google Scholar] [CrossRef]
- Park, J.S.; Park, J.B.; Park, I.J.; Park, E.Y. Accelerated premature stress-induced senescence of young annulus fibrosus cells of rats by high glucose-induced oxidative stress. Int. Orthop. 2014, 38, 1311–1320. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Jiang, C.; Jin, L.; Chen, Z.; Feng, Z.; Jiang, X.; Cao, Y. In vitro and in vivo effects of hyperglycemia and diabetes mellitus on nucleus pulposus cell senescence. J. Orthop. Res. 2022, 40, 2350–2361. [Google Scholar] [CrossRef]
- Sakellaridis, N. The influence of diabetes mellitus on lumbar intervertebral disk herniation. Surg. Neurol. 2006, 66, 152–154. [Google Scholar] [CrossRef] [PubMed]
- Hou, G.; Zhao, H.; Teng, H.; Li, P.; Xu, W.; Zhang, J.; Lv, L.; Guo, Z.; Wei, L.; Yao, H.; et al. N-Cadherin Attenuates High Glucose-Induced Nucleus Pulposus Cell Senescence Through Regulation of the ROS/NF-kappaB Pathway. Cell Physiol. Biochem. 2018, 47, 257–265. [Google Scholar] [CrossRef]
- Wang, W.; Li, P.; Xu, J.; Wu, X.; Guo, Z.; Fan, L.; Song, R.; Wang, J.; Wei, L.; Teng, H. Resveratrol attenuates high glucose-induced nucleus pulposus cell apoptosis and senescence through activating the ROS-mediated PI3K/Akt pathway. Biosci. Rep. 2018, 38, BSR20171454. [Google Scholar] [CrossRef]
- Chen, J.; Huang, X.; Halicka, D.; Brodsky, S.; Avram, A.; Eskander, J.; Bloomgarden, N.A.; Darzynkiewicz, Z.; Goligorsky, M.S. Contribution of p16INK4a and p21CIP1 pathways to induction of premature senescence of human endothelial cells: Permissive role of p53. Am. J. Physiol. Heart Circ. Physiol. 2006, 290, H1575–H1586. [Google Scholar] [CrossRef] [PubMed]
- Roger, L.; Tomas, F.; Gire, V. Mechanisms and Regulation of Cellular Senescence. Int. J. Mol. Sci. 2021, 22, 13173. [Google Scholar] [CrossRef]
- Mijit, M.; Caracciolo, V.; Melillo, A.; Amicarelli, F.; Giordano, A. Role of p53 in the Regulation of Cellular Senescence. Biomolecules 2020, 10, 420. [Google Scholar] [CrossRef]
- Kumari, R.; Jat, P. Mechanisms of Cellular Senescence: Cell Cycle Arrest and Senescence Associated Secretory Phenotype. Front. Cell Dev. Biol. 2021, 9, 645593. [Google Scholar] [CrossRef]
- Sheekey, E.; Narita, M. p53 in senescence—It’s a marathon, not a sprint. FEBS J. 2023, 290, 1212–1220. [Google Scholar] [CrossRef]
- Chandra, A.; Lagnado, A.B.; Farr, J.N.; Doolittle, M.; Tchkonia, T.; Kirkland, J.L.; LeBrasseur, N.K.; Robbins, P.D.; Niedernhofer, L.J.; Ikeno, Y.; et al. Targeted clearance of p21- but not p16-positive senescent cells prevents radiation-induced osteoporosis and increased marrow adiposity. Aging Cell 2022, 21, e13602. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Wang, L.; Gasek, N.S.; Zhou, Y.; Kim, T.; Guo, C.; Jellison, E.R.; Haynes, L.; Yadav, S.; Tchkonia, T.; et al. An inducible p21-Cre mouse model to monitor and manipulate p21-highly-expressing senescent cells in vivo. Nat. Aging 2021, 1, 962–973. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.H.; Gao, J.W.; Bao, J.P.; Zhu, L.; Xie, Z.Y.; Chen, L.; Peng, X.; Zhang, C.; Wu, X.T. GATA4 promotes the senescence of nucleus pulposus cells via NF-kappaB pathway. Arch. Gerontol. Geriatr. 2022, 101, 104676. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Chen, Z.J. The cGAS-cGAMP-STING pathway connects DNA damage to inflammation, senescence, and cancer. J. Exp. Med. 2018, 215, 1287–1299. [Google Scholar] [CrossRef] [PubMed]
- Acosta, J.C.; Banito, A.; Wuestefeld, T.; Georgilis, A.; Janich, P.; Morton, J.P.; Athineos, D.; Kang, T.W.; Lasitschka, F.; Andrulis, M.; et al. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat. Cell Biol. 2013, 15, 978–990. [Google Scholar] [CrossRef] [PubMed]
- Novais, E.J.; Diekman, B.O.; Shapiro, I.M.; Risbud, M.V. p16(Ink4a) deletion in cells of the intervertebral disc affects their matrix homeostasis and senescence associated secretory phenotype without altering onset of senescence. Matrix Biol. 2019, 82, 54–70. [Google Scholar] [CrossRef]
- Ronkina, N.; Gaestel, M. MAPK-Activated Protein Kinases: Servant or Partner? Annu. Rev. Biochem. 2022, 91, 505–540. [Google Scholar] [CrossRef]
- Dong, C.; Davis, R.J.; Flavell, R.A. MAP kinases in the immune response. Annu. Rev. Immunol. 2002, 20, 55–72. [Google Scholar] [CrossRef]
- Zhang, S.; Liu, W.; Wang, P.; Hu, B.; Lv, X.; Chen, S.; Wang, B.; Shao, Z. Activation of HSP70 impedes tert-butyl hydroperoxide (t-BHP)-induced apoptosis and senescence of human nucleus pulposus stem cells via inhibiting the JNK/c-Jun pathway. Mol. Cell Biochem. 2021, 476, 1979–1994. [Google Scholar] [CrossRef]
- Zhao, R.; Yang, L.; He, S.; Xia, T. Nucleus pulposus cell senescence is regulated by substrate stiffness and is alleviated by LOX possibly through the integrin beta1-p38 MAPK signaling pathway. Exp. Cell Res. 2022, 417, 113230. [Google Scholar] [CrossRef]
- Yu, H.; Lin, L.; Zhang, Z.; Zhang, H.; Hu, H. Targeting NF-kappaB pathway for the therapy of diseases: Mechanism and clinical study. Signal Transduct. Target. Ther. 2020, 5, 209. [Google Scholar] [CrossRef] [PubMed]
- Nasto, L.A.; Seo, H.Y.; Robinson, A.R.; Tilstra, J.S.; Clauson, C.L.; Sowa, G.A.; Ngo, K.; Dong, Q.; Pola, E.; Lee, J.Y.; et al. ISSLS prize winner: Inhibition of NF-kappaB activity ameliorates age-associated disc degeneration in a mouse model of accelerated aging. Spine (Phila Pa 1976) 2012, 37, 1819–1825. [Google Scholar] [CrossRef] [PubMed]
- Qiu, H.B.; Bian, W.G.; Zhang, L.J.; Mei, N.; Wu, Y.; Wei, Y.Q.; Han, X.Z. Inhibition of p53/p21 by TWIST alleviates TNF-alpha induced nucleus pulposus cell senescence in vitro. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 12645–12654. [Google Scholar] [CrossRef]
- Zhang, L.; Li, X.; Kong, X.; Jin, H.; Han, Y.; Xie, Y. Effects of the NF-kappaB/p53 signaling pathway on intervertebral disc nucleus pulposus degeneration. Mol. Med. Rep. 2020, 22, 1821–1830. [Google Scholar] [CrossRef] [PubMed]
- Li, X.C.; Wang, M.S.; Liu, W.; Zhong, C.F.; Deng, G.B.; Luo, S.J.; Huang, C.M. Co-culturing nucleus pulposus mesenchymal stem cells with notochordal cell-rich nucleus pulposus explants attenuates tumor necrosis factor-alpha-induced senescence. Stem. Cell Res. Ther. 2018, 9, 171. [Google Scholar] [CrossRef]
- Anderson, K.A.; Madsen, A.S.; Olsen, C.A.; Hirschey, M.D. Metabolic control by sirtuins and other enzymes that sense NAD(+), NADH, or their ratio. Biochim. Biophys. Acta Bioenerg. 2017, 1858, 991–998. [Google Scholar] [CrossRef]
- Wu, Q.J.; Zhang, T.N.; Chen, H.H.; Yu, X.F.; Lv, J.L.; Liu, Y.Y.; Liu, Y.S.; Zheng, G.; Zhao, J.Q.; Wei, Y.F.; et al. The sirtuin family in health and disease. Signal Transduct. Target. Ther. 2022, 7, 402. [Google Scholar] [CrossRef]
- Kim, J.K.; Silwal, P.; Jo, E.K. Sirtuin 1 in Host Defense during Infection. Cells 2022, 11, 2921. [Google Scholar] [CrossRef]
- Tanno, M.; Sakamoto, J.; Miura, T.; Shimamoto, K.; Horio, Y. Nucleocytoplasmic shuttling of the NAD+-dependent histone deacetylase SIRT1. J. Biol. Chem. 2007, 282, 6823–6832. [Google Scholar] [CrossRef]
- Wu, L.; Shen, J.; Zhang, X.; Hu, Z. LDHA-Mediated Glycolytic Metabolism in Nucleus Pulposus Cells Is a Potential Therapeutic Target for Intervertebral Disc Degeneration. Biomed. Res. Int. 2021, 2021, 9914417. [Google Scholar] [CrossRef]
- He, J.; Zhang, A.; Song, Z.; Guo, S.; Chen, Y.; Liu, Z.; Zhang, J.; Xu, X.; Liu, J.; Chu, L. The resistant effect of SIRT1 in oxidative stress-induced senescence of rat nucleus pulposus cell is regulated by Akt-FoxO1 pathway. Biosci. Rep. 2019, 39, BSR20190112. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Shao, M.; Lu, F.; Jiang, J.; Xia, X. Role of Sirt1 Plays in Nucleus Pulposus Cells and Intervertebral Disc Degeneration. Spine (Phila Pa 1976) 2017, 42, E757–E766. [Google Scholar] [CrossRef] [PubMed]
- Sarikhani, M.; Maity, S.; Mishra, S.; Jain, A.; Tamta, A.K.; Ravi, V.; Kondapalli, M.S.; Desingu, P.A.; Khan, D.; Kumar, S.; et al. SIRT2 deacetylase represses NFAT transcription factor to maintain cardiac homeostasis. J. Biol. Chem. 2018, 293, 5281–5294. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Park, S.H.; Imbesi, M.; Nathan, W.J.; Zou, X.; Zhu, Y.; Jiang, H.; Parisiadou, L.; Gius, D. Loss of NAD-Dependent Protein Deacetylase Sirtuin-2 Alters Mitochondrial Protein Acetylation and Dysregulates Mitophagy. Antioxid. Redox Signal. 2017, 26, 849–863. [Google Scholar] [CrossRef]
- Gomes, P.; Fleming Outeiro, T.; Cavadas, C. Emerging Role of Sirtuin 2 in the Regulation of Mammalian Metabolism. Trends Pharmacol. Sci. 2015, 36, 756–768. [Google Scholar] [CrossRef]
- Yang, M.; Peng, Y.; Liu, W.; Zhou, M.; Meng, Q.; Yuan, C. Sirtuin 2 expression suppresses oxidative stress and senescence of nucleus pulposus cells through inhibition of the p53/p21 pathway. Biochem. Biophys. Res. Commun. 2019, 513, 616–622. [Google Scholar] [CrossRef]
- Gomes, P.; Viana, S.D.; Nunes, S.; Rolo, A.P.; Palmeira, C.M.; Reis, F. The yin and yang faces of the mitochondrial deacetylase sirtuin 3 in age-related disorders. Ageing Res. Rev. 2020, 57, 100983. [Google Scholar] [CrossRef]
- Kanfi, Y.; Naiman, S.; Amir, G.; Peshti, V.; Zinman, G.; Nahum, L.; Bar-Joseph, Z.; Cohen, H.Y. The sirtuin SIRT6 regulates lifespan in male mice. Nature 2012, 483, 218–221. [Google Scholar] [CrossRef]
- Li, X.; Liu, L.; Li, T.; Liu, M.; Wang, Y.; Ma, H.; Mu, N.; Wang, H. SIRT6 in Senescence and Aging-Related Cardiovascular Diseases. Front. Cell Dev. Biol. 2021, 9, 641315. [Google Scholar] [CrossRef]
- Grootaert, M.O.J.; Finigan, A.; Figg, N.L.; Uryga, A.K.; Bennett, M.R. SIRT6 Protects Smooth Muscle Cells From Senescence and Reduces Atherosclerosis. Circ. Res. 2021, 128, 474–491. [Google Scholar] [CrossRef]
- Zhao, G.; Wang, H.; Xu, C.; Wang, P.; Chen, J.; Wang, P.; Sun, Z.; Su, Y.; Wang, Z.; Han, L.; et al. SIRT6 delays cellular senescence by promoting p27Kip1 ubiquitin-proteasome degradation. Aging 2016, 8, 2308–2323. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Xiao, Q.; Xiao, J.; Niu, C.; Li, Y.; Zhang, X.; Zhou, Z.; Shu, G.; Yin, G. Wnt/beta-catenin signalling: Function, biological mechanisms, and therapeutic opportunities. Signal Transduct. Target. Ther. 2022, 7, 3. [Google Scholar] [CrossRef] [PubMed]
- Zhu, D.; Wang, Z.; Zhang, G.; Ma, C.; Qiu, X.; Wang, Y.; Liu, M.; Guo, X.; Chen, H.; Deng, Q.; et al. Periostin promotes nucleus pulposus cells apoptosis by activating the Wnt/beta-catenin signaling pathway. FASEB J. 2022, 36, e22369. [Google Scholar] [CrossRef]
- Yun, Z.; Wang, Y.; Feng, W.; Zang, J.; Zhang, D.; Gao, Y. Overexpression of microRNA-185 alleviates intervertebral disc degeneration through inactivation of the Wnt/beta-catenin signaling pathway and downregulation of Galectin-3. Mol. Pain 2020, 16, 1744806920902559. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Jian, Y.; Fu, H.; Li, B. MiR-532 downregulation of the Wnt/beta-catenin signaling via targeting Bcl-9 and induced human intervertebral disc nucleus pulposus cells apoptosis. J. Pharmacol. Sci. 2018, 138, 263–270. [Google Scholar] [CrossRef]
- Kondo, N.; Yuasa, T.; Shimono, K.; Tung, W.; Okabe, T.; Yasuhara, R.; Pacifici, M.; Zhang, Y.; Iwamoto, M.; Enomoto-Iwamoto, M. Intervertebral disc development is regulated by Wnt/beta-catenin signaling. Spine (Phila Pa 1976) 2011, 36, E513–E518. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Sun, Y.; Li, X.; Zhang, W. Rac1 regulates nucleus pulposus cell degeneration by activating the Wnt/beta-catenin signaling pathway and promotes the progression of intervertebral disc degeneration. Am. J. Physiol. Cell Physiol. 2022, 322, C496–C507. [Google Scholar] [CrossRef]
- Wang, X.; Zou, M.; Li, J.; Wang, B.; Zhang, Q.; Liu, F.; Lu, G. LncRNA H19 targets miR-22 to modulate H2O2 -induced deregulation in nucleus pulposus cell senescence, proliferation, and ECM synthesis through Wnt signaling. J. Cell Biochem. 2018, 119, 4990–5002. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Chen, S.; Chen, S.; Huang, D.; Ma, K.; Shao, Z. Moderate activation of Wnt/beta-catenin signaling promotes the survival of rat nucleus pulposus cells via regulating apoptosis, autophagy, and senescence. J. Cell Biochem. 2019, 120, 12519–12533. [Google Scholar] [CrossRef]
- Xiao, Q.; Teng, Y.; Xu, C.; Pan, W.; Yang, H.; Zhao, J.; Zhou, Q. Role of PI3K/AKT Signaling Pathway in Nucleus Pulposus Cells. Biomed. Res. Int. 2021, 2021, 9941253. [Google Scholar] [CrossRef]
- Zhao, Q.; Wang, X.Y.; Yu, X.X.; Zhai, Y.X.; He, X.; Wu, S.; Shi, Y.A. Expression of human telomerase reverse transcriptase mediates the senescence of mesenchymal stem cells through the PI3K/AKT signaling pathway. Int. J. Mol. Med. 2015, 36, 857–864. [Google Scholar] [CrossRef] [PubMed]
- Hemmings, B.A.; Restuccia, D.F. PI3K-PKB/Akt pathway. Cold Spring Harb. Perspect. Biol. 2012, 4, a011189. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Gan, Y.; Xu, Y.; Song, L.; Wang, L.; Ouyang, B.; Zhang, C.; Zhou, Q. The inflammatory cytokine TNF-alpha promotes the premature senescence of rat nucleus pulposus cells via the PI3K/Akt signaling pathway. Sci. Rep. 2017, 7, 42938. [Google Scholar] [CrossRef] [PubMed]
- Gong, C.; Pan, W.; Hu, W.; Chen, L. Bone morphogenetic protein-7 retards cell subculture-induced senescence of human nucleus pulposus cells through activating the PI3K/Akt pathway. Biosci. Rep. 2019, 39, BSR20182312. [Google Scholar] [CrossRef] [PubMed]
- Kakiuchi, Y.; Yurube, T.; Kakutani, K.; Takada, T.; Ito, M.; Takeoka, Y.; Kanda, Y.; Miyazaki, S.; Kuroda, R.; Nishida, K. Pharmacological inhibition of mTORC1 but not mTORC2 protects against human disc cellular apoptosis, senescence, and extracellular matrix catabolism through Akt and autophagy induction. Osteoarthr. Cartil. 2019, 27, 965–976. [Google Scholar] [CrossRef]
- Ito, M.; Yurube, T.; Kakutani, K.; Maeno, K.; Takada, T.; Terashima, Y.; Kakiuchi, Y.; Takeoka, Y.; Miyazaki, S.; Kuroda, R.; et al. Selective interference of mTORC1/RAPTOR protects against human disc cellular apoptosis, senescence, and extracellular matrix catabolism with Akt and autophagy induction. Osteoarthr. Cartil. 2017, 25, 2134–2146. [Google Scholar] [CrossRef]
- Yurube, T.; Ito, M.; Kakiuchi, Y.; Kuroda, R.; Kakutani, K. Autophagy and mTOR signaling during intervertebral disc aging and degeneration. JOR Spine 2020, 3, e1082. [Google Scholar] [CrossRef]
- Suh, N. MicroRNA controls of cellular senescence. BMB Rep. 2018, 51, 493–499. [Google Scholar] [CrossRef]
- Munk, R.; Panda, A.C.; Grammatikakis, I.; Gorospe, M.; Abdelmohsen, K. Senescence-Associated MicroRNAs. Int. Rev. Cell Mol. Biol. 2017, 334, 177–205. [Google Scholar] [CrossRef]
- Yan, J.; Wu, L.G.; Zhang, M.; Fang, T.; Pan, W.; Zhao, J.L.; Zhou, Q. miR-328-5p Induces Human Intervertebral Disc Degeneration by Targeting WWP2. Oxid. Med. Cell Longev. 2022, 2022, 3511967. [Google Scholar] [CrossRef]
- Xie, L.; Huang, W.; Fang, Z.; Ding, F.; Zou, F.; Ma, X.; Tao, J.; Guo, J.; Xia, X.; Wang, H.; et al. CircERCC2 ameliorated intervertebral disc degeneration by regulating mitophagy and apoptosis through miR-182-5p/SIRT1 axis. Cell Death Dis. 2019, 10, 751. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zhang, S.; Zhao, Y.; Qu, Z.; Zhuang, X.; Song, Q.; Leng, J.; Liu, Y. MicroRNA-140-3p alleviates intervertebral disc degeneration via KLF5/N-cadherin/MDM2/Slug axis. RNA Biol. 2021, 18, 2247–2260. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Li, D.; Wu, H.; Liu, F.; Liu, F.; Zhang, Q.; Li, J. LncRNA TRPC7-AS1 regulates nucleus pulposus cellular senescence and ECM synthesis via competing with HPN for miR-4769-5p binding. Mech. Ageing Dev. 2020, 190, 111293. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Han, Y.; Deng, C.; Chen, W.; Jin, L.; Chen, H.; Wang, K.; Shen, H.; Qian, L. Inflammation-dependent downregulation of miR-194-5p contributes to human intervertebral disc degeneration by targeting CUL4A and CUL4B. J. Cell Physiol. 2019, 234, 19977–19989. [Google Scholar] [CrossRef]
- Chai, X.; Si, H.; Song, J.; Chong, Y.; Wang, J.; Zhao, G. miR-486-5p Inhibits Inflammatory Response, Matrix Degradation and Apoptosis of Nucleus Pulposus Cells through Directly Targeting FOXO1 in Intervertebral Disc Degeneration. Cell Physiol. Biochem. 2019, 52, 109–118. [Google Scholar] [CrossRef]
- Li, Z.; Chen, X.; Xu, D.; Li, S.; Chan, M.T.V.; Wu, W.K.K. Circular RNAs in nucleus pulposus cell function and intervertebral disc degeneration. Cell Prolif. 2019, 52, e12704. [Google Scholar] [CrossRef]
- Hao, Y.; Zhu, G.; Yu, L.; Ren, Z.; Zhang, P.; Zhu, J.; Cao, S. Extracellular vesicles derived from mesenchymal stem cells confer protection against intervertebral disc degeneration through a microRNA-217-dependent mechanism. Osteoarthr. Cartil. 2022, 30, 1455–1467. [Google Scholar] [CrossRef]
- Zhan, S.; Wang, K.; Xiang, Q.; Song, Y.; Li, S.; Liang, H.; Luo, R.; Wang, B.; Liao, Z.; Zhang, Y.; et al. lncRNA HOTAIR upregulates autophagy to promote apoptosis and senescence of nucleus pulposus cells. J. Cell Physiol. 2020, 235, 2195–2208. [Google Scholar] [CrossRef]
- Li, G.; Ma, L.; He, S.; Luo, R.; Wang, B.; Zhang, W.; Song, Y.; Liao, Z.; Ke, W.; Xiang, Q.; et al. WTAP-mediated m(6)A modification of lncRNA NORAD promotes intervertebral disc degeneration. Nat. Commun. 2022, 13, 1469. [Google Scholar] [CrossRef]
- Guan, Y.; Zhang, C.; Lyu, G.; Huang, X.; Zhang, X.; Zhuang, T.; Jia, L.; Zhang, L.; Zhang, C.; Li, C.; et al. Senescence-activated enhancer landscape orchestrates the senescence-associated secretory phenotype in murine fibroblasts. Nucleic. Acids Res. 2020, 48, 10909–10923. [Google Scholar] [CrossRef]
- Horvath, S. DNA methylation age of human tissues and cell types. Genome Biol. 2013, 14, R115. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Petroni, G.; Amaravadi, R.K.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Bravo-San Pedro, J.M.; Cadwell, K.; Cecconi, F.; Choi, A.M.K.; et al. Autophagy in major human diseases. EMBO J. 2021, 40, e108863. [Google Scholar] [CrossRef] [PubMed]
- Kritschil, R.; Scott, M.; Sowa, G.; Vo, N. Role of autophagy in intervertebral disc degeneration. J. Cell Physiol. 2022, 237, 1266–1284. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.J.; Yang, W.; Wang, C.; He, W.S.; Deng, H.Y.; Yan, Y.G.; Zhang, J.; Xiang, Y.X.; Wang, W.J. Autophagy: A double-edged sword in intervertebral disk degeneration. Clin. Chim. Acta 2016, 457, 27–35. [Google Scholar] [CrossRef]
- Ito, M.; Yurube, T.; Kanda, Y.; Kakiuchi, Y.; Takeoka, Y.; Takada, T.; Kuroda, R.; Kakutani, K. Inhibition of Autophagy at Different Stages by ATG5 Knockdown and Chloroquine Supplementation Enhances Consistent Human Disc Cellular Apoptosis and Senescence Induction rather than Extracellular Matrix Catabolism. Int. J. Mol. Sci. 2021, 22, 3965. [Google Scholar] [CrossRef]
- Shao, Z.; Ni, L.; Hu, S.; Xu, T.; Meftah, Z.; Yu, Z.; Tian, N.; Wu, Y.; Sun, L.; Wu, A.; et al. RNA-binding protein HuR suppresses senescence through Atg7 mediated autophagy activation in diabetic intervertebral disc degeneration. Cell Prolif. 2021, 54, e12975. [Google Scholar] [CrossRef]
- Tsujimoto, R.; Yurube, T.; Takeoka, Y.; Kanda, Y.; Miyazaki, K.; Ohnishi, H.; Kakiuchi, Y.; Miyazaki, S.; Zhang, Z.; Takada, T.; et al. Involvement of autophagy in the maintenance of rat intervertebral disc homeostasis: An in-vitro and in-vivo RNA interference study of Atg5. Osteoarthr. Cartil. 2022, 30, 481–493. [Google Scholar] [CrossRef]
- Zheng, G.; Pan, Z.; Zhan, Y.; Tang, Q.; Zheng, F.; Zhou, Y.; Wu, Y.; Zhou, Y.; Chen, D.; Chen, J.; et al. TFEB protects nucleus pulposus cells against apoptosis and senescence via restoring autophagic flux. Osteoarthr. Cartil. 2019, 27, 347–357. [Google Scholar] [CrossRef]
- Zhou, W.; Shi, Y.; Wang, H.; Chen, L.; Yu, C.; Zhang, X.; Yang, L.; Zhang, X.; Wu, A. Exercise-induced FNDC5/irisin protects nucleus pulposus cells against senescence and apoptosis by activating autophagy. Exp. Mol. Med. 2022, 54, 1038–1048. [Google Scholar] [CrossRef]
- Nguyen, T.N.; Padman, B.S.; Lazarou, M. Deciphering the Molecular Signals of PINK1/Parkin Mitophagy. Trends Cell Biol. 2016, 26, 733–744. [Google Scholar] [CrossRef]
- Wang, Y.; Shen, J.; Chen, Y.; Liu, H.; Zhou, H.; Bai, Z.; Hu, Z.; Guo, X. PINK1 protects against oxidative stress induced senescence of human nucleus pulposus cells via regulating mitophagy. Biochem. Biophys. Res. Commun. 2018, 504, 406–414. [Google Scholar] [CrossRef]
- Hu, S.; Chen, L.; Al Mamun, A.; Ni, L.; Gao, W.; Lin, Y.; Jin, H.; Zhang, X.; Wang, X. The therapeutic effect of TBK1 in intervertebral disc degeneration via coordinating selective autophagy and autophagic functions. J. Adv. Res. 2021, 30, 1–13. [Google Scholar] [CrossRef]
- Oakes, J.A.; Davies, M.C.; Collins, M.O. TBK1: A new player in ALS linking autophagy and neuroinflammation. Mol. Brain 2017, 10, 5. [Google Scholar] [CrossRef]
- Zhang, Z.; Xu, T.; Chen, J.; Shao, Z.; Wang, K.; Yan, Y.; Wu, C.; Lin, J.; Wang, H.; Gao, W.; et al. Parkin-mediated mitophagy as a potential therapeutic target for intervertebral disc degeneration. Cell Death Dis. 2018, 9, 980. [Google Scholar] [CrossRef]
- Jeon, O.H.; Kim, C.; Laberge, R.M.; Demaria, M.; Rathod, S.; Vasserot, A.P.; Chung, J.W.; Kim, D.H.; Poon, Y.; David, N.; et al. Local clearance of senescent cells attenuates the development of post-traumatic osteoarthritis and creates a pro-regenerative environment. Nat. Med. 2017, 23, 775–781. [Google Scholar] [CrossRef]
- Baker, D.J.; Wijshake, T.; Tchkonia, T.; LeBrasseur, N.K.; Childs, B.G.; van de Sluis, B.; Kirkland, J.L.; van Deursen, J.M. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature 2011, 479, 232–236. [Google Scholar] [CrossRef]
- Farr, J.N.; Xu, M.; Weivoda, M.M.; Monroe, D.G.; Fraser, D.G.; Onken, J.L.; Negley, B.A.; Sfeir, J.G.; Ogrodnik, M.B.; Hachfeld, C.M.; et al. Targeting cellular senescence prevents age-related bone loss in mice. Nat. Med. 2017, 23, 1072–1079. [Google Scholar] [CrossRef]
- Schafer, M.J.; White, T.A.; Iijima, K.; Haak, A.J.; Ligresti, G.; Atkinson, E.J.; Oberg, A.L.; Birch, J.; Salmonowicz, H.; Zhu, Y.; et al. Cellular senescence mediates fibrotic pulmonary disease. Nat. Commun. 2017, 8, 14532. [Google Scholar] [CrossRef]
- Alessio, N.; Squillaro, T.; Lettiero, I.; Galano, G.; De Rosa, R.; Peluso, G.; Galderisi, U.; Di Bernardo, G. Biomolecular Evaluation of Piceatannol’s Effects in Counteracting the Senescence of Mesenchymal Stromal Cells: A New Candidate for Senotherapeutics? Int. J. Mol. Sci. 2021, 22, 11619. [Google Scholar] [CrossRef]
- Zhu, Y.; Tchkonia, T.; Pirtskhalava, T.; Gower, A.C.; Ding, H.; Giorgadze, N.; Palmer, A.K.; Ikeno, Y.; Hubbard, G.B.; Lenburg, M.; et al. The Achilles’ heel of senescent cells: From transcriptome to senolytic drugs. Aging Cell 2015, 14, 644–658. [Google Scholar] [CrossRef]
- Novais, E.J.; Tran, V.A.; Johnston, S.N.; Darris, K.R.; Roupas, A.J.; Sessions, G.A.; Shapiro, I.M.; Diekman, B.O.; Risbud, M.V. Long-term treatment with senolytic drugs Dasatinib and Quercetin ameliorates age-dependent intervertebral disc degeneration in mice. Nat. Commun. 2021, 12, 5213. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Yao, S.; Fu, F.; Bian, Y.; Zhang, Z.; Zhang, H.; Luo, H.; Ge, Y.; Chen, Y.; Ji, W.; et al. Morroniside attenuates nucleus pulposus cell senescence to alleviate intervertebral disc degeneration via inhibiting ROS-Hippo-p53 pathway. Front. Pharmacol. 2022, 13, 942435. [Google Scholar] [CrossRef] [PubMed]
- Ghasemi, F.; Shafiee, M.; Banikazemi, Z.; Pourhanifeh, M.H.; Khanbabaei, H.; Shamshirian, A.; Amiri Moghadam, S.; ArefNezhad, R.; Sahebkar, A.; Avan, A.; et al. Curcumin inhibits NF-kB and Wnt/beta-catenin pathways in cervical cancer cells. Pathol. Res. Pract. 2019, 215, 152556. [Google Scholar] [CrossRef]
- Mortezaee, K.; Salehi, E.; Mirtavoos-Mahyari, H.; Motevaseli, E.; Najafi, M.; Farhood, B.; Rosengren, R.J.; Sahebkar, A. Mechanisms of apoptosis modulation by curcumin: Implications for cancer therapy. J. Cell Physiol. 2019, 234, 12537–12550. [Google Scholar] [CrossRef] [PubMed]
- Kang, L.; Xiang, Q.; Zhan, S.; Song, Y.; Wang, K.; Zhao, K.; Li, S.; Shao, Z.; Yang, C.; Zhang, Y. Restoration of Autophagic Flux Rescues Oxidative Damage and Mitochondrial Dysfunction to Protect against Intervertebral Disc Degeneration. Oxid. Med. Cell Longev. 2019, 2019, 7810320. [Google Scholar] [CrossRef] [PubMed]
- Cherif, H.; Bisson, D.G.; Jarzem, P.; Weber, M.; Ouellet, J.A.; Haglund, L. Curcumin and o-Vanillin Exhibit Evidence of Senolytic Activity in Human IVD Cells In Vitro. J. Clin. Med. 2019, 8, 433. [Google Scholar] [CrossRef]
- Cherif, H.; Bisson, D.G.; Mannarino, M.; Rabau, O.; Ouellet, J.A.; Haglund, L. Senotherapeutic drugs for human intervertebral disc degeneration and low back pain. eLife 2020, 9, e54693. [Google Scholar] [CrossRef]
- Ren, C.; Jin, J.; Li, C.; Xiang, J.; Wu, Y.; Zhou, Y.; Sun, L.; Zhang, X.; Tian, N. Metformin inactivates the cGAS-STING pathway through autophagy and suppresses senescence in nucleus pulposus cells. J. Cell Sci. 2022, 135, jcs259738. [Google Scholar] [CrossRef]
- Huang, X.; Chen, C.; Chen, Y.; Xu, J.; Liu, L. Omentin-1 alleviate interleukin-1beta(IL-1beta)-induced nucleus pulposus cells senescence. Bioengineered 2022, 13, 13849–13859. [Google Scholar] [CrossRef]
- Li, P.; Gan, Y.; Xu, Y.; Wang, L.; Ouyang, B.; Zhang, C.; Luo, L.; Zhao, C.; Zhou, Q. 17beta-estradiol Attenuates TNF-alpha-Induced Premature Senescence of Nucleus Pulposus Cells through Regulating the ROS/NF-kappaB Pathway. Int. J. Biol. Sci. 2017, 13, 145–156. [Google Scholar] [CrossRef]
- Wang, X.Y.; Jiao, L.Y.; He, J.L.; Fu, Z.A.; Guo, R.J. Parathyroid hormone 1-34 inhibits senescence in rat nucleus pulposus cells by activating autophagy via the m-TOR pathway. Mol. Med. Rep. 2018, 18, 2681–2688. [Google Scholar] [CrossRef] [PubMed]
- Che, H.; Ma, C.; Li, H.; Yu, F.; Wei, Y.; Chen, H.; Wu, J.; Ren, Y. Rebalance of the Polyamine Metabolism Suppresses Oxidative Stress and Delays Senescence in Nucleus Pulposus Cells. Oxid. Med. Cell Longev. 2022, 2022, 8033353. [Google Scholar] [CrossRef] [PubMed]
- Shi, P.Z.; Wang, J.W.; Wang, P.C.; Han, B.; Lu, X.H.; Ren, Y.X.; Feng, X.M.; Cheng, X.F.; Zhang, L. Urolithin a alleviates oxidative stress-induced senescence in nucleus pulposus-derived mesenchymal stem cells through SIRT1/PGC-1alpha pathway. World J. Stem Cells 2021, 13, 1928–1946. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Li, J.; Li, Y.; Che, H.; Chen, Y.; Dong, J.; Xian, C.J.; Miao, D.; Wang, L.; Ren, Y. Bmi deficiency causes oxidative stress and intervertebral disc degeneration which can be alleviated by antioxidant treatment. J. Cell Mol. Med. 2020, 24, 8950–8961. [Google Scholar] [CrossRef]
- Li, X.; Lin, F.; Wu, Y.; Liu, N.; Wang, J.; Chen, R.; Lu, Z. Resveratrol attenuates inflammation environment-induced nucleus pulposus cell senescence in vitro. Biosci. Rep. 2019, 39, BSR20190126. [Google Scholar] [CrossRef]
- Harrison, D.E.; Strong, R.; Sharp, Z.D.; Nelson, J.F.; Astle, C.M.; Flurkey, K.; Nadon, N.L.; Wilkinson, J.E.; Frenkel, K.; Carter, C.S.; et al. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature 2009, 460, 392–395. [Google Scholar] [CrossRef]
- Kim, K.W.; Jeong, S.W.; Park, H.Y.; Heu, J.Y.; Jung, H.Y.; Lee, J.S. The effect of prolonged rhBMP-2 treatment on telomerase activity, replicative capacity and senescence of human nucleus pulposus cells. Biotech. Histochem. 2020, 95, 490–498. [Google Scholar] [CrossRef]
- Murray Stewart, T.; Dunston, T.T.; Woster, P.M.; Casero, R.A., Jr. Polyamine catabolism and oxidative damage. J. Biol. Chem. 2018, 293, 18736–18745. [Google Scholar] [CrossRef]
- Nasto, L.A.; Robinson, A.R.; Ngo, K.; Clauson, C.L.; Dong, Q.; St Croix, C.; Sowa, G.; Pola, E.; Robbins, P.D.; Kang, J.; et al. Mitochondrial-derived reactive oxygen species (ROS) play a causal role in aging-related intervertebral disc degeneration. J. Orthop. Res. 2013, 31, 1150–1157. [Google Scholar] [CrossRef]
- Tomas-Barberan, F.A.; Gonzalez-Sarrias, A.; Garcia-Villalba, R.; Nunez-Sanchez, M.A.; Selma, M.V.; Garcia-Conesa, M.T.; Espin, J.C. Urolithins, the rescue of “old” metabolites to understand a “new” concept: Metabotypes as a nexus among phenolic metabolism, microbiota dysbiosis, and host health status. Mol. Nutr. Food Res. 2017, 61, 1500901. [Google Scholar] [CrossRef]
- Zhang, G.Z.; Chen, H.W.; Deng, Y.J.; Liu, M.Q.; Wu, Z.L.; Ma, Z.J.; He, X.G.; Gao, Y.C.; Kang, X.W. BRD4 Inhibition Suppresses Senescence and Apoptosis of Nucleus Pulposus Cells by Inducing Autophagy during Intervertebral Disc Degeneration: An In Vitro and In Vivo Study. Oxid. Med. Cell Longev. 2022, 2022, 9181412. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Zeng, Q.; Wu, J.; Li, D.; Wang, H.; Lu, W.; Jiang, Z.; Xu, G. Caveolin-1 regulates oxidative stress-induced senescence in nucleus pulposus cells primarily via the p53/p21 signaling pathway in vitro. Mol. Med. Rep. 2017, 16, 9521–9527. [Google Scholar] [CrossRef] [PubMed]
- Ottone, O.K.; Kim, C.; Collins, J.A.; Risbud, M.V. The cGAS-STING Pathway Affects Vertebral Bone but Does Not Promote Intervertebral Disc Cell Senescence or Degeneration. Front. Immunol. 2022, 13, 882407. [Google Scholar] [CrossRef]
- Han, M.; Pandey, D. ZMPSTE24 Regulates SARS-CoV-2 Spike Protein-enhanced Expression of Endothelial PAI-1. Am. J. Respir. Cell Mol. Biol. 2021, 65, 300–308. [Google Scholar] [CrossRef] [PubMed]
- Galant, D.; Gaborit, B.; Desgrouas, C.; Abdesselam, I.; Bernard, M.; Levy, N.; Merono, F.; Coirault, C.; Roll, P.; Lagarde, A.; et al. A Heterozygous ZMPSTE24 Mutation Associated with Severe Metabolic Syndrome, Ectopic Fat Accumulation, and Dilated Cardiomyopathy. Cells 2016, 5, 21. [Google Scholar] [CrossRef] [PubMed]
- Navarro, C.L.; Cadinanos, J.; De Sandre-Giovannoli, A.; Bernard, R.; Courrier, S.; Boccaccio, I.; Boyer, A.; Kleijer, W.J.; Wagner, A.; Giuliano, F.; et al. Loss of ZMPSTE24 (FACE-1) causes autosomal recessive restrictive dermopathy and accumulation of Lamin A precursors. Hum. Mol. Genet. 2005, 14, 1503–1513. [Google Scholar] [CrossRef]
- Li, X.; Wu, A.; Han, C.; Chen, C.; Zhou, T.; Zhang, K.; Yang, X.; Chen, Z.; Qin, A.; Tian, H.; et al. Bone marrow-derived mesenchymal stem cells in three-dimensional co-culture attenuate degeneration of nucleus pulposus cells. Aging 2019, 11, 9167–9187. [Google Scholar] [CrossRef]
- Bai, X.D.; Li, X.C.; Chen, J.H.; Guo, Z.M.; Hou, L.S.; Wang, D.L.; He, Q.; Ruan, D.K. Coculture with Partial Digestion Notochordal Cell-Rich Nucleus Pulposus Tissue Activates Degenerative Human Nucleus Pulposus Cells. Tissue Eng. Part A 2017, 23, 837–846. [Google Scholar] [CrossRef]
- Lei, C.; Colangelo, D.; Patil, P.; Li, V.; Ngo, K.; Wang, D.; Dong, Q.; Yousefzadeh, M.J.; Lin, H.; Lee, J.; et al. Influences of circulatory factors on intervertebral disc aging phenotype. Aging 2020, 12, 12285–12304. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Non-Coding RNAs | Expression in IDD | In Vitro Study Model | In Vivo Study Model | Senescence Markers | Key Findings | Ref. |
|---|---|---|---|---|---|---|
| circERCC2 | ↓ | Human IDD tissue and NP cells + TBHP siRNAs for miR-182-5p and SIRT1 | Rat tail puncture model of IDD, intraperitoneal injection of circERCC2 vectors | SA-β-gal staining | circERCC2 targets miR-182-5p/SIRT1 signaling axis to regulate mitophagy and apoptosis, and reduces senescence to alleviate IDD | [131] |
| miR-182-5p | ↑ | |||||
| LncRNA H19 | ↑ | Human NP cells +H2O2 | - | SA-β-gal staining, Telomerase activity | Regulation of miR-22 expression by H19 to upregulate the Wnt/β-catenin pathway and promote CS and degenerative changes | [118] |
| LncRNA TRPC7-AS1 | ↑ | Human disc tissues and NP cells from IDD patients, TRPC7-AS1 siRNA, miR-4769−5p mimics and inhibitor, Hepsin overexpression | - | SA-β-gal staining, p21 and p16 expression | lncRNA TRPC7-AS1 inhibits miR-4769-5p which targets Hepsin to upregulate CS, and regulates NP cell viability and ECM synthesis | [133] |
| miR-4769-5p | ↓ | |||||
| LncRNA NORAD | ↓ | Human disc tissues and NP cells + TNF-α stimulation | NORAD knockout mice | SA-β-gal staining, p53, p21 and p16 expression | WTAP mediated m6A modification of the lncRNA NORAD leading to degradation of E2F3 transcript to promote NP cell senescence | [139] |
| LncRNA HOTAIR | ↑ | Human disc tissues and NP cells, HOTAIR overexpression | Rat tail puncture model of IDD, intradiscal injection of HOTAIR siRNA | SA-β-gal staining, p53, p21 and p16 expression | AMPK/mTOR/ULK1 pathway mediated autophagy activation to promote CS and apoptosis | [138] |
| miR-130b-3p | ↑ | Human disc tissues and NP cells + TBHP stimulation | Rat tail puncture model of IDD, intradiscal injection of mir-130b-3p inhibitor | SA-β-gal staining, p16 expression | Inhibition of autophagy by miR-130b-3p via ATG14 and AMPK to promote CS miR-130b-3p inhibition alleviates IDD in rat | [43] |
| miR-217 | ↓ | Human disc tissues and NP cells + TNF-α stimulation MSC-EVs | Rat tail puncture model of IDD, tail vein injection of MSC-EV | SA-β-gal staining, p16 expression | miR-217 targets EZH2 and elevates FOXO3 to activate autophagy and inhibits senescence, apoptosis, and degradation of ECM in NP cells, MSC-EVs carrying miR-217 inhibit IDD in vivo. | [137] |
| Senotherapy | In Vivo Study Model | In Vitro Study Model | Key Findings | Ref. |
|---|---|---|---|---|
| Senolytics | ||||
| Dasatinib+ Quercitin | C57BL/6 mice aged 6, 14, 18 and 23 months, Weekly i.p. injection of vehicle or 5 mg/kg Dasatinib + 50 mg/kg Quercetin up until 23 months. | - | Inhibition of disc p16, p19, p21 Prevention of age-associated systemic increase in pro-inflammatory mediators and Th-17 related proteins Reduction in disc ECM degradation and NP fibrosis | [161] |
| Dasatinib+ Quercitin | Ercc1−/Δ mice, Weekly oral treatment with 5 mg/kg Dasatinib and 50 mg/kg Quercetin or vehicle up to 10–12 weeks. | - | Reduction in physical signs of aging Increased glycosaminoglycan expression in NP tissue of male but not of female mice | [160] |
| Quercitin | Puncture-induced rat IDD model, Intragastric administration of vehicle (every day) or 100 mg/kg of Quercetin (every other day) for 4 weeks | human NP cells stimulated with 0, 10, and 20 μM IL-1β | Induction of Nrf2 expression leading to inhibition of NF-κB pathway | [52] |
| Genetic interventions | ||||
| p16INK4a | Young (6 months) and old (22 months) C57BL/6 mice, p16-3MR transgenic mice ± Ganciclovir | - | Increased level of disc p53, p21 and p16Ink4a in old mice, Clearance of SnCs by glaciclovir leading to improved disc health | [1] |
| p16INK4a | Cdkn2a (p16) knockout mice—mouse tail suspension-induced IVDD model | Human NP cells stimulated with 10 mg/mL IL-1β, 50 nM rapamycin, and p16 siRNA transfection | Reduction in NF-κB activation, ROS levels, SASPs in p16 deficient models Inhibition of p16 by rapamycin | [41] |
| p16INK4a | INK-ATTAC transgenic BubR1H/H mouse model | - | Reduction in p16Ink4a-positive SnCs via FKBP-Casp8 activation Prevention of adipose tissue and muscle loss Delay of age-related phenotypes | [156] |
| Small biologic drugs inhibiting disc CS | ||||
| Curcumin and o-vanillin | - | human NP cells cultured with 5 μM curcumin, 100 μM o-Vanillin, or vehicle (DMSO) for 1 h and 6 h | Reduction in Nrf2 expression via inhibition of NF- κB pathway Decreased expression of p16INK4a | [166] |
| o-vanillin | - | human NP cells treated with 100 μM o-vanillin or vehicle 0.01% DMSO for 4 days then cultured for 21 days | Reduction in TLR-2 expression Reduction in p16, IL-1β, IL-8, NGF, IL-6, and TNF-α expression | [59] |
| RG-7112 and o-Vanillin | - | human NP cells treated with DMSO vehicle, 100 μM o-Vanillin, or 5 μM RG-7112 for 6 h | Decrease of pro-inflammatory factors INF-γ, IL-6, IL-8, CCL24, and cytokines Reduction in SnCs | [167] |
| Morroniside | C57BL/6 mice aged 8 weeks Weekly i.p. injection of 20 and 100 mg/kg morroniside up to 8 weeks | rat NP cells treated with 200 and 400 μM morroniside for 2 h before 200 μM H2O2 exposure | Inhibition of ROS-Hippo-p53 and p21 Reduction in p53 and p21 expression | [162] |
| Honokiol | Puncture-induced rat IDD model, Oral administration of 40 mg/kg honokiol or 0.5% CMC-Na for 1 week | rat NP cells treated with 0.1–20 μM honokiol for 24 h | Activation of SIRT3 leading to suppression of apoptosis via AMPK-PGC-1α-SIRT3 pathway | [45] |
| Resveratrol | - | rat NP cells treated with 20 μM resveratrol prior to 100 μM H2O2 exposure | Activation of SIRT1 via Akt-FoxO1-SIRT1 pathway Reduction in pro-inflammatory cytokines TNF-α, IL-1β, IL-6, IL-8 Reduction in p53, p16, p21 | [101] |
| Metformin | Acupuncture-induced rat IVDD model, i.p. injection of 50 mg/kg metformin for 4 weeks | rat NP cells treated with 10, 50, 100, and 200 μM metformin for 24 h prior to 100 μM TBHP | Inhibition of p16INK4a, p53, and p21 via autophagy activation and cGAS-STING-NF- κB pathway inactivation | [168] |
| Rapamycin | - | rabbit AF cells treated with 50 μg/mL bleomycin + 25 nM rapamycin or 50 μg/mL bleomycin + 50 nM rapamycin for 6 days | Reduction in p16 and p21 expression with rapamycin Reduction in pro-inflammatory factors TNF-α, IL-1β, IL-6, IL-8 | [33] |
| Other interventions | ||||
| Omentin-1 | - | human NP cells treated with 150 or 300 ng/mL omentin-1 and 10 ng/mL IL-1β for 24 h | Prevention of IL-1β-induced senescence via SIRT1 activation Decreased p16 and p53 expression | [169] |
| E2 | - | rat NP cells treated with 10 ng/mL TNF-α + 10−7 M E2 for 24 h and 48 h | Inhibition of ROS/NF- κB activity Increased telomerase activity Reduction in p53 and p16 expression | [170] |
| Parathyroid hormone | - | rat NP cells treated with 1, 25, and 50 μg/mL dexamethasone for prior to 10 nM PTH for 48 h | Autophagy activation via inhibition of mTOR pathway | [171] |
| Spermidine | Natural IDD aging mouse model, Daily oral treatment of 25 mg/kg spermidine | human NP cells treated 50 μM spermidine for 24 h prior to 10 ng/mL IL-1β | Prevention of H2O2 and ROS accumulation via decreased p16 expression | [172] |
| SB203580 | - | rat NP cells cultured at pH 6.2 for 10 days with SB203580 | Increased telomerase activity Reduction in p16 and p53 expression Inhibition of p38 MAPK pathway | [68] |
| Urolithin A | IDD rat model, Daily oral treatment of 25 mg/kg UA for 4 weeks | rat NP cells treated with 80 μM H2O2 + 20 μM UA | Reduction in oxidative stress via activation of SIRT1/PGC-1α pathway Reduction of p16 and p21 expression | [173] |
| NAC | Bmi-1−/− mouse Oral treatment of 1 mg/mL NAC | mouse disc cells treated with 2.5 mmol/L NAC for 7–14 days | Prevention of ECM degradation Reduction in oxidative stress via p16INK4a/Rb and p19/p53 pathway | [174] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Silwal, P.; Nguyen-Thai, A.M.; Mohammad, H.A.; Wang, Y.; Robbins, P.D.; Lee, J.Y.; Vo, N.V. Cellular Senescence in Intervertebral Disc Aging and Degeneration: Molecular Mechanisms and Potential Therapeutic Opportunities. Biomolecules 2023, 13, 686. https://doi.org/10.3390/biom13040686
Silwal P, Nguyen-Thai AM, Mohammad HA, Wang Y, Robbins PD, Lee JY, Vo NV. Cellular Senescence in Intervertebral Disc Aging and Degeneration: Molecular Mechanisms and Potential Therapeutic Opportunities. Biomolecules. 2023; 13(4):686. https://doi.org/10.3390/biom13040686
Chicago/Turabian StyleSilwal, Prashanta, Allison M. Nguyen-Thai, Haneef Ahamed Mohammad, Yanshan Wang, Paul D. Robbins, Joon Y. Lee, and Nam V. Vo. 2023. "Cellular Senescence in Intervertebral Disc Aging and Degeneration: Molecular Mechanisms and Potential Therapeutic Opportunities" Biomolecules 13, no. 4: 686. https://doi.org/10.3390/biom13040686